

Abstract
The type I AAA ATPase Vps4 and its co-factor Vta1p/LIP5 function in membrane remodeling events that accompany cytokinesis, multivesicular body biogenesis, and retrovirus budding, apparently by driving disassembly and recycling of membrane-associated ESCRT-III complexes. Here, we present cryo-EM reconstructions of dodecameric yeast Vps4p complexes with and without their MIT N-terminal domains and Vta1p co-factors. The ATPase domains of Vps4p form a bowl-like structure composed of stacked hexameric rings. The two rings adopt dramatically different conformations, with the “upper” ring forming an open assembly that defines the sides of the bowl and the lower ring forming a closed assembly that forms the bottom of the bowl. The N-terminal MIT domains of the upper ring localize on the symmetry axis above the cavity of the bowl, and the binding of six extended Vta1p monomers causes additional density to appear both above and below the bowl. The structures suggest models in which Vps4p MIT and Vta1p domains engage ESCRT-III substrates above the bowl and help transfer them into the bowl to be pumped through the center of the dodecameric assembly.
Keywords: Vps4, Vta1, AAA ATPase, MVB pathway, HIV budding, Cryo-EM
Introduction
A common set of cellular machinery, the ‘Class E’ vacuolar protein sorting (Vps) proteins, function in vesiculation processes that accompany the final abscission stage of cytokinesis1; 2, retrovirus budding3; 4; 5; 6; 7; 8; 9; 10; 11; 12, and protein sorting at the late endosomal multivesicular body (MVB)13; 14; 15; 16. Most Class E proteins function as subunits of three hetero-oligomeric endosomal sorting complexes required for transport (ESCRT) that are recruited to different membranes to function in vesicle formation. Although their precise functions are not yet fully understood, ESCRT-I and ESCRT-II form stable, discrete complexes that help to concentrate ubiquitylated protein cargoes and recruit ESCRT-III subunits, whereas ESCRT-III (and ESCRT-III-like proteins) co-assemble on membranes and appear to function directly in protein sorting, membrane remodeling, and/or fission events. Finally, the ESCRT machinery is apparently recycled by Vps4, an ATPase that binds ESCRT-III subunits and is required for their release from the membrane17; 18. As the only Class E protein with enzymatic activity, Vps4 imparts directionality to the budding process and may also play a direct role in protein sorting and membrane fission. Vps4 ATPase activity is enhanced by association with a cofactor, Vta1p (called LIP5 in higher eukaryotes).
Vps4 proteins belong to the large family of AAA ATPases (ATPase associated with a variety of cellular activities) that are spread widely throughout all kingdoms of life (reviewed in 19; 20; 21; 22). These enzymes typically assemble into oligomeric rings that use the energy generated by ATP hydrolysis to drive conformational changes that cause macromolecular assemblies to be remodeled. This activity has been adapted for numerous diverse biological functions including vesicle formation; membrane fusion and transport; protein unfolding, disaggregation and refolding; DNA replication, recombination, and repair; transcription; apoptosis; cytoskeletal regulation; and organelle biogenesis. The hallmark of AAA proteins is a 200-250 amino-acid ATP-binding domain named the AAA “domain” or “cassette.”
The two human Vps4 proteins (VPS4A and VPS4B) and the unique Vps4 protein of yeast (Vps4p) all share a common domain organization. The proteins start with a three-helical bundle termed the MIT (microtuble interacting and transport) domain that binds the C-termini of a subset of the ESCRT-III/CHMP proteins23; 24; 25; 26; 27 and is connected to the AAA cassette by what appears to be a semi-flexible linker28. Crystal structures have revealed that the monomeric VPS4B and Vps4p AAA cassettes are composed of two domains, a large mixed α/β domain and a smaller four helix bundle28; 29. These domains resemble other AAA cassettes, except that the four helix bundle of Vps4 is split by the insertion of a small domain composed of a three-stranded beta-sheet (termed the ‘beta domain”) that forms the binding site for the Vta1p/LIP5 cofactor28.
Vps4 proteins contain a single AAA cassette and are therefore classified as type I AAA proteins, whereas family members that contain two AAA cassettes in a single polypeptide, such as p97, VAT, NSF, ClpA and ClpB, are classified as type II AAA proteins. Most type I AAA proteins, including ClpX, HslU and katanin, form a single ring of six protomers 22. Vps4 is unusual, however, in that it assembles into even larger complexes in the presence of ATP. Chemical crosslinking data suggested that the fully assembled Vps4 complex contained 10 subunits 18, whereas gel filtration chromatography and equilibrium analytical ultracentrifugation experiments suggested a complex of 10-12 subunits, with 12 being the best fit 28. It has therefore been proposed that Vps4 may form pentameric or hexameric double ring structures, but structural data has been lacking. In the presence of ATP, Vta1p/LIP5 binds directly to the fully assembled Vps4 complex to form an even larger complex of unknown stoichoimetry, with an estimated molecular weight of ~1 MDa 28. Vta1p/LIP5 enhances the stability and ATPase activity of assembled Vps4 30 and can also interact directly with the Vps60p/CHMP5 subset of ESCRT-III-like proteins 10; 31; 32.
Over the last decade, major progress has been made in structural and functional studies of other AAA proteins that act on protein substrates33, including p97 34; 35; 36; 37; 38; 39; 40, VAT41; 42, NSF 43; 44, ClpB 45; 46; 47, HslU 48; 49; 50, ClpX, and ClpA 51; 52; 53; 54; 55; 56; 57. These studies have revealed that AAA ATPase rings are typically hexameric, but that other arrangements are also possible. For example, crystal structures have revealed that the clamp loader AAA ATPases form open pentameric rings58. Cryo-EM studies of ClpB indicated 6-fold symmetry in all nucleotide states 47, but there are also reports based on sedimentation equilibrium experiments that ClpB is a heptamer in the apo state 59. EM studies of HslU have reported both hexameric and heptameric rings 60, while all available crystal studies suggest hexameric rings 48; 49; 50. The Lon protease was reported to be a seven-membered ring 61 by EM, but hexameric in subsequent biochemical, biophysical, and crystallographic studies 62; 63; 64. EM reconstructions of the apoptosome protein Apaf-1 indicated both hexameric 65 and heptameric66 configurations. RuvB is reported to be a heptamer in the absence of substrate DNA duplex, but is converted to a hexameric ring when it binds short strands of duplex DNA, implying that substrate binding changes its oligomeric state 67. Finally, dynein forms asymmetric single polypeptide rings with seven lobes 68. Thus, the architecture of different AAA ATPases can vary considerably, and must be determined experimentally.
To determine the architecture of Vps4 and to provide a basis for understanding how this complex enzyme functions, we imaged three different Vps4p complexes by electron cryomicroscopy (cryo-EM). The reconstruction of the smallest complex, formed by an N-terminally truncated Vps4p (ΔN-Vps4p) comprising just the AAA ATPase domain, reveals a core structure of two stacked hexameric rings that adopt strikingly different conformations and together form a bowl. While this core is preserved in the larger structures of full-length Vps4p and a Vta1p-Vps4p complex, additional densities reveal the locations of the substrate-binding N-terminal domains and shed light on the interactions of Vps4p with Vta1p.
Results and Discussion
Sample preparation
The S.cerevisiae Vps4p E233Q point mutant was used for all cryo-EM reconstructions because this mutant protein binds but does not hydrolyse ATP, and it therefore forms stable higher-order Vps4p and Vta1p-Vps4p complexes in the presence of ATP18. Purified ΔN-Vps4p, full length Vps4p, and mixtures of Vta1p and Vps4p formed oligomeric complexes in the presence of ATP and MgCl2, as analyzed by size–exclusion chromatography (Fig. 1). Vta1p and Vps4p were previously shown to form a large complex of undetermined stoichiometry28. To investigate the stoichiometry, we mixed purified Vta1p and Vps4p in different ratios, purified the resulting complexes by gel filtration, and analyzed the molar ratios of the two proteins in each fraction using SDS-PAGE and standard curves of known proteins concentrations (Fig. 2). As shown in Fig. 2a, Vta1p and Vps4p formed a single discrete complex when mixed in a 1:2 molar ratio (black curve). The expected 1:2 Vta1p:Vps4p ratio in this purified complex was confirmed by SDS-PAGE and densitometry (Fig. 2b). An equimolar mixture of Vta1p and Vps4p also produced the same 1:2 complex (Fig. 2a, red curve), but in this case there was also a second peak composed of unbound Vta1p (Figs. 2a and 2c). Furthermore, the apparent size of the Vta1p-Vps4p complex did not shift to higher mass, even when the two proteins were mixed in a 3:1 ratio. We therefore conclude that Vta1p and Vps4p form a complex of ~1:2 stoichiometry, even when Vta1p is present in excess.
Fig. 1. Gel filtration analyses of multimeric Vps4p complexes.

Multimeric ΔN-Vps4p, full-length Vps4p and Vta1p-Vps4p complexes were isolated by size – exclusion chromatography (Superose 6) in buffer A (100 mM NaCl, 25 mM Tris–HCl (pH 7.5), 2 mM MgCl2, 1 mM ATP, and 1mM DTT). Elution positions of molecular weight standards are shown below each chromatogram.
Fig. 2. The Vta1p-Vps4p complex exhibits 1:2 stoichiometry.

(a) Size-exclusion chromatography of the complexes formed when Vta1p and Vps4p were mixed at molar ratios of 1:2 (black trace), 1:1 (red trace) and 3:1 (green trace). Samples were chromatographed on a Superdex 200 HR column in the presence of 1 mM ATP. For reference, elution positions of size standards and of free Vps4 and Vta1 proteins are shown above (arrows). (b) and (c) SDS-PAGE analyses (above) and associated protein quantities (below) for fractions spanning the Vps4p-Vta1p complex peaks shown in part (a) for the 1:2 and 1:1 mixtures, respectively.
In our initial attempts to visualize Vps4p complexes using cryo-EM, the samples were simply plunge-frozen and imaged. Unfortunately, the process of plunge-freezing the samples in thin films across EM grids destabilized the native protein complexes, presumably when they came in contact with the air-water interface. Consequently, most regions of the cryo-EM grids displayed clear ice devoid of protein while a few were crowded with what appeared to be protein aggregates, and no intact, regular, large protein complexes were seen (Fig. 3a). To stabilize the protein assemblies for cryo-EM, the complexes were chemically cross-linked with 0.02% glutaraldehyde and repurified by size-exclusion chromatography (Fig. S1a). The elution profiles were only slightly perturbed, indicating that, at least for the peak fractions used for imaging, the crosslinking procedure did not change the oligomeric state or essential structure of the complexes. SDS-PAGE analysis of the three protein complexes showed that the cross-linking was sufficient to covalently link nearly all the subunits of the complexes (Fig. S1b and c). After crosslinking, individual, regular, large protein complexes were routinely observed on the EM grids (Fig. 3b).
Fig. 3. Cryo-EM images of full-length Vps4p.
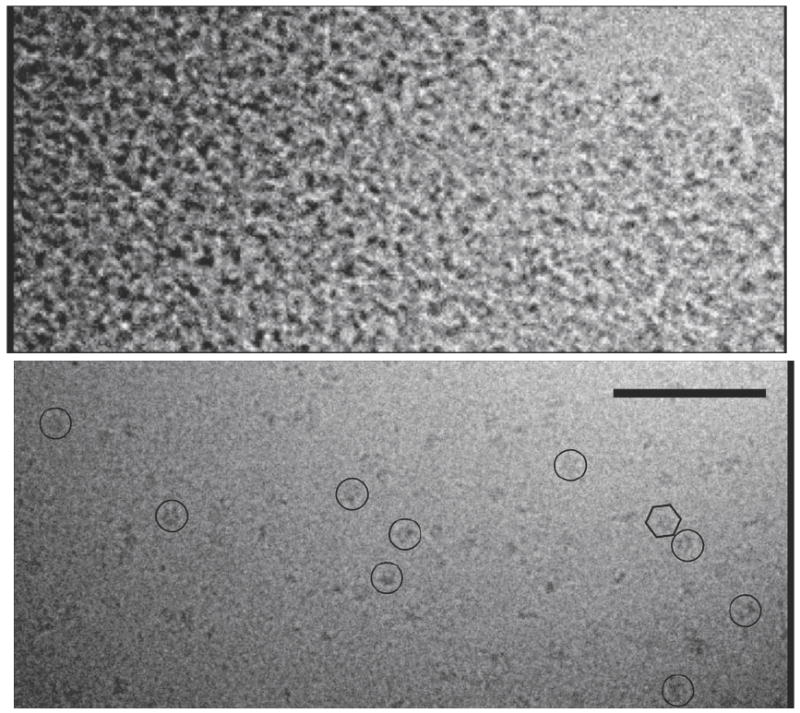
Before crosslinking, most regions of the cryo-grid had clear ice devoid of protein while other regions were filled with what appeared to be protein aggregates (top). After crosslinking (bottom), individual large protein complexes were routinely seen (example particles are circled). The hexagon encloses one complex with visible hexagonal symmetry. Scale bar 100 nm.
The Vps4p oligomer displays six-fold symmetry
The symmetry of the fully assembled Vps4p complex has not been unambiguously determined, and both double ring pentamers (i.e., decamers) 18 and hexamers (i.e., dodecamers) 28 have been proposed. We therefore used three independent methods to look for rotational symmetries in the projection images of the three separate complexes. First, a two-dimensional classification procedure (the EMAN program refine2d.py) was used to separate each set of images into 50 different classes such that related projection images were assigned to the same class 69; 70. Fig. 4 shows the 18 resultant class averages most closely corresponding to “top” views for the three complexes. Six protruding densities can be recognized around the periphery in all 18 class averages of each complex, whereas none of the single class averages indicated five- or seven-fold symmetry.
Fig. 4. Top views reveal 6-fold symmetry.

Select class averages of (top) ΔN-Vps4p, (middle) full-length Vps4p, and (bottom) Vta1p-Vps4p complexes are shown. All the raw images were iteratively classified based on their similarity using singular value decomposition by refine2d.py. Particles within the same class were aligned and averaged to generate 50 class averages. The 18 class averages most closely corresponding to “top” views are shown, all of which suggest 6-fold symmetry.
The second method to determine the symmetry of the complexes used a different EMAN program (startcsym), which deliberately searches for particles that exhibit a particular symmetry. The underlying algorithm simply rotates a particle by 360 / n degrees and cross-correlates it with the original particle. For n = 5, 6, and 7, the 90 most-n-fold-symmetric particles were picked, aligned, and averaged, but without applying any symmetry. The insets (a)-(c) in Fig. 5 show these averages for the full-length Vps4p images. By visual inspection, even the most 5- and 7-fold-like particles produced averages that appeared 6-fold symmetric. For a more quantitative comparison, the rotational power spectrum of each 5-, 6-, and 7-fold-like average was calculated. The only clear peak occurred in the spectra of the 6-fold-like average (data not shown). The same analysis for the most 5-fold-like or the most 7-fold-like particles showed only small signals, with 5- and 6-fold or 7- and 6-fold values displaying approximately equal magnitudes (not shown). Finally the average rotational power spectrum of the combined set of all 270 particles picked for the 5-, 6-, and 7-fold-like averages was calculated, which showed a single strong peak corresponding to 6-fold symmetry (shown for the full-length Vps4p complex in Fig. 5). Similar results were obtained for the ΔN-Vps4p and Vta1p-Vps4p complexes (data not shown).
Fig. 5. Directed symmetry search.
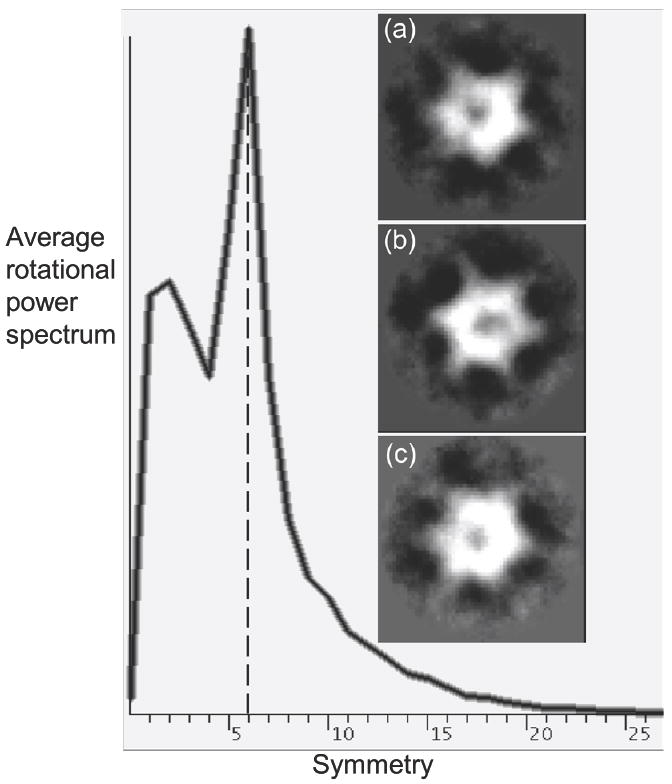
The EMAN program startcsym was used to search the particle gallery to find the images that were most apparently Cn-fold (n=5, 6, 7) symmetric. Three classes containing the 90 most 5- (panel a), 6- (panel b), and 7-fold (panel c) symmetric particles, respectively, were aligned and averaged. Even the most 5- and 7-fold symmetric particles appear 6-fold symmetric. The averaged rotational power spectrum of all 270 particles is also shown with a clear peak indicating 6-fold symmetry. Further statistical analysis by Rotastat also overwhelmingly favored 6-fold symmetry (not shown).
As a third independent method, the program RotaStat 71 was used, which reports the relative likelihood of different symmetries within a set of images in different radial bands. Applying Rotastat to the combined dataset of 270 particles in Fig. 5(a)-(c) showed clearly that 6-fold symmetry was the most significant (the “spectral ratio product” was 33 orders of magnitude higher for C6 symmetry than the next-most-likely symmetry). Similar results were obtained for the ΔN-Vps4p and Vta1p-Vps4p images. These three analyses demonstrate that, like most AAA proteins, Vps4p complexes are six-fold symmetric, and that if any Vps4p particles assembled with either five- or seven-fold symmetry in this analysis, their numbers were negligible.
3-D reconstructions
Three-dimensional reconstructions were generated from the images using standard “single particle” methods and imposing six-fold symmetry (see Fig. S2 for Euler angle distributions and Figs. S3-5 for comparisons of final class averages and corresponding model projections for all three reconstructions). The Fourier shell correlation (FSC) curves are shown in Fig. S6 and the estimated resolutions (FSC = 0.5) are 25 Å, 34 Å and 38 Å for the ΔN-Vps4p, full-length Vps4p and Vta1p-Vps4p complexes, respectively. The N-terminal region of Vps4p contains an MIT domain (residues 1-79) followed by a linker region that is poorly ordered in solution (residues 80-121)26; 28; 29. This region of the protein is therefore likely to be at least partially disordered in the reconstruction. The adaptor protein Vta1p consists of three regions, an N-terminal segment that binds Vps60p10; 31; 32, a C-terminal VSL domain that dimerizes and binds Vps4p30, and an intervening linker that is likely to be in a disordered or extended conformation (W. Sundquist, unpublished). Thus the resolutions obtained for the different complexes correlate with the expected degree of disorder, though it is unclear what effect cross-linking may have had on domain flexibility, since intrinsically flexible domains could have been stabilized by cross-linking and their visibility in averages would depend on how uniform their (cross-linked) positions were. FSC analysis requires splitting a dataset into halves and comparing the corresponding half-data-set reconstructions. To check whether the resolutions obtained were limited by particle numbers or heterogeneity, we further split the Vta1p-Vps4p dataset into quarters and carried out four independent reconstructions. These reconstructions (Fig. S7) and their FSC curves show that the heterogeneity of the particles, rather than the total number of images included, is the principle resolution limitation.
Structure of ΔN-Vps4p, full-length Vps4p, and Vta1p-Vps4p complexes
As shown in Fig. 6, the ΔN-Vps4p, full-length Vps4p and Vta1p-Vps4p complexes all display distinctive bowl-like structures with a central cavity in the upper half. The dimensions of the Vps4p assemblies match the double-hexameric ring structures of NSF 44 and ClpB 47 reconstructed by cryo-EM as well as the crystal structure of p97 37. These morphological similarities indicate that, unlike the other single-ring type I AAA ATPases, Vps4p assembles into a stack of two rings like type II AAA ATPases. Taken together with the 6-fold symmetry, Vps4p is thus a dodecamer, and its complex with Vta1p must have a stoichiometry of 6:12. When contoured to enclose their predicted molecular weights, (446, 578 and 824 kDa for the ΔN-Vps4p, full-length Vps4p, and Vta1p-Vps4p complex, respectively), all three complexes appeared as single continuous objects without detached densities. As explained and shown later, the boundaries of the inner cavities of the three complexes at these contours matched remarkably well, especially in the bottom, suggesting that the presence of the N-terminus and Vta1p had little impact on that feature.
Fig. 6. 3-D cryo-EM reconstructions of ΔN-Vps4p, full-length Vps4p and Vta1p-Vps4p complexes.

Isosurfaces are shown, contoured to enclose the full expected molecular weights of the ΔN-Vps4p, full-length Vps4p, and Vta1p-Vps4p complexes. All three complexes form bowl-like structures with a central cavity (asterisk) in the upper half, suggesting that the top and bottom rings are in very different conformations. The presence of the N-terminal MIT domain in full-length Vps4p causes a nipple (arrow) to appear above the cavity as well as six small fin-like densities (arrowheads) around the lower ring. The binding of Vta1p produces additional density mainly over and around the nipple and beneath the bottom ring. Scale bar 50 Å.
The ΔN-Vps4p complex was ~140 Å across at its broadest point and ~80 Å high. Remarkably, the top and bottom rings were in strikingly different conformations: the top ring contained a large central cavity ~50 Å wide (asterisk, Fig. 6) and the bottom ring appeared closed, at least at this contour. To compare relative dimensions and shapes, the proposed VPS4B hexamer model 28, which was based on the crystal structures of a VPS4B monomer and the p97 D1 ring, was placed into the lower half of the ΔN-Vps4p reconstruction (Fig. 7). The Vps4p reconstruction was approximately twice as tall as the model, and the shape of the lower but not the upper ring in the reconstruction was consistent with the model’s “closed” conformation. No further conclusions (including for instance the possible orientation of the atomic model in the lower ring) were justified at this resolution.
Fig. 7. Isosurface comparisons.

(a) Full-length Vps4p contoured at 578 (solid purple) and 446 kDa (dashed purple) superimposed on ΔN-Vps4p contoured at 446 kDa (blue). The additional densities above the bowl and in the fins persist at both contours, indicating they are significant. (b) Vta1p-Vps4p complex contoured at 824 kDa (solid yellow line) and 578 kDa (innermost dashed yellow line) superimposed over the full-length Vps4p at 578 kDa (purple). Compared with the full-length Vps4p, the Vta1p-Vps4p complex has significant extra densities around and over the nipple above the top ring and beneath the center of the bottom ring. (c) and (d) Bottom and side view of the previously proposed model of hexameric Vps4B placed into the bottom ring of the full-length Vps4p reconstruction contoured at 578 kDa.
The full-length Vps4p complex had roughly the same dimensions as ΔN-Vps4p. The most obvious difference was that the full-length Vps4p contained an ~35-Å long nipple-like structure (arrow, Fig. 6) above the inner cavity. Compared to the ΔN structure, there were also six extra fin-like densities (arrowhead, Fig. 6) protruding from the bottom ring. The widest diameter of the bottom ring (fin tip to fin tip) in the full-length Vps4p complex was ~145 Å.
The Vta1p-Vps4p complex exhibited an inner cavity that matched that of the full-length Vps4p complex almost perfectly in both size and shape. Three peripheral features were, however, significantly enlarged: the nipple-like structure grew taller and thicker, the central portion of the bottom ring was thicker, and the fins on the bottom ring were thicker and extended further so that the tip-to-tip distance of the bottom ring was ~165 Å. The combined height of the top ring and the bottom ring (excluding the height of the nipple) was ~95 Å and the height of the nipple was ~55 Å.
Isosurface comparisons
When the ΔN- and full-length Vps4p complexes were contoured to enclose their predicted molecular weights, aligned, and superimposed, the additional densities were clearly seen to be in the nipple (Fig. 7a). An important control in analyses of sub-complexes and decorated structures is, however, a comparison of the more massive complex contoured so conservatively that it only encloses the volume expected for the less massive sub-complex. New features in a “decorated” structure that persist even at the unrealistically conservative contour of the “core” structure are more likely to be significant than features that shrink back gradually to match the original surface of the core. When the full-length structure was contoured more conservatively to enclose only the expected volume of the ΔN-Vps4p structure, both the nipple and the fins persisted, though decreased in size. Importantly, the thickness of the bottom of the “bowl” was very nearly the same, indicating that steep gradients exist there in the 3-D reconstructions and that these boundaries are therefore useful landmarks for aligning the two structures.
Similarly, two different contours of the Vta1p-Vps4p complex reconstruction were considered in detail (Fig. 7b) with one contour enclosing the full predicted molecular weight (824 kDa) and the other contour being extremely conservative, corresponding to the molecular weight of just the full-length Vps4p complex (578 kDa). Compared with full-length Vps4p, the Vta1p-Vps4p complex had significant extra densities around and over the nipple above the top ring, and beneath the center of the bottom ring. These extra densities diminished but did not disappear even when the Vta1p-Vps4p complex was contoured at the exceedingly conservative level of 578 kDa.
Difference maps
Difference maps were also inspected. First, the ΔN-Vps4p, full-length Vps4p, and Vta1p-Vps4p maps were each normalized to have a mean of zero and a standard deviation of one. Three difference maps were then obtained by subtracting ΔN-Vps4p from the full-length Vps4p, full-length Vps4p from the Vta1p-Vps4p, and ΔN-Vps4p from the Vta1p-Vps4p complex (Fig. 8). As a control, the “internal” differences between the two half-maps of each reconstruction were also calculated (Fig. S8). Inspection of the difference map histograms (Fig. S9) suggested that the positive differences beyond ~+3.5σ did in fact reflect real new protein densities. Strong positive differences between the full-length and ΔN-Vps4p maps were seen in the location of the nipple, the bottom-ring fins, above the top-ring shoulders, and high above the nipple disconnected from any other density (Fig. 8a). Of these, the appearance of the nipple is most likely to represent real new ordered protein, since it was large, continuous with the main body of the complex, enclosed by all reasonable isosurfaces for the full-length complex, and there was no comparable noise in that location in the “control” maps (Fig. S8). As a further internal control, there were a few voxels with negative differences at the (-)3.5 σ significance level (not shown), but none were enclosed by any reasonable isosurface of either the ΔN or full-length Vps4p complex.
Fig. 8. Difference maps.

(a) Full-length Vps4p minus ΔN-Vps4p, (b) Vta1p-Vps4p minus full-length Vps4p, and (c) Vta1p-Vps4p minus ΔN-Vps4p maps contoured at 3.5σ above the mean, superimposed on the 3-D reconstructions of the corresponding subcomplex. The largest difference densities are colored in purple (a) and yellow in (b) and (c). Additional smaller (shaded purple) or disconnected (gray) difference densities are also seen.
The differences between the Vta1p-Vps4p complex and the full-length Vps4p complex were simpler (Fig. 8b). Only two major densities appeared above +3.5σ, one above and around the nipple and the other beneath the bottom ring. Both were likely to be real and significant because they were connected to the main body of the complex, enclosed by all reasonable isosurfaces of the Vta1p-Vps4p complex, and there was no comparable noise in the control maps. The small extra densities on the fins and shoulders predicted by the isosurface comparisons (i.e. Fig. 7b) were missing in the difference maps at this significance level (+3.5σ). Indeed they did not appear until the significance level was dropped to +1.5σ (data not shown), at which point so many other voxels were also enclosed that the map was judged to be noise. As the internal control, no densities were seen below -3.5σ.
The differences between the Vta1p-Vps4p complex and the ΔN-Vps4p complex (Fig. 8c) resemble the differences between the Vta1p-Vps4p complex and the full-length Vps4p complex (Fig 8b), with strong nipple-like density above the cup in the top ring and strong density underneath the center of the bottom ring. The size of the extra nipple density in Fig. 8c is like the sum of extra nipple densities in Fig. 8a and b, as expected. Fig 8c also serves as an independent check on which differences in Fig. 8a are most likely to be meaningful: in principle, the differences between Fig. 8c and Fig. 8b should be the same as those shown in Fig. 8a. In both cases the emergence of the nipple was consistent, but the smaller differences did not match and are therefore less reliable.
Comparison with other AAA ATPases
The oligomeric state of most type I (e.g., katanin, ClpX and HslU) and type II (e.g., p97, NSF, ClpA and ClpB) AAA ATPases contains six subunits, with type I hexamers forming a single ring and type II hexamers adopting a double-ring structure in which each of the two AAA domains forms a separate ring. Fig. 9 shows published cryo-EM reconstructions of ClpB 47, NSF 44, p97-p47 39, and p9735 together with our full-length Vps4p complex. (Note we have reversed the original assignment of rings in the Wilson-Kubalek p97 structure to make it consistent with the others 35; 36; 39.) The five complexes were presumably all in the ATP or its analogue AMP-PNP binding state, although it is unclear which if any of the ATPs have already been hydrolyzed in the stable hexameric complexes 38. Note that the p97-p47 complex in Fig. 9 is composed of a p97 hexamer and a p47 trimer, and the p47 trimer is suggested to be the plug-like density located around the symmetry axis over the top ring 39. Similarly, the NSF structure also contains the NSF adaptor protein α-SNAP and substrate SNARE, which are all attributed to be the big bump-like density around the central axis over the top ring 44. In the ClpB structure, the long spokes extending sideways out of the top ring are believed to be the coiled-coil M domain, unique to ClpB and absent in other AAA proteins 45. The clear morphological similarities between Vps4p and the type II AAA ATPases constitute compelling evidence that the Vps4p complex is also a double ring.
Fig. 9. Comparison of full-length Vps4p with cryo-EM reconstructions of type II AAA proteins.

Full-length Vps4p, ClpB (EMDB ID 1243), NSF-αSNAP-SNARE (EMDB ID 1059), p97-p47 (EMDB ID 1191) and p97 (from E. M. Wilson-Kubalek) maps, all in presumably ATP-bound states, contoured at their full estimated molecular weights. The D2 rings of the type II AAA proteins face the viewer in the top row and comprise the lower rings in the bottom row.
As seen for Vps4p, the three type II AAA ATPases shown in Fig. 9 display very different conformations for their top and bottom rings. ClpB most closely resembles Vps4p with the bottom ring closed and the top ring wide open with a large cavity. Because the top and bottom rings of type II AAA ATPases are formed by two different AAA domains, some structural differences between the rings is to be expected. Indeed, all of the structures for type II AAA hexamers determined by either cryo-EM, X-ray crystallography, or SAXS 34; 35; 36; 37; 38; 44; 45; 57; 72; 73 confirm that the two rings are structurally different, though the extent of this difference varies. In contrast, the two rings of the Vps4p complex are formed by exactly the same protein protomer, so the pronounced difference in the two Vps4p rings is noteworthy. It is possible, however, that the different conformations of the two rings reflect that the two rings are in different nucleotide conditions, despite the presence of a 1000-fold molar excess of ATP-γS during the crosslinking step.
N-terminal domain
The N-terminal domain(s) that precede AAA domain(s) are usually responsible for substrate recognition and binding 74. The Vps4 N-terminus consists of a flexible linker and an MIT domain that is dispensable for ATP-dependent oligomerization and ATP hydrolysis in vitro. The MIT domain is required for recruitment of Vps4p to endosomal membranes 18 and it forms a three α-helix bundle that interacts directly with the C-terminal helices found on a subset of ESCRT-III proteins 25 26; 27. While the common bowl-like “core” structure was seen in both the ΔN- and full-length Vps4p reconstructions, the presence of the N-terminal domains in the full-length Vps4p structure caused a prominent nipple-like density to appear above the top ring on the symmetry axis. We interpret this new density to consist mainly of the MIT domains of the top ring, which would be positioned to bind ESCRT-III substrates directly above the cavity.
The six N-terminal domains of double-ring type II AAA ATPases are flexible and typically do not show up in averaged cryo-EM reconstructions. In those cases where the N-terminal domains have been immobilized, however, they were located above or around the D1 (top) ring. For example, the NSF N-terminal domain was suggested to lie above the D1 ring in a cryo-EM reconstruction 44 and the N-terminal domain of the P97 hexamer was found both above the D1 ring 36 and around the D1 ring 35; 37 in cryo-EM and crystal structures. Similarly, cryo-EM reconstructions of ClpB found that its N-terminal domain was flexible, but nevertheless six small protrusions around the central pore above the D1 ring were suggested to be the beginning of the flexible linker connecting the N-terminal domain to the D1 ring47. Thus, our assignment of the nipple-like density as the N-terminal domain over the top ring in the full-length Vps4p is consistent with these earlier reports. It is possible, however, that the flexible N-domains diffusing around the symmetry axis were crosslinked together, giving them an unnaturally well-ordered appearance. At 3.5σ, the molecular weight of the nipple-like density in Fig 8(a) was estimated to be ~45 kDa, corresponding to ~68% of the mass of six N-terminal domains (each N-domain is ~11 kDa).
Vta1p
The Vta1p cofactor interacts directly with Vps60p and with Vps4p, promoting Vps4p oligomerization and stimulating ATP hydrolysis 10; 30; 31; 32; 75. The amino acid sequence of Vta1p and recent NMR experiments indicate that purified Vta1p is highly extended, with ordered N- and C-terminal regions joined by a long flexible linker (W. Sundquist, unpublished data). The C-terminal VSL domain (40 residues) forms a dimer of helical hairpins30 (W. Sundquist, unpublished data), and this domain alone is necessary and sufficient for Vps4p binding, although it does not stimulate ATP hydrolysis as well as full-length Vta1p 30, indicating that other Vta1p regions likely also contact Vps4p. Mutations in Vps4p β domain inhibit Vta1p binding28, suggesting that the VSL dimer may contact the Vps4p β domain.
Unfortunately the resolution of our Vta1p-Vps4p reconstruction was not sufficient to locate or identify specific domains of Vta1p, but it did show that as when alone, in complex with Vps4p the six Vta1p’s are also not highly-ordered, globular proteins because new globular densities large enough to account for the entire mass of Vta1p were not observed. Instead, addition of Vta1p increased the height and width of the nipple-like density above the bowl and added strong new density below the bottom ring ~85 Å away. At 3.5σ, the additional molecular weight of the nipple-like density in Fig 7(b) was estimated to be ~40 kDa, which accounts for only 32% of the mass of six Vta1p’s (each Vta1p is ~41 kDa). One possibility is that the C-terminal VSL domains account for some of the extra density seen below and around the bottom ring of the Vta1p-Vps4p complex, near where the β domains of the bottom ring are expected (Fig. 7), where they might help stabilize the complex and stimulate ATP hydrolysis (see discussion of the role of the two rings below). The N-termini of Vta1p, which are known to interact with the ESCRT-III-like protein Vps60p, might then account for the additional density above the bowl, in a position to help the Vps4p MIT domains recruit and direct substrate into the central cavity. In any case, our finding that there are only 6 Vta1p subunits in the Vta1p-Vps4p complex is consistent with the notion that the two Vps4p rings are in different conformations, and that only half the Vps4p molecules present the correct interface to bind Vta1p. The three symmetric Vta1p dimers expected could not, however, conform strictly to the six-fold symmetry seen at this resolution, which might have obscured the relatively small contribution of their ordered domains. Higher resolution structures will clearly be needed to resolve these ambiguities.
Implications for mechanism
The two rings in type II AAA ATPases may play different functional roles. While the D1 ring (top ring in Fig. 9) of NSF is the main site of ATP hydrolysis, the D2 ring (bottom ring in Fig. 9) exhibits little hydrolysis activity 76; 77. It has therefore been suggested that the D2 ring plays a mainly structural role by holding the complex together and the D1 ring disassembles SNARE complexes through ATP-hydrolysis-driven conformational changes (for a review see Whiteheart et al 78). Interestingly, the roles of the two rings in p97 appear to be reversed with respect to NSF 37. In the various cryo-EM reconstructions of ClpB throughout the ATP hydrolysis cycle, the top (D1) ring undergoes significant conformational changes while the bottom (D2) ring stays relatively uniform47. The roles of the two rings in Vps4p are still unknown, but the asymmetric nature of the two rings shared by Vps4p and the type II AAA proteins suggests that their mechanisms may be similar. The ClpA, ClpB, and ClpX rings unfold protein substrates by translocating them through the central pore of the hexamer 46; 51; 52; 53; 55; 79. Similarly, key residues around the core of the p97 D2 hexameric ring blocked activity40, and alanine substitution mutations in the pore I residues predicted to reside in the central channel of Vps4 impair HIV budding in vivo 28. It is therefore likely that Vps4 also disassembles ESCRT III assemblies by feeding them through the central cavity of the lower ring.
As noted above, a plausible model for the bottom ring of Vps4 has been proposed based on comparisons with the structurally characterized D1 ring of p9728. In contrast, the conformation of the upper ring must be quite different. Comparison of upper and lower rings gives the impression of large scale domain/subunit reorientations, as have been seen previously for the protein rings in the group I chaperonin, GroEL, where the apical domains of each identical subunit twist by 90° from the open, substrate-binding state to the closed, substrate-folding state 80; 81; 82;83. Although the details remain to be determined, the open conformation of the upper ring of Vps4 suggests that this ring may function to create a large central chamber that can envelope ESCRT-III subunits in the course of removing them from their membrane-bound lattice.
Materials and Methods
Protein expression and purification
Full-length S.cerevisiae Vps4p E233Q, ΔN-Vps4p (residues 104-437) E233Q, and Vta1p were expressed and purified as described 28. Briefly, proteins were expressed from a pET151 vector (Invitrogen) and autoinduced in BL21 Codon + E. coli cells in ZY media (37°C for 6 h and 21°C overnight). All three proteins were purified by Ni2+ sepharose, Q-sepharose, and Superdex200 column chromatography. The histidine tag was removed by incubation with TEV protease (~1mg/100mg protein, 20 h, at 4°C), and the mixture of cleaved and uncleaved Vps4p or Vta1p proteins was separated by Ni2+ sepharose chromatography. The cleaved protein was collected in the flow through, dialyzed into 25 mM Tris pH.4, 100 mM NaCl, 1 mM DTT, concentrated and used directly for crosslinking.
Analysis of the Vta1p-Vps4p Complex
Vps4p and Vta1p were mixed at molar ratios of 2:1, 1:1 and 1:3 in the presence of ATP and the resulting complexes were isolated by size-exclusion chromatography (as described above). Fractions corresponding to the Vta1p-Vps4p and free Vta1p complexes were subjected to SDS-PAGE, and the component proteins were visualized by Coomassie Blue staining and their band intensities were quantified and analyzed by densitometry on an Odyssey fluorescence scanner (Li-Cor, Lincoln, NE) using the Odyssey scanning software. Absolute protein concentrations were determined by comparing band intensities with standard curves made from known concentrations of pure Vps4p and Vta1p.
Cross-linking
Oligomeric Vps4p and Vta1p-Vps4p complexes were isolated by size – exclusion chromatography (Superdex-200HR) in buffer A (100 mM NaCl, 25 mM Tris–HCl pH 7.5, 2 mM MgCl2, 1 mM ATP, and 1mM DTT) and crosslinked with 0.02 % glutaraldehyde at a protein concentration of 20 μg/ml in 0.1 M potassium acetate, 2 mM magnesium acetate, 20 mM HEPES pH 7.4, and 1 mM ATP following the protocol reported for ClpB45; 47. After 20 min incubation at room temperature, the reaction was quenched by addition of an equal volume of 1 M glycine. The cross-linked complexes were concentrated and re-analyzed by size-exclusion chromatography in buffer A without ATP and MgCl2, and peak fractions were collected for EM analysis.
Cryo EM Data collection
4 μl of Vps4p solutions at a concentration of 0.3 mg/ml were applied to holey-carbon-film-covered copper grids and excess solution was blotted away with filter paper in a 100% relative humidity chamber. Grids were plunged into liquid ethane using a Vitrobot and transferred to a 300 kV FEI G2 Polara transmission electron microscope equipped with a field emission gun and an energy filter. Projection images were recorded on a 2k × 2k CCD under low dose conditions at a nominal magnification of 50,000, which resulted in a pixel size of 4.6 Å on the CCD. The images were taken with a range of defocuses from 2.5 to 5.5 μm underfocus.
Image processing and 3D reconstruction
Particle selection, CTF-correction, and 3-D reconstruction was done using the EMAN software suite69. First, the power spectrum of each digital image was calculated and only those images without drift were used for further analysis. Particles were picked manually using boxer. 9593, 4190 and 4445 particles were picked for ΔN-Vps4p, full-length Vps4p and Vta1-Vps4p complexes, respectively. The defocus of each image and the corresponding contrast transfer function were determined using ctfit. Phase corrections were applied to all particles. Symmetry analyses were performed as described in the main text.
The first 3-D reconstruction was of the full-length Vps4p complex. A reference-free initial model was generated using startnrclasses and startAny. Specifically, all the projection images were divided into 50 classes based on their similarity and particles within each class were aligned and averaged to generate class averages. Eight representative class averages were chosen to build an initial 3-D model using the cross common line method. Projections of this initial model were calculated to re-classify the particles and generate new class averages and a new 3-D model. This process was repeated iteratively until the model converged. Six-fold symmetry was imposed throughout the reconstruction process. A second reference-free initial model was produced using a different set of class averages and converged to essentially the same result. The first refined full-length Vps4p model was then used as the initial model to reconstruct the ΔN-Vps4p and Vta1p-Vps4p complexes. As controls, multiple reference-free models of the ΔN-Vps4p and Vta1p-Vps4p complexes were also calculated as described above for full-length Vps4p, and an additional model of the full-length Vps4p structure was produced using one of these “ab-initio” models of ΔN-Vps4p as an initial reference. All the various models of ΔN-Vps4p, full-length Vps4p, and Vta1p-Vps4p confirmed that the complexes assembled as two stacked hexameric rings with a large central cavity in the top ring. In the final reconstructions 4775, 1506 and 3497 particles were included for the ΔN-Vps4p, full-length Vps4p and Vta1p-Vps4p complexes, respectively. The contour levels (thresholds) used for surface representations were estimated by assuming 0.81 Da/Å3 (1.35 g/ml). The final ΔN-Vps4p, full-length Vps4p, and Vta1p-Vps4p structures will be deposited in the EMDB.
Supplementary Material
Acknowledgments
This work was supported in part by NIH Grant P50 GM082545 to WIS, CPH, and GJJ, the Beckman Institute at Caltech, and gifts to Caltech from the Ralph M. Parsons Foundation, the Agouron Institute, and the Gordon and Betty Moore Foundation. We thank E. Kubalek for sharing her cryo-EM structure of p97.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–12. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 2.Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. Embo J. 2007;26:4215–27. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 4.Gottwein E, Bodem J, Muller B, Schmechel A, Zentgraf H, Krausslich HG. The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J Virol. 2003;77:9474–85. doi: 10.1128/JVI.77.17.9474-9485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci U S A. 2003;100:12414–9. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shehu-Xhilaga M, Ablan S, Demirov DG, Chen C, Montelaro RC, Freed EO. Late domain-dependent inhibition of equine infectious anemia virus budding. J Virol. 2004;78:724–32. doi: 10.1128/JVI.78.2.724-732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–99. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 8.von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI. The protein network of HIV budding. Cell. 2003;114:701–13. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 9.Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 10.Ward DM, Vaughn MB, Shiflett SL, White PL, Pollock AL, Hill J, Schnegelberger R, Sundquist WI, Kaplan J. The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J Biol Chem. 2005;280:10548–55. doi: 10.1074/jbc.M413734200. [DOI] [PubMed] [Google Scholar]
- 11.Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128:841–52. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 12.Usami Y, Popov S, Gottlinger HG. Potent Rescue of Human Immunodeficiency Virus Type 1 Late Domain Mutants by ALIX/AIP1 that Depends on its CHMP4 Binding Site. J Virol. 2007 doi: 10.1128/JVI.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–23. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- 14.Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 15.Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8:355–68. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- 16.Hurley JH, Emr SD. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct. 2006;35:277–98. doi: 10.1146/annurev.biophys.35.040405.102126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Babst M, Sato TK, Banta LM, Emr SD. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. Embo J. 1997;16:1820–31. doi: 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babst M, Wendland B, Estepa EJ, Emr SD. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. Embo J. 1998;17:2982–93. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 20.Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure--diverse function. Genes Cells. 2001;6:575–97. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 21.Vale RD. AAA proteins. Lords of the ring. J Cell Biol. 2000;150:F13–9. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 23.Yeo SC, Xu L, Ren J, Boulton VJ, Wagle MD, Liu C, Ren G, Wong P, Zahn R, Sasajala P, Yang H, Piper RC, Munn AL. Vps20p and Vta1p interact with Vps4p and function in multivesicular body sorting and endosomal transport in Saccharomyces cerevisiae. J Cell Sci. 2003;116:3957–70. doi: 10.1242/jcs.00751. [DOI] [PubMed] [Google Scholar]
- 24.Reid E, Connell J, Edwards TL, Duley S, Brown SE, Sanderson CM. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum Mol Genet. 2005;14:19–38. doi: 10.1093/hmg/ddi003. [DOI] [PubMed] [Google Scholar]
- 25.Scott A, Gaspar J, Stuchell-Brereton MD, Alam SL, Skalicky JJ, Sundquist WI. Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc Natl Acad Sci U S A. 2005;102:13813–8. doi: 10.1073/pnas.0502165102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obita T, Saksena S, Ghazi-Tabatabai S, Gill DJ, Perisic O, Emr SD, Williams RL. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 2007;449:735–9. doi: 10.1038/nature06171. [DOI] [PubMed] [Google Scholar]
- 27.Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, Sundquist WI. ESCRT-III recognition by VPS4 ATPases. Nature. 2007;449:740–4. doi: 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- 28.Scott A, Chung HY, Gonciarz-Swiatek M, Hill GC, Whitby FG, Gaspar J, Holton JM, Viswanathan R, Ghaffarian S, Hill CP, Sundquist WI. Structural and mechanistic studies of VPS4 proteins. Embo J. 2005;24:3658–69. doi: 10.1038/sj.emboj.7600818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao J, Xia H, Yoshino-Koh K, Zhou J, Xu Z. Structural characterization of the ATPase reaction cycle of endosomal AAA protein Vps4. J Mol Biol. 2007;374:655–70. doi: 10.1016/j.jmb.2007.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azmi I, Davies B, Dimaano C, Payne J, Eckert D, Babst M, Katzmann DJ. Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J Cell Biol. 2006;172:705–17. doi: 10.1083/jcb.200508166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowers K, Lottridge J, Helliwell SB, Goldthwaite LM, Luzio JP, Stevens TH. Protein-protein interactions of ESCRT complexes in the yeast Saccharomyces cerevisiae. Traffic. 2004;5:194–210. doi: 10.1111/j.1600-0854.2004.00169.x. [DOI] [PubMed] [Google Scholar]
- 32.Shiflett SL, Ward DM, Huynh D, Vaughn MB, Simmons JC, Kaplan J. Characterization of Vta1p, a class E Vps protein in Saccharomyces cerevisiae. J Biol Chem. 2004;279:10982–90. doi: 10.1074/jbc.M312669200. [DOI] [PubMed] [Google Scholar]
- 33.Pye VE, Dreveny I, Briggs LC, Sands C, Beuron F, Zhang X, Freemont PS. Going through the motions: the ATPase cycle of p97. J Struct Biol. 2006;156:12–28. doi: 10.1016/j.jsb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS. Structure of the AAA ATPase p97. Mol Cell. 2000;6:1473–84. doi: 10.1016/s1097-2765(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 35.Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson-Kubalek EM. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol. 2002;9:950–7. doi: 10.1038/nsb872. [DOI] [PubMed] [Google Scholar]
- 36.Beuron F, Flynn TC, Ma J, Kondo H, Zhang X, Freemont PS. Motions and negative cooperativity between p97 domains revealed by cryo-electron microscopy and quantised elastic deformational model. J Mol Biol. 2003;327:619–29. doi: 10.1016/s0022-2836(03)00178-5. [DOI] [PubMed] [Google Scholar]
- 37.DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–63. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- 38.DeLaBarre B, Brunger AT. Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol. 2005;347:437–52. doi: 10.1016/j.jmb.2005.01.060. [DOI] [PubMed] [Google Scholar]
- 39.Beuron F, Dreveny I, Yuan X, Pye VE, McKeown C, Briggs LC, Cliff MJ, Kaneko Y, Wallis R, Isaacson RL, Ladbury JE, Matthews SJ, Kondo H, Zhang X, Freemont PS. Conformational changes in the AAA ATPase p97-p47 adaptor complex. Embo J. 2006;25:1967–76. doi: 10.1038/sj.emboj.7601055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell. 2006;22:451–62. doi: 10.1016/j.molcel.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 41.Gerega A, Rockel B, Peters J, Tamura T, Baumeister W, Zwickl P. VAT, the Thermoplasma homolog of mammalian p97/VCP, is an N domain-regulated protein unfoldase. Journal of Biological Chemistry. 2005;280:42856–42862. doi: 10.1074/jbc.M510592200. [DOI] [PubMed] [Google Scholar]
- 42.Rockel B, Jakana J, Chiu W, Baumeister W. Electron cryomicroscopy of VAT, the archaeal p97/CDC48 homologue from Thermoplasma acidophilum. Journal of Molecular Biology. 2002;317:673–681. doi: 10.1006/jmbi.2002.5448. [DOI] [PubMed] [Google Scholar]
- 43.Yu RC, Hanson PI, Jahn R, Brunger AT. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol. 1998;5:803–11. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]
- 44.Furst J, Sutton RB, Chen J, Brunger AT, Grigorieff N. Electron cryomicroscopy structure of N-ethyl maleimide sensitive factor at 11 A resolution. Embo J. 2003;22:4365–74. doi: 10.1093/emboj/cdg420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, Tsai FT. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–40. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 46.Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber-Ban EU, Dougan DA, Tsai FT, Mogk A, Bukau B. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–65. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 47.Lee S, Choi JM, Tsai FT. Visualizing the ATPase cycle in a protein disaggregating machine: structural basis for substrate binding by ClpB. Mol Cell. 2007;25:261–71. doi: 10.1016/j.molcel.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403:800–5. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 49.Sousa MC, Trame CB, Tsuruta H, Wilbanks SM, Reddy VS, McKay DB. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 2000;103:633–43. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Song JJ, Seong IS, Franklin MC, Kamtekar S, Eom SH, Chung CH. Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure. 2001;9:1107–16. doi: 10.1016/s0969-2126(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 51.Weber-Ban EU, Reid BG, Miranker AD, Horwich AL. Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature. 1999;401:90–3. doi: 10.1038/43481. [DOI] [PubMed] [Google Scholar]
- 52.Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell. 2000;5:639–48. doi: 10.1016/s1097-2765(00)80243-9. [DOI] [PubMed] [Google Scholar]
- 53.Singh SK, Grimaud R, Hoskins JR, Wickner S, Maurizi MR. Unfolding and internalization of proteins by the ATP-dependent proteases ClpXP and ClpAP. Proc Natl Acad Sci U S A. 2000;97:8898–903. doi: 10.1073/pnas.97.16.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishikawa T, Beuron F, Kessel M, Wickner S, Maurizi MR, Steven AC. Translocation pathway of protein substrates in ClpAP protease. Proc Natl Acad Sci U S A. 2001;98:4328–33. doi: 10.1073/pnas.081543698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reid BG, Fenton WA, Horwich AL, Weber-Ban EU. ClpA mediates directional translocation of substrate proteins into the ClpP protease. Proc Natl Acad Sci U S A. 2001;98:3768–72. doi: 10.1073/pnas.071043698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo F, Maurizi MR, Esser L, Xia D. Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J Biol Chem. 2002;277:46743–52. doi: 10.1074/jbc.M207796200. [DOI] [PubMed] [Google Scholar]
- 57.Ishikawa T, Maurizi MR, Steven AC. The N-terminal substrate-binding domain of ClpA unfoldase is highly mobile and extends axially from the distal surface of ClpAP protease. J Struct Biol. 2004;146:180–8. doi: 10.1016/j.jsb.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 58.O’Donnell M, Kuriyan J. Clamp loaders and replication initiation. Curr Opin Struct Biol. 2006;16:35–41. doi: 10.1016/j.sbi.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Akoev V, Gogol EP, Barnett ME, Zolkiewski M. Nucleotide-induced switch in oligomerization of the AAA+ ATPase ClpB. Protein Sci. 2004;13:567–74. doi: 10.1110/ps.03422604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rohrwild M, Pfeifer G, Santarius U, Muller SA, Huang HC, Engel A, Baumeister W, Goldberg AL. The ATP-dependent HslVU protease from Escherichia coli is a four-ring structure resembling the proteasome. Nat Struct Biol. 1997;4:133–9. doi: 10.1038/nsb0297-133. [DOI] [PubMed] [Google Scholar]
- 61.Stahlberg H, Kutejova E, Suda K, Wolpensinger B, Lustig A, Schatz G, Engel A, Suzuki CK. Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc Natl Acad Sci U S A. 1999;96:6787–90. doi: 10.1073/pnas.96.12.6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rudyak SG, Brenowitz M, Shrader TE. Mg2+-linked oligomerization modulates the catalytic activity of the Lon (La) protease from Mycobacterium smegmatis. Biochemistry. 2001;40:9317–23. doi: 10.1021/bi0102508. [DOI] [PubMed] [Google Scholar]
- 63.Lee AY, Hsu CH, Wu SH. Functional domains of Brevibacillus thermoruber lon protease for oligomerization and DNA binding: role of N-terminal and sensor and substrate discrimination domains. J Biol Chem. 2004;279:34903–12. doi: 10.1074/jbc.M403562200. [DOI] [PubMed] [Google Scholar]
- 64.Botos I, Melnikov EE, Cherry S, Khalatova AG, Rasulova FS, Tropea JE, Maurizi MR, Rotanova TV, Gustchina A, Wlodawer A. Crystal structure of the AAA+ alpha domain of E. coli Lon protease at 1.9A resolution. J Struct Biol. 2004;146:113–22. doi: 10.1016/j.jsb.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 65.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–32. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 66.Yu X, Acehan D, Menetret JF, Booth CR, Ludtke SJ, Riedl SJ, Shi Y, Wang X, Akey CW. A structure of the human apoptosome at 12.8 A resolution provides insights into this cell death platform. Structure. 2005;13:1725–35. doi: 10.1016/j.str.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 67.Miyata T, Yamada K, Iwasaki H, Shinagawa H, Morikawa K, Mayanagi K. Two different oligomeric states of the RuvB branch migration motor protein as revealed by electron microscopy. J Struct Biol. 2000;131:83–9. doi: 10.1006/jsbi.2000.4290. [DOI] [PubMed] [Google Scholar]
- 68.Samso M, Koonce MP. 25 angstrom resolution structure of a cytoplasmic dynein motor reveals a seven-member planar ring. Journal of Molecular Biology. 2004;340:1059–1072. doi: 10.1016/j.jmb.2004.05.063. [DOI] [PubMed] [Google Scholar]
- 69.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 70.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 71.Kocsis E, Cerritelli ME, Trus BL, Cheng N, Steven AC. Improved methods for determination of rotational symmetries in macromolecules. Ultramicroscopy. 1995;60:219–28. doi: 10.1016/0304-3991(95)00070-2. [DOI] [PubMed] [Google Scholar]
- 72.Huyton T, Pye VE, Briggs LC, Flynn TC, Beuron F, Kondo H, Ma J, Zhang X, Freemont PS. The crystal structure of murine p97/VCP at 3.6A. J Struct Biol. 2003;144:337–48. doi: 10.1016/j.jsb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 73.Davies JM, Tsuruta H, May AP, Weis WI. Conformational changes of p97 during nucleotide hydrolysis determined by small-angle X-Ray scattering. Structure. 2005;13:183–95. doi: 10.1016/j.str.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 74.Dougan DA, Mogk A, Zeth K, Turgay K, Bukau B. AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett. 2002;529:6–10. doi: 10.1016/s0014-5793(02)03179-4. [DOI] [PubMed] [Google Scholar]
- 75.Lottridge JM, Flannery AR, Vincelli JL, Stevens TH. Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc Natl Acad Sci U S A. 2006;103:6202–7. doi: 10.1073/pnas.0601712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126:945–54. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nagiec EE, Bernstein A, Whiteheart SW. Each domain of the N-ethylmaleimide-sensitive fusion protein contributes to its transport activity. J Biol Chem. 1995;270:29182–8. doi: 10.1074/jbc.270.49.29182. [DOI] [PubMed] [Google Scholar]
- 78.Whiteheart SW, Schraw T, Matveeva EA. N-ethylmaleimide sensitive factor (NSF) structure and function. Int Rev Cytol. 2001;207:71–112. doi: 10.1016/s0074-7696(01)07003-6. [DOI] [PubMed] [Google Scholar]
- 79.Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW, Horwich AL. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell. 2005;121:1029–41. doi: 10.1016/j.cell.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 80.Rye HS, Burston SG, Fenton WA, Beechem JM, Xu Z, Sigler PB, Horwich AL. Distinct actions of cis and trans ATP within the double ring of the chaperonin GroEL. Nature. 1997;388:792–8. doi: 10.1038/42047. [DOI] [PubMed] [Google Scholar]
- 81.Xu Z, Horwich AL, Sigler PB. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature. 1997;388:741–50. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 82.Xu Z, Sigler PB. GroEL/GroES: structure and function of a two-stroke folding machine. J Struct Biol. 1998;124:129–41. doi: 10.1006/jsbi.1998.4060. [DOI] [PubMed] [Google Scholar]
- 83.Gutsche I, Essen LO, Baumeister W. Group II chaperonins: new TRiC(k)s and turns of a protein folding machine. J Mol Biol. 1999;293:295–312. doi: 10.1006/jmbi.1999.3008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.