

Abstract
Objective
Cytomegalovirus (CMV) is one of the most common causes of major birth defects in humans. Of the approximately 8400 children born each year in the U.S. with CMV-induced birth defects, more than 1/3 of these children exhibit hypoplasia and hypocalcification of tooth enamel. Our objective was to initiate the investigation of the pathogenesis of CMV-induced tooth defects.
Design
Mouse Cap stage mandibular first molars were infected with mouse CMV (mCMV) in vitro in a chemically-defined organ culture system and analysed utilising histological and immunolocalisation methodologies. The antiviral, acyclovir, was used to inhibit mCMV replication and comparisons made between mCMV-infected and acyclovir-treated, mCMV-infected teeth.
Results
Active infection of Cap stage molars for up to 15 days in vitro results in smaller, developmentally-delayed and dysmorphic molars characterised by shallow, broad and misshapen cusps, infected and affected dental papilla mesenchyme, poorly differentiated odontoblasts and ameloblasts, and no dentin matrix. Initial protein localisation studies suggest that the pathogenesis is mediated through NF-κB signaling and that there appears to be an unusual interaction between abnormal mesenchymal cells and surrounding matrix. Rescue with acyclovir indicates that mCMV replication is necessary to initiate and sustain progressive tooth dysmorphogenesis.
Conclusions
Our results indicate that mCMV-induced changes in signaling pathways severely delays, but does not completely interrupt, tooth morphogenesis. Importantly, our results demonstrate that this well-defined embryonic mouse organ culture system can be utilised to delineate the molecular mechanism underlying the CMV-induced tooth defects that characterise the amelogenesis imperfecta phenocopy seen in many CMV-infected children.
Keywords: Odontogenesis, Cytomegalovirus, Amelogenesis imperfecta, Fibronectin, NF-κB, β-Catenin
1. Introduction
Human clinical studies and mouse models clearly demonstrate that cytomegalovirus (CMV) is dysmorphogenic to organ and tissue development. CMV, an enveloped, double-stranded DNA betaherpesvirus, is species-specific and has a slow replication cycle. In infected newborns, CMV establishes a long-lasting persistence in salivary glands and the virus is shed in saliva for months to years before termination of productive infection and establishment of latency.1
It is established that about 2% of live born infants are congenitally infected with active CMV. About 10% of this group has newborn symptoms, and most of these infants will exhibit subsequent abnormalities of the central nervous system (CNS): microcephaly, mental retardation, deafness and blindness.2,3 Thus, at least 1 in every 500 newborns will exhibit major CMV-induced congenital pathology, making CMV one of the most common causes of major birth defects in humans (http://www.cdc.gov/cmv).2,3
It has been established that about 36% of children with CMV-induced birth defects and 5% of CMV-infected asymptomatic infants also exhibit enamel hypoplasia and hypocalcification of the teeth.4–7 In many cases, the enamel is absent and affected teeth tend to wear down rapidly or to fracture. These distinct defects of amelogenesis are mostly reported in primary teeth. However, given the persistence of active CMV infection in some infants for 6–18 months postnatal, enamel defects may be expected in the permanent dentition as well. It can be estimated that each year in the U.S. there are an additional 3000 children with CMV-induced amelogenesis imperfecta (AI), and that the prevalence to age 12 is more than 30,000. This is a significant problem in that children have incisal and cuspal attrition, as well as rampant dental caries.6 Further, these children may require orthodontic therapy due to caries-induced loss of primary teeth, as well as abnormalities of growth and development of the oral–facial complex secondary to microcephaly and growth retardation.4
Since mouse CMV (mCMV) has many features in common with human CMV (hCMV), the mouse model has been widely employed for studying the pathogenesis associated with acute, latent, and recurrent infections.8 Baskar et al.9–11 have extensively investigated the effects of CMV on embryonic development. They consistently observed substantial foetal loss, foetal growth retardation, and foetal dysmorphogenesis, particularly of the craniofacial complex (brain and branchial arches). Subsequently, Tsutsui12 reported that viral-antigen positive cells were abundant in the mesenchyme of the oral and nasal cavities, and in the mesenchyme of the brain. He postulated that mesenchymal infection is the critical step in disrupting organogenesis. If so, oral–facial organogenesis, which is highly dependent on mesenchymal integrity and epithelial–mesenchymal interactions, would be particularly vulnerable to CMV infection. Indeed, recent studies in our laboratory has demonstrated that first branchial arch derivatives [submandibular salivary gland (SMG)13 and mandible (unpublished)] are vulnerable to CMV infection during critical stages of their organogenesis, and that CMV has a particular tropism for neural-crest-derived ectomesenchyme (EM).
Active mCMV infection of embryonic day 15 (E15) mouse SMGs for up to 12 days in vitro13 results in a remarkable pathology, characterised by significantly smaller SMGs, atypical ductal epithelial hyperplasia, apparent epithelial–mesenchymal transformation, and oncocytic-like stromal cell metaplasia. Expression studies indicate that molecular pathogenesis centers around the activation of the canonical and noncanonical NF-κB pathways. At the cellular level, there appears to be a consequential interplay between the transformed SMG cells and the surrounding extracellular matrix, resulting in significant changes in fibronectin and β-catenin distribution. While much is still unknown regarding CMV-induced embryopathology, these experimental results served as a framework for investigating mCMV-induced AI.
The objective of the present research was to begin to delineate the pathogenesis of CMV-induced tooth defects. Using a chemically-defined organ culture system, we investigated the effect of mCMV infection on mouse Cap stage mandibular first molar morphogenesis and differentiation. We also determined whether mCMV infection induces changes in cell proliferation and components of relevant signal transduction pathways.
2. Materials and methods
2.1. Embryonic culture system and mCMV infection
Female B10A/SnSg mice, obtained from Jackson Laboratories (Bar Harbor, ME), were maintained under standard laboratory conditions and mated as previously described;13,14 plug day = day 0 of gestation. Timed-pregnant females were sacrificed on gestation day 15 (E15) by carbon dioxide narcosis and cervical dislocation. Embryos were dissected in cold phosphate-buffered saline (PBS). All procedures are performed in accordance with the Institutional Animal Care and Use Committee of USC in accordance with the Panel on Euthanasia of the American Veterinary Medical Association. E15 mandibular molar regions were cultured under chemically-defined conditions using a modified Trowell method as previously described for up to 15 days in culture.15,16
2.1.1. mCMV infection
On day 0, E15 tooth organs were incubated with 50,000 or 100,000 plaque-forming units (PFU)/ml of lacZ-tagged mCMV RM427+17 for 24 h and then cultured in virus-free BGJb-defined media for a total of 12 (E15 + 12) and 15 (E15 + 15) days; controls consisted of E15 mandibular molar organs cultured in BGJb-defined media for the entire culture period. Explants were collected and processed for whole mount morphology, routine histology, viral expression, and immunolocalisation as previously described.13 For each experimental protocol, 3–20 tooth organs/treatment/day were analysed for each assay. Since no marked difference was seen in tooth organs infected with 50,000 or 100,000 PFU mCMV, tooth organs were routinely cultured in 100,000 PFU except where noted. For histological analyses, tooth organs were fixed for 4 h in Carnoy's fixative at 4 °C or overnight in 10% neutral buffered formalin at room temperature, embedded in paraffin, serially-sectioned at 8 μm and stained with haematoxylin and eosin as previously described.13
2.2. mCMV analysis
We assayed β-galactosidase (lacZ) activity, and localisation of viral immediate early (IE1) proteins as described in Melnick et al.13
2.2.1. β-Galactosidase (β-gal) staining
Briefly, mCMV-infected E15 + 12 and E15 + 15 tooth organs were processed, stained for 4–6 h and photographed. Whole mounts were then dehydrated through graded alcohols, embedded in paraffin, serially-sectioned at 8 μm and counterstained with eosin. A minimum of three explants per day were analysed.
2.2.2. IE1 distribution
mCMV-infected E15 + 12 and E15 + 15 tooth organs were fixed in Carnoy's fixative, serially-sectioned at 8 μm, and incubated overnight with anti-IE; controls consisted of sections incubated with mouse IgG alone. A minimum of three explants per day were analysed.
2.3. Cell proliferation assay
Cell proliferation in E15 + 12 control and mCMV-infected tooth organs was determined by the cell-specific localisation and quantitation of proliferating cell nuclear antigen (PCNA) using the Zymed mouse PCNA kit (Invitrogen Corp.) and counterstained with haematoxylin as previously described.13 In this experiment, the cytoplasm appears blue and PCNA-positive nuclei appear dark brown. The mesenchymal and epithelial cell proliferation index was determined in mCMV-infected and uninfected (control) tooth organs (n = 3). To quantitate cell proliferation differences, three sections per explant and three explants per group were analysed. Cuspal region per tooth was selected and the proliferation index (PCNA-positive epithelial cells/total epithelial cells/mm2 and PCNA-positive mesenchymal cells/total mesenchymal cells/mm2) in three sections per explant was determined. The mean ratios per explant and mean ratios per group were calculated. The cell proliferation index is presented as the ratio of PCNA-positive cells/total cells. Since percent data are not normally distributed, the data were arcsin transformed before the mean ratios were compared by t-test.
2.4. Antibodies and immunostaining
Immunolocalisation was conducted essentially as previously described13,14,18,19 using the following polyclonal (Pab) antibodies: β-catenin (SC-7199), RelB (SC-30887), NF-κB2(p52) (SC-298)(Santa Cruz Biotechnology, Santa Cruz, CA); FN (F3648, Sigma–Aldrich Corp., St. Louis, MO); RelA(p65) (Ab-435) (ABM Inc., Vancouver, CA). Nuclei were counterstained with DAPI (Invitrogen Corporation, Carlsbad, CA).
2.5. CMV replication and pathology
Acyclovir, a synthetic purine nucleoside analogue, is a highly selective agent for CMV with low toxicity to the host cell.20 Acyclovir sodium (100 mg/20 ml) was purchased from American Pharmaceutical Partners, Inc. (Schaumberg, Il). We employed 10 μg/ml acyclovir, a dose shown in our laboratory to be nonteratogenic to embryonic salivary glands13 and tooth organs in vitro (unpublished). E15 Cap stage mandibular molar regions were infected with 50,000 or 100,000 PFU mCMV for 24 h and then cultured in control medium with or without 10 μg/ml acyclovir (CMV vs. CMV + Acy) for a total of 12 or 15 days in culture; controls consisted of E15 mandibular molar regions cultured in control medium for 24 h and then with or without 10 μg/ml acyclovir (CONT vs. Acy) for a total of 12 or 15 days. Tooth organs were collected and analysed for whole mount morphology, mCMV infection (β-gal staining), and histopathology. For each assay, 3–5 tooth organs/group/day were analysed.
3. Results
To determine the affect of mCMV infection on embryonic mouse tooth development, we cultured embryonic Cap stage (E15) mandibular first molars (Fig. 1A, insert) with lacZ-labelled mCMV13 or control medium for up to 15 days in vitro using a chemically-defined organ culture system.13,15,16,18 Exposure of embryonic mouse teeth to mCMV in vitro substantially disrupted tooth morphogenesis and differentiation.
Fig. 1.
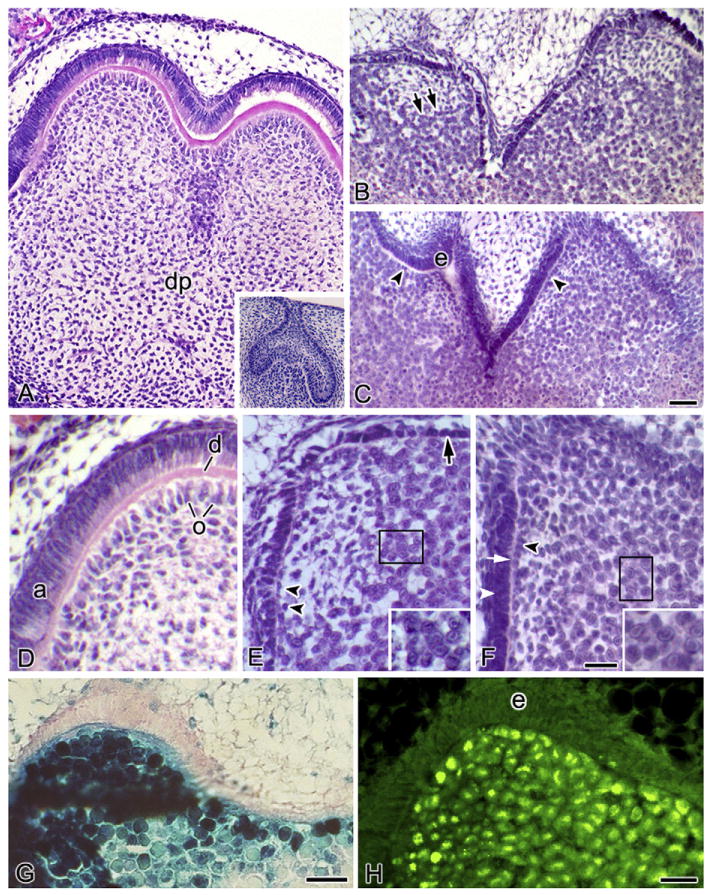
mCMV-induced histopathology and viral distribution. (A–F) Histological analysis of control (A and D) and mCMV-infected (B and C, E and F) E15 Cap stage first mandibular molars cultured in serumless, chemically-defined medium. (A and D) After 12 days in culture, control molars have undergone cuspal morphogenesis and reached the Bell stage, with polarised odontoblast (o) and ameloblasts (a), as well as dentin matrix (d), being seen. dp, dental papilla. (Insert A) Starting E15 Cap stage mandibular first molar. (B and E) After 12 days in culture, the mCMV-infected molars are markedly smaller, developmentally-delayed, and severely abnormal, having only achieving the Early Bell stage. mCMV infection results in shallow, broad and misshapen cusps composed of abnormally short, undifferentiated dental epithelium (arrow), disorganised undifferentiated odontoblasts (double arrowheads), and no dentin matrix. The dental papilla is composed of clusters of large basophilic, pleiomorphic infected [with viral inclusion bodies (insert)] and affected cells being seen (double arrows). (C and F) After 15 days of infection, the smaller, underdeveloped and abnormal tooth phenotype persists. The presence of aligned (but not polarised) preodontoblasts (arrowhead), secreted predentin matrix (white arrow), and elongated (but not polarised) epithelia (white arrowhead) indicates developmental advancement. (Insert F) Viral inclusion bodies. (G and H) mCMV distribution in E15 + 12 tooth organs. Expression of β-galactosidase staining of lacZ (mCMV) (G) and viral IE1 protein (H) demonstrates the presence of viral infection throughout dental papilla EM cells but not in dental epithelia (e). Bar (A–C), 30 μm; (A insert), 60 μm; (D–F, H), 20 μm; (E and F insert), 12 μm; (G), 50 μm.
3.1. Histopathology and viral expression
After 12 days in culture, the uninfected (control) E15 mandibular first molars have undergone cuspal morphogenesis and reached the Bell stage. At this stage, the odontoblasts (dentin forming cells) have differentiated from the neural-crest-derived ectomesenchyme and have secreted dentin matrix and the ameloblasts (enamel forming cells) have differentiated from the inner dental epithelium (Fig. 1A and D). With mCMV infection, E15 + 12 mandibular first molars are severely dysmorphic, smaller and developmentally-delayed compared to controls, having only reached the Early Bell stage (compare Fig. 1B to Fig. 1A). The mCMV-infected molars are characterised by shallow, broad and misshapen cusps, abnormally-short dental epithelia, undifferentiated odontoblasts, and the absence of dentin matrix (Fig. 1 B and E). The major cytological differences between mCMV-infected and control molars are seen in the dental papilla, with disorganised odontoblasts, stromal hypercellularity and altered mesenchymal cytology being seen (compare Fig. 1B and E to Fig. 1A and D). Clusters of large basophilic, pleiomorphic cells (some with inclusion bodies) are found in the dental papilla, as well as in the peripherally-localised EM surrounding the tooth germ (i.e., the presumptive dental sac).
After 15 days in culture, developmental delay and cuspal dysmorphogenesis persist in mCMV-infected mandibular first molars (Fig. 1C and F). Although an additional 3 days in culture is not sufficient to reach the Bell stage phenotype seen in control E15 + 12 molars (compare Fig. 1C and F to Fig. 1A and D), morphogenetic advances are seen between mCMV-infected E15 + 12 and E15 + 15 molars (compare Fig. 1C and F to Fig. 1B and E). Notably, E15 + 15 molars exhibit preodontoblast alignment at the epithelial junction, predentin matrix, and epithelial cells having elongated to become preameloblasts. This more developed phenotype is similar to that seen in Cap stage mandibular first molars cultured for 9 days in control medium (data not shown). Since predentin induces ameloblast differentiation, the presence of predentin matrix likely accounts for improved epithelial morphology.
To determine which cells are infected by mCMV, we analysed the distribution of mCMV in E15 + 12 Cap stage mandibular first molars by the cell-specific localisation of viral inclusion body (Fig. 1E and F), lacZ (β-galactosidase) expression (Fig. 1G) and viral immediate early protein 1 (Fig. 1H). mCMV is found in dental papilla ectomesenchymal cells and is absent from inner dental epithelia.
3.2. mCMV infection alters cell proliferation
mCMV infection of embryonic SMG induced substantial changes in the cell-specific distribution of cell proliferation, from epithelia to stroma.13 To determine whether mCMV infection of Cap stage tooth organs alters cell proliferation, we compared the spatial distribution of PCNA-positive cells (compare Fig. 2A and Fig. 2B), as well as quantitative differences in proliferation index (PCNA-positive cells/total cells/mm2), in mCMV-infected and uninfected (cont) teeth. In controls, PCNA-positive nuclei are detected primarily in the ameloblasts and, to a far lesser extent, in some dental papilla EM (Fig. 2A). Since withdrawal from the cell cycle is a prerequisite for odontoblast terminal differentiation and differentiation progresses from the tip of the cusp downward,21 the absence of PCNA-positive nuclei in odontoblasts clearly indicates that they are terminally differentiated. In contrast, mCMV-infected molars exhibit a very different proliferation pattern. PCNA-positive nuclei are found in infected and affected EM throughout the dental papilla and, to a lesser extent, in the short dental epithelial cells (Fig. 2B). mCMV infection induced a significant 161% increase (P < 0.003) in mesenchymal cell proliferation (PCNA-positive mesenchymal cells/total mesenchymal cells/mm2: CMV 81 ± 13 vs. CONT 31 ± 8.3), as well as a significant 50% decrease (P < 0.0065) in epithelial cell proliferation (PCNA-positive epithelial cells/total epithelial cells/mm2: CMV 41 ± 3.2 vs. CONT 83 ± 4.7). The increase in EM proliferation is correlated with stromal hypercellularity.
Fig. 2.
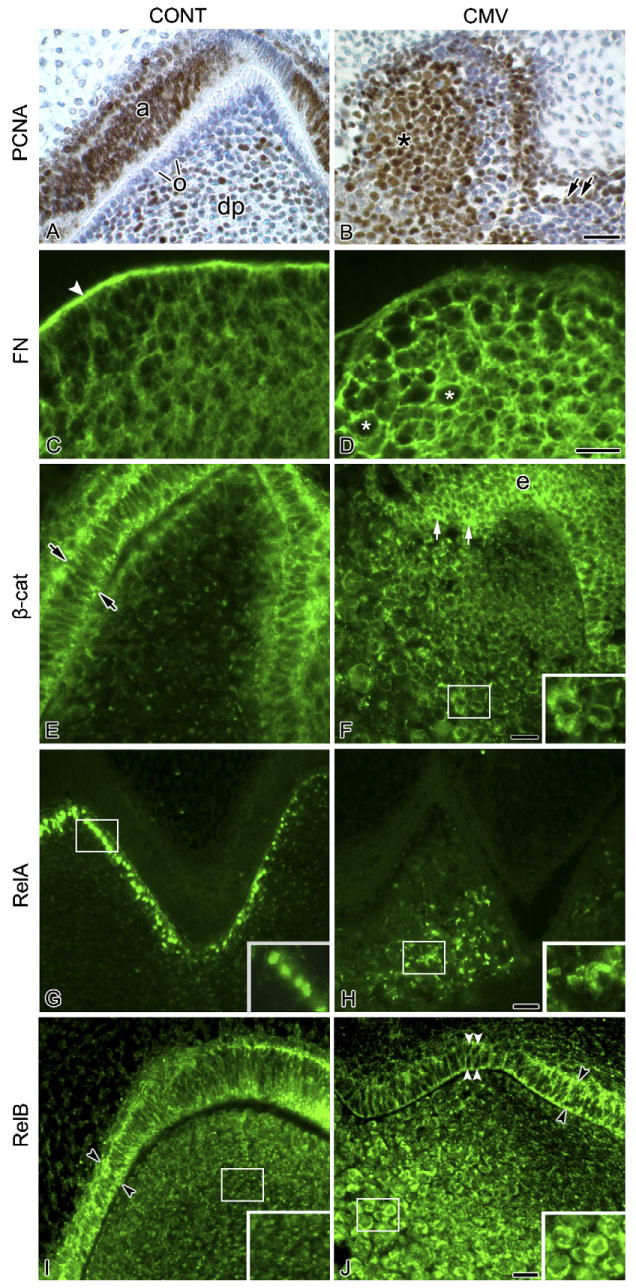
Characterisation of mCMV-induced cellular changes. (A and B) Cell proliferation. Cell proliferation was determined by the distribution of PCNA-positive nuclei (brown colour). In control molars (A), PCNA-positive nuclei are seen in ameloblasts (a) and, to a lesser extent, in dental papilla (dp) mesenchyme. Note the absence of PCNA-positive nuclei in polarised odontoblasts (o) in the cuspal tip, indicating differentiation. With mCMV infection (B), PCNA-positive nuclei are seen throughout the cytomegalic mesenchymal cell population (*) and, to a lesser extent, in short, nonpolarised epithelial cells (double arrows). (C and D) FN distribution. In control molars (C), FN is strongly immunolocalised in basement membrane (white arrowhead) and weakly throughout the dental papilla extracellular matrix. With mCMV infection (D), FN intensely surrounds individual cytomegalic dental papilla cells (white asterisks) and is relatively absent from the basement membrane. (E and F) β-Catenin distribution. In control molars (E), β-catenin is seen in the apical and basal regions (arrows) of polarised ameloblasts and more weakly in aligned odontoblasts. In mCMV-infected molars (F), β-catenin is strongly accumulated in the cytoplasm of dental papilla mesenchymal (insert) and short, nonpolarised epithelial (e) cells (white arrows); β-catenin is also detected in cell membranes. (G and H) RelA distribution. In control molars (G), polarised odontoblasts exhibit nuclear-localised RelA (insert). With mCMV infection (H), RelA is seen in the cytoplasm of centrally-localised cytomegalic dental papilla cells (insert) and is absent from presumptive odontoblasts. (I and J) RelB distribution. In control molars (I), RelB is seen in the apical and basal regions of polarised ameloblasts (black arrowheads). A similar cytoplasmic distribution pattern (black arrowheads) is seen in the mCMV-infected epithelia (D) which consists of nonpolarised cuboidal epithelial cells (white arrowheads). Note that mCMV infection induced a marked increase in RelB immunostain in the cytoplasm of centrally-localised dental papilla mesenchyme (insert D) compared to control (insert C). Bar (A and B, E–J), 20 μm; (C and D), 10 μm. (F–J inserts), 13 μm.
3.3. Cell characterisation
To begin to delineate the mechanisms underlying mCMV-induced tooth dysmorphology, we analysed spatial differences in the distribution of components of several cell signaling pathways important for tooth morphogenesis and shown to be altered by mCMV infection of embryonic SMGs.13 mCMV infection of embryonic mouse SMGs-induced marked differences in the localisation of fibronectin (FN),22 a basement membrane component important for odontogenesis.23,24 Thus, we postulated that changes in FN distribution would also be seen in mCMV-infected molars. This is exactly what we found: FN is seen surrounding individual cytomegalic dental papilla cells and is relatively absent from the basement membrane (Fig. 2D). This FN distribution pattern markedly differs from that seen in control Bell stage molars: FN was most prominent in the basement membrane at the epithelial–mesenchymal interface and, to a lesser extent, throughout dental papilla extracellular matrix (compare Fig. 2C to Fig. 2D). Given that (1) FN may be involved in odontoblast polarisation24,25 and (2) preameloblasts adhere to FN,23 this substantial change in FN localisation in mCMV-infected tooth organs may account for the disrupted odontoblast and ameloblast alignment and differentiation.
Moreover, given the relationship between FN and β-catenin expression26 and our observation that mCMV infection induced β-catenin translocation in embryonic SMGs,13 we analysed the spatial distribution of β-catenin in control and mCMV-infected E15 + 12 mandibular first molars. In Bell stage controls, β-catenin (both an adherens junction constituent and a transcription factor) is primarily localised in the apical and basal regions of ameloblasts and in aligned odontoblasts, and barely in centrally-localised dental papilla mesenchyme (Fig. 2E); this distribution pattern is consistent with previous reports.27,28 Importantly, mCMV infection induces the accumulation of β-catenin in the cytoplasm of cytomegalic mesenchymal cells found throughout the dental papilla and in abnormal epithelia, as well as in cell membranes (Fig. 2F). Cytoplasmic accumulation of β-catenin is indicative of its potential function as a transcription factor.
3.4. mCMV and NF-κB expression
NF-κB signaling plays an important role during morphogenesis, including tooth development.29–31 Given that CMV infection has been shown to induce the canonical [NF-κB1/RelA; NF-κB1/RelB] and noncanonical (NF-κB2/RelB) NF-κB pathways,13,32–38 we analysed the spatial distribution of RelA(p65) and RelB (components of both the canonical and noncanonical pathways) in control and mCMV-infected molars. In Bell stage control molars, RelA(p65) is nuclear localised in polarised odontoblasts (Fig. 2G) whereas RelB is primarily detected in the apical and basal regions of polarised ameloblasts (Fig. 2I). Since activation of the NF-κB/Rel complex requires translocation into the nucleus, our observation of nuclear-localised RelA and cytoplastic-localised RelB indicates that, at this stage, RelA (but not RelB) has been activated. mCMV infection induces marked differences in RelA and RelB protein expression. Specifically, there is de novo cytoplasmic localisation of both RelA and RelB proteins in infected and affected centrally-localised cytomegalic dental papilla mesenchymal cells (Fig. 2H and J); RelB also exhibits an epithelial distribution similar to that seen in controls (compare Fig. 2J to Fig. 2I). Since the noncanonical NF-κB2/RelB pathway had appeared to be important for mCMV-induced SMG embryopathology,13 we also determined the distribution of NF-κB2 in control and mCMV-infected molars. The absence of immunodetectable NF-κB2 protein in both control and mCMV-infected mandibular first molars (data not shown) suggests that the noncanonical NF-κB2/RelB pathway is not involved in normal and abnormal odontogenesis.
3.5. Active mCMV infection required for tooth pathology
To determine if active mCMV infection is necessary to initiate and sustain progressive tooth pathogenesis, we utilised acyclovir, an antiherpesviral nucleoside active against CMV,20 to inhibit mCMV replication. Since acyclovir has been shown at high doses to be teratogenic to rat embryos,39,40 we used a dose shown in our laboratory to be nonteratogenic to embryonic mouse SMGs13 and tooth organs (data not shown) in vitro. E15 (Cap stage) mandibular first molars were infected for 24 h with mCMV and then cultured in the presence or absence of 10 μg/ml acyclovir sodium for up to 15 days in vitro (Fig. 3); controls consisted of tooth organs cultured in chemically-defined medium alone. Acyclovir treatment suppresses mCMV replication and spread, as indicated by the absence of lacZ (mCMV) expression (compare Fig. 3C and D to Fig. 3A and B), and promoted differentiation (Fig. 3). Acyclovir-treated, mCMV-infected molars are characterised by improved cuspal formation compared to mCMV-infected organs, with differentiated, polarised odontoblasts and ameloblasts, as well as secreted dentin matrix, being seen (compare Fig. 3D to Fig. 3B, Fig. 3G to Fig. 3F). The acyclovir-treated tooth phenotype resembles that seen in control (compare Fig. 3G to E). Since the mCMV replication cycle is incomplete after only 24 h of infection when our antiviral treatment is initiated, it appears that completion of the viral replication cycle beyond DNA replication is critical to the initiation of tooth pathogenesis.
Fig. 3.
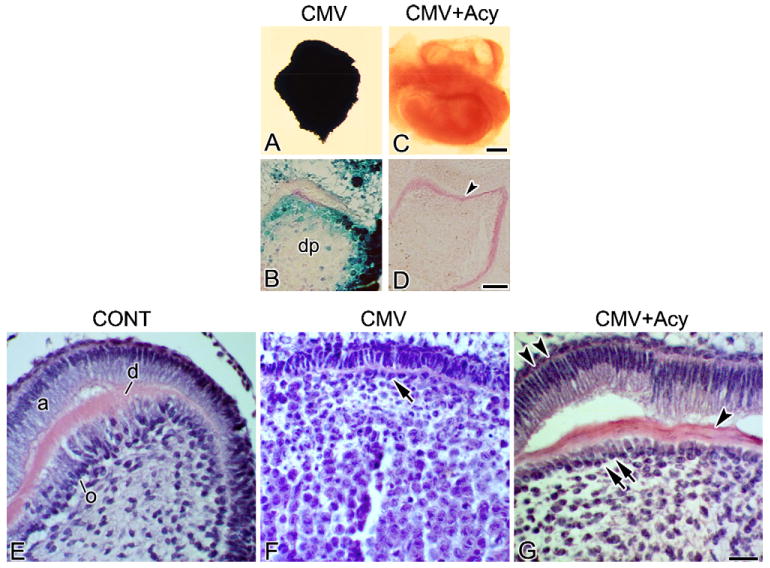
Acyclovir treatment ameliorates mCMV-induced pathology. (A–D) β-Galactosidase (lacZ) expression. LacZ is detected in EM dental papilla cells in mCMV-infected tooth organs (A and B) and was absent from acyclovir-treated, mCMV-infected (CMV + Acy) tooth organs (C and D). (E–G) Histological sections of control (E), mCMV-infected (F) and mCMV + Acy (G) molars. mCMV infection induces abnormal tooth development (F), characterised by unaligned, disorganised odontoblasts (arrow), undifferentiated ameloblasts, and abnormal mesenchymal cellularity. In contrast, acyclovir-treated, mCMV-infected molar phenotypes (G) are similar to that seen in control (E). Note, the presence of aligned, polarised odontoblasts (double arrows) and ameloblasts (double arrowheads), increased dentin matrix deposition (arrowhead), and dental papilla cells exhibiting a normal, fibroblastic appearance. Bar (A and C), 30 μm; (B, D–G), 20 μm.
4. Discussion
It is well established that reciprocal interactions between the epithelium and mesenchyme are critical for the development of many tissues and organs, tooth being a classic example.41,42 Tooth development is a continuous process during which the oral epithelium, a derivative of surface ectoderm, thickens and buds into the underlying ectomesenchyme, and then grows and folds to form several crown shapes of varying complexity. This progressive differentiation is regulated by sequential and reciprocal epithelial–mesenchymal interactions.41–43 These communications are effected by paracrine signaling molecules from the TGFβ, FGF, Hedgehog, Wnt, and TNF families and their cognate receptors and transcription factors. Typically, during Mid to Late Bell stage, the odontoblasts and ameloblasts differentiate at the interface of the epithelium and mesenchyme and deposit dentin and enamel matrixes, respectively. Recombination experiments have shown that the dental epithelium governs tooth development prior to the Bud stage and odontoblast differentiation44–47 and the dental papilla regulates tooth shape and ameloblast differentiation in the Cap and Bell stages.48,49
In this study, mCMV infection of Cap stage mouse mandibular first molars-induced smaller, developmentally-delayed and severely dysmorphic tooth organs. After 12 days in culture, the mCMV-infected molars exhibit shallow, misshapen cusps, abnormal EM cellularity, undifferentiated odontoblasts and ameloblasts, and the absence of dentin matrix. Analysis of viral distribution indicates that mCMV exclusively infects dental papilla EM. These results are consistent with our prior observations in first branchial arch derivatives (SMG, mandible) that mCMV has a particular tropism for neural-crest-derived EM (Melnick et al., 2006; unpublished). The absence of mCMV infection in affected abnormal dental epithelia suggests that, as in mCMV-infected SMGs, paracrine factors likely mediate the viral effect on dental epithelia. The same may reasonably be concluded for affected, but not infected, dental papilla EM.
Terminal differentiation of odontoblasts involves withdrawal from the cell cycle, polarisation and secretion of predentin/dentin matrix.50 Since the dental epithelium induces odontoblast differentiation,44,47 the presence of infected and affected mesenchyme and absence of differentiated odontoblasts in mCMV-infected teeth suggest that mCMV infection interrupts the reciprocal epithelial–mesenchymal interactions critical for their differentiation. Our observation of cell proliferation in unaligned, nonpolarised EM adjacent to the inner dental epithelium provides additional evidence suggesting that mCMV infection interferes with differentiation. Moreover, since odontoblast secretion of predentin, the first layer of the dentin matrix, induces ameloblast differentiation,51 the absence of polarised odontoblasts and the marked delay in predentin secretion in mCMV-infected tooth organs likely results in the substantial abnormality of ameloblast differentiation. Taken together, our results indicate that defects in the dental epithelium are secondary to defects in dental papilla mesenchyme.
Further, since mCMV-infected tooth organs exhibit morphogenetic advancement between day 12 and 15 of culture, with the mCMV-infected E15 + 15 molar phenotype resembling that seen in control E15 + 9 molars, it is reasonable to expect that odontoblast and ameloblast differentiation would progress with additional days in culture. It appears then that mCMV infection severely delays, but does not completely interrupt, tooth morphogenesis. This is consistent with the AI phenocopy seen in children who exhibit active perinatal and postnatal CMV infection.4–7
Over the last decades, it has become increasingly clear that basement membrane components are important for tooth development.24,25,50,52,53 The functional network of matrix molecules, including FN, mediates the complex series of cell–cell interactions between the epithelium and mesenchyme. We demonstrate that FN is intensely localised to the basement membrane (Fig. 2C); this observation is similar to previous reports.24,52,53 Importantly, viral infection of Cap stage molars induces a marked decrease in basement membrane localisation; it also induces FN localisation surrounding individual infected and affected dental papilla EM (Fig. 2D). Given that FN may be involved in odontoblast polarisation24,25 and preameloblasts must adhere to FN prior to differentiation,23 the relative absence of FN in the basement membrane in mCMV-infected molars may account for the disrupted odontoblast and ameloblast alignment and differentiation.
β-catenin has dual roles as both a cell adhesion molecule and a transcription factor and appears to be an important molecule linking cell adhesion and cell signaling during organogenesis.54–56 β-catenin stabilizes cell–cell adhesions by anchoring cadherins to the actin cytoskeleton and transduces the Wnt signal by interacting with transcription factor TCF/Lef. In the absence of Wnt signaling, cytoplasmic levels of β-catenin are kept low through continuous ubiquitination and proteosome-mediated degradation. The presence of Wnt signaling inhibits the degradation pathway, resulting in β-catenin accumulation in the cytoplasm and translocation into the nucleus where it binds to the transcription factor TCF/LEF to regulate gene expression. The canonical Wnt/β-catenin pathway has been shown to be essential for normal odontogenesis51,57 and may possibly link the differentiation of odontoblasts and cusp morphogenesis.58
Herein, we demonstrate that mCMV infection of Cap stage tooth organs induces a substantial increase in cytoplasmic accumulation of β-catenin in cytomegalic mesenchyme and abnormal epithelia (Fig. 2F). This cytoplasmic localisation is indicative of its potential function as a transcription factor. Given that (1) β-catenin can induce Fn gene transcription in fibroblasts26 and (2) FN can activate tyrosine kinase receptors and activated tyrosine kinases can increase β-catenin signaling,22,56 the concomitant increase of β-catenin and FN in dental papilla cells is likely correlated to tooth pathology.
NF-κB/Rel signaling is important to normal odontogenesis.29–31,59 Abnormal molar phenotypes were seen in IKBαΔN and Ikkα mutant mice with reduced NF-κB/Rel signaling.30–31 Sharpe and coworkers29 found similar abnormal molar phenotypes in Eda, Edar, Traf6 and NEMO (IKKγ) mutant mice with decreased NF-κB/Rel function.
NF-κB is composed of homo- and hetero-dimeric complexes of Rel family polypeptides. Mammals express five NF-κB subunits: NF-κB1(p50), NF-κB2(p52), RelA(p65), RelB and c-Rel.60–62 Since NF-κB1(p50) and NF-κB2(p52) lack a transactivation domain, they can only promote transcription when heterodimerised with transactivating Rel units. The IκB family of inhibitory proteins keeps inactive NF-κB/Rel dimers in the cytoplasm. Activation of the NF-κB/Rel complex requires degradation of IκB which allows nuclear translocation, followed by binding to NF-κB and/or Rel recognition sites to regulate gene transcription. Two NF-κB pathways, the canonical NF-κB1(p50)/Rel pathway and noncanonical NF-κB2(p52)/Rel pathways, have been identified and their functions investigated.60–62 Since nuclear localisation indicates NF-κB/Rel activation, our observation in uninfected (control) Bell stage molars of nuclear-localised RelA (but not RelB) suggests that the canonical NF-κB1/RelA pathway mediates cuspal morphogenesis. In contrast, the absence of NF-κB2 protein in developing teeth suggests that the noncanonical NF-κB2(p52)/Rel pathway does not play a role during normal odontogenesis.
It is well established that CMV infection induces the canonical and noncanonical NF-κB pathways.32–38,63 mCMV and hCMV activation of the canonical NF-κB pathway during infection of fibroblasts and other cell types facilitates viral replication.32–34,37,38 In addition, viral IE1 induces nuclear translocation (activation) of NF-κB and upregulates RelB transcription.32,35,36,63 IE1 is also a NF-κB response gene, with NF-κB binding sites being identified in the promoter and enhancer regions of i.e. genes.64 Moreover, we have previously demonstrated that mCMV-induced SMG embryopathology was centred around both the canonical and the noncanonical NF-κB pathways.13
In this study, we report viral IE1 expression in dental papilla mesenchymal cells (Fig. 1G), with a concomitant upregulation of RelA and RelB protein expression being seen in these infected and affected dental papilla EM cells (Fig. 1G–J). Our observation that mCMV infection induced a shift in RelA localisation from odontoblast nuclei to the cytoplasm of undifferentiated dental papilla mesenchyme suggests that viral-induced pathology is mediated by reduction of canonical NF-κB1/RelA signaling. Our results are consistent with the previous findings that (1) reduced NF-κB/Rel signaling is associated with abnormal odontogenesis29–31 and (2) activation of the canonical NF-κB1(p50)/RelA pathway and binding of p50/RelA to the major i.e. gene promoter is not required for CMV replication in cultured fibroblasts.32 Finally, the absence of NF-κB2 protein (a required component of the noncanonical pathway) in mCMV-infected tooth organs indicates that the noncanonical NF-κB2/RelB pathway is not involved in mCMV-induced tooth pathogenesis.
5. Conclusions
mCMV infection of embryonic mouse Cap stage mandibular first molars results in significant inhibition of progressive differentiation. mCMV infection of neural crest-derived ectomesenchyme appears to interrupt the sequence of reciprocal interactions between epithelial and mesenchymal components. Initial localisation studies suggest that the pathogenesis is mediated by the canonical NF-κB pathway and there appears to be an unusual interaction between abnormal mesenchymal cells and surrounding matrix. Our results indicate that mCMV-induced changes in signaling pathways severely delays, but does not completely interrupt, tooth morphogenesis. Importantly, we demonstrate that this well-defined embryonic mouse organ culture system can be utilised to delineate the molecular pathogenesis of CMV-induced tooth defects, ultimately providing a rational basis for the future discovery of a safe and effective therapy to ameliorate the enamel defects seen in approximately 3000 children born each year with congenital CMV infection. This is of considerable clinical importance since CMV is shed in saliva for months to years before termination of active infection, and currently there is no acceptable treatment of congenital CMV infection in children.65
Acknowledgments
We would like to thank Dr. Edward Mocarski for providing mCMV and IE1 antibody. This research was supported by NIH grant RO1 DE014535 (T.J./M.M.).
References
- 1.Pass RF. Cytomegalovirus. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, et al., editors. Fields virology. 4th. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 2675–705. [Google Scholar]
- 2.Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355–63. doi: 10.1002/rmv.544. [DOI] [PubMed] [Google Scholar]
- 3.Ross DS, Dollard SC, Victor M, Sumartojo E, Cannon MJ. The epidemiology and prevention of congenital cytomegalovirus infection and disease: activities of the Centers for Disease Control and Prevention Workgroup. J Womens Health (Larchmt) 2006;15:224–9. doi: 10.1089/jwh.2006.15.224. [DOI] [PubMed] [Google Scholar]
- 4.Stagno S, Pass RF, Dworsky ME, Alford CA. Congenital and perinatal cytomegalovirus infections. Semin Perinatol. 1983;7:31–42. [PubMed] [Google Scholar]
- 5.Stagno S, Pass RF, Dworsky ME, Britt WJ, Alford CA. Congenital and perinatal cytomegalovirus infections: clinical characteristics and pathogenic factors. Birth Defects Orig Article Ser. 1984;20:65–85. [PubMed] [Google Scholar]
- 6.Stagno S, Pass RF, Thomas JP, Navia JM, Dworsky ME. Defects of tooth structure in congenital cytomegalovirus infection. Pediatrics. 1982;69:646–8. [PubMed] [Google Scholar]
- 7.Stehel EK, Sanchez PJ. Cytomegalovirus infection in the fetus and neonate. NeoReviews. 2005;6:e38–45. [Google Scholar]
- 8.Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 2003;5:1263–77. doi: 10.1016/j.micinf.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Baskar JF, Furnari B, Huang ES. Demonstration of developmental anomalies in mouse fetuses by transfer of murine cytomegalovirus DNA-injected eggs to surrogate mothers. J Infect Dis. 1993;167:1288–95. doi: 10.1093/infdis/167.6.1288. [DOI] [PubMed] [Google Scholar]
- 10.Baskar JF, Peacock J, Sulik KK, Huang ES. Early-stage developmental abnormalities induced by murine cytomegalovirus. J Infect Dis. 1987;155:661–6. doi: 10.1093/infdis/155.4.661. [DOI] [PubMed] [Google Scholar]
- 11.Baskar JF, Stanat SC, Sulik KK, Huang ES. Murine cytomegalovirus-induced congenital defects and fetal maldevelopment. J Infect Dis. 1983;148:836–43. doi: 10.1093/infdis/148.5.836. [DOI] [PubMed] [Google Scholar]
- 12.Tsutsui Y. Developmental disorders of the mouse brain induced by murine cytomegalovirus: animal models for congenital cytomegalovirus infection. Pathol Int. 1995;45:91–102. doi: 10.1111/j.1440-1827.1995.tb03428.x. [DOI] [PubMed] [Google Scholar]
- 13.Melnick M, Mocarski ES, Abichaker G, Huang J, Jaskoll T. Cytomegalovirus-induced embryopathology: mouse submandibular salivary gland epithelial–mesenchymal ontogeny as a model. BMC Dev Biol. 2006;6:42. doi: 10.1186/1471-213X-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaskoll T, Leo T, Witcher D, Ormestad M, Astorga J, Bringas P, Jr, et al. Sonic hedgehog signaling plays an essential role during embryonic salivary gland epithelial branching morphogenesis. Dev Dyn. 2004;229:722–32. doi: 10.1002/dvdy.10472. [DOI] [PubMed] [Google Scholar]
- 15.Bringas P, Jr, Nakamura M, Nakamura E, Evans J, Slavkin HC. Ultrastructural analysis of enamel formation during in vitro development using chemically-defined medium. Scanning Microsc. 1987;1:1103–8. [PubMed] [Google Scholar]
- 16.Slavkin HC, Brownell AG, Bringas P, Jr, MacDougall M, Bessem C. Basal lamina persistence during epithelial–mesenchymal interactions in murine tooth development in vitro. J Craniofac Genet Dev Biol. 1983;3:387–407. [PubMed] [Google Scholar]
- 17.Saederup N, Lin YC, Dairaghi DJ, Schall TJ, Mocarski ES. Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc Natl Acad Sci USA. 1999;96:10881–6. doi: 10.1073/pnas.96.19.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaskoll T, Abichaker G, Witcher D, Sala FG, Bellusci S, Hajihosseini MK, et al. FGF10/FGFR2b signaling plays essential roles during in vivo embryonic submandibular salivary gland morphogenesis. BMC Dev Biol. 2005;5:11–22. doi: 10.1186/1471-213X-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melnick M, Witcher D, Bringas P, Jr, Carlsson P, Jaskoll T. Meckel's cartilage differentiation is dependent on hedgehog signaling. Cells Tissues Organ. 2005;179:146–57. doi: 10.1159/000085950. [DOI] [PubMed] [Google Scholar]
- 20.Burns WH, Wingard JR, Bender WJ, Saral R. Thymidine kinase not required for antiviral activity of acyclovir against mouse cytomegalovirus. J Virol. 1981;39:889–93. doi: 10.1128/jvi.39.3.889-893.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruch JV. Patterned distribution of differentiating dental cells: facts and hypotheses. J Biol Buccale. 1990;18:91–8. [PubMed] [Google Scholar]
- 22.Matsuo M, Sakurai H, Ueno Y, Ohtani O, Saiki I. Activation of MEK/ERK and PI3K/Akt pathways by fibronectin requires integrin alpha-mediated ADAM activity in hepatocellular carcinoma: a novel functional target for gefitinib. Cancer Sci. 2006;97:155–62. doi: 10.1111/j.1349-7006.2006.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukumoto S, Yamada Y. Review: extracellular matrix regulates tooth morphogenesis. Connect Tissue Res. 2005;46:220–6. doi: 10.1080/03008200500344017. [DOI] [PubMed] [Google Scholar]
- 24.Ruch JV. Odontoblast commitment and differentiation. Biochem Cell Biol. 1998;76:923–38. [PubMed] [Google Scholar]
- 25.Ruch JV, Lesot H, Begue-Kirn C. Odontoblast differentiation. Int J Dev Biol. 1995;39:51–68. [PubMed] [Google Scholar]
- 26.Gradl D, Kuhl M, Wedlich D. The Wnt/Wg signal transducer beta-catenin controls fibronectin expression. Mol Cell Biol. 1999;19:5576–87. doi: 10.1128/mcb.19.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fausser JL, Schlepp O, Aberdam D, Meneguzzi G, Ruch JV, Lesot H. Localization of antigens associated with adherens junctions, desmosomes, and hemidesmosomes during murine molar morphogenesis. Differentiation. 1998;63:1–11. doi: 10.1046/j.1432-0436.1998.6310001.x. [DOI] [PubMed] [Google Scholar]
- 28.Obara N, Lesot H. Subcellular localization of beta-catenin and cadherin expression in the cap-stage enamel organ of the mouse molar. Histochem Cell Biol. 2004;121:351–8. doi: 10.1007/s00418-004-0637-5. [DOI] [PubMed] [Google Scholar]
- 29.Courtney JM, Blackburn J, Sharpe PT. The ectodysplasin and NFkappaB signalling pathways in odontogenesis. Arch Oral Biol. 2005;50:159–63. doi: 10.1016/j.archoralbio.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt-Ullrich R, Aebischer T, Hülsken J, Birchmeier W, Klemm U, Scheidereit C. Requirement of NF-κB/Rel for the development of hair follicles and other epidermal appendices. Development. 2001;128:3843–53. doi: 10.1242/dev.128.19.3843. [DOI] [PubMed] [Google Scholar]
- 31.Ohazama A, Hu Y, Schmidt-Ullrich R, Cao Y, Scheidereit C, Karin M, et al. A dual role for Ikkα in tooth development. Dev Cell. 2004;6:219–27. doi: 10.1016/s1534-5807(04)00024-3. [DOI] [PubMed] [Google Scholar]
- 32.Benedict CA, Angulo A, Patterson G, Ha S, Huang H, Messerle M, et al. Neutrality of the canonical NF-kappaB-dependent pathway for human and murine cytomegalovirus transcription and replication in vitro. J Virol. 2004;78:741–50. doi: 10.1128/JVI.78.2.741-750.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeMeritt IB, Milford LE, Yurochko AD. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J Virol. 2004;78:4498–507. doi: 10.1128/JVI.78.9.4498-4507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeMeritt IB, Podduturi JP, Tilley AM, Nogalski MT, Yurochko AD. Prolonged activation of NF-kappaB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virology. 2006;346:15–31. doi: 10.1016/j.virol.2005.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang HY, Petrovas C, Sonenshein GE. RelB-p50 NF-kappa B complexes are selectively induced by cytomegalovirus immediate-early protein 1: differential regulation of Bcl-x(L) promoter activity by NF-kappa B family members. J Virol. 2002;76:5737–47. doi: 10.1128/JVI.76.11.5737-5747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Sonenshein GE. Induction of the RelB NF-kappaB subunit by the cytomegalovirus IE1 protein is mediated via Jun kinase and c-Jun/Fra-2 AP-1 complexes. J Virol. 2005;79:95–105. doi: 10.1128/JVI.79.1.95-105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yurochko AD, Hwang ES, Rasmussen L, Keay S, Pereira L, Huang ES. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J Virol. 1997;71:5051–9. doi: 10.1128/jvi.71.7.5051-5059.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yurochko AD, Kowalik TF, Huong SM, Huang ES. Human cytomegalovirus upregulates NF-kappa B activity by transactivating the NF-kappa B p105/p50 and p65 promoters. J Virol. 1995;69:5391–400. doi: 10.1128/jvi.69.9.5391-5400.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chahoud I, Stahlmann R, Bochert G, Dillmann I, Neubert D. Gross-structural defects in rats after acyclovir application on day 10 of gestation. Arch Toxicol. 1988;62:8–14. doi: 10.1007/BF00316250. [DOI] [PubMed] [Google Scholar]
- 40.Stahlmann R, Klug S, Lewandowski C, Bochert G, Chahoud I, Rahm U, et al. Prenatal toxicity of acyclovir in rats. Arch Toxicol. 1988;61:468–79. doi: 10.1007/BF00293693. [DOI] [PubMed] [Google Scholar]
- 41.Thesleff I. Epithelial–mesenchymal signalling regulating tooth morphogenesis. J Cell Sci. 2003;116:1647–8. doi: 10.1242/jcs.00410. [DOI] [PubMed] [Google Scholar]
- 42.Thesleff I, Sharpe P. Signalling networks regulating dental development. Mech Dev. 1997;67:111–23. doi: 10.1016/s0925-4773(97)00115-9. [DOI] [PubMed] [Google Scholar]
- 43.Miletich I, Sharpe PT. Normal and abnormal dental development. Hum Mol Genet. 2003;12:R69–73. doi: 10.1093/hmg/ddg085. [DOI] [PubMed] [Google Scholar]
- 44.Kollar EJ. Epithelio-mesenchymal interactions in the mammalian integument: tooth development as a model for instructive induction. In: Sawyer RM, Fallon JF, editors. Epithelial–mesenchymal interactions in development. New York: Praeger Press; 1983. pp. 87–102. [Google Scholar]
- 45.Lumsden AG. Spatial organization of the epithelium and the role of neural crest cells in the initiation of the mammalian tooth germ. Development. 1988 103:155–69. doi: 10.1242/dev.103.Supplement.155. [DOI] [PubMed] [Google Scholar]
- 46.Mina M, Kollar EJ. The induction of odontogenesis in non-dental mesenchyme combined with early murine mandibular arch epithelium. Arch Oral Biol. 1987;32:123–7. doi: 10.1016/0003-9969(87)90055-0. [DOI] [PubMed] [Google Scholar]
- 47.Thesleff I, Keranen S, Jernvall J. Enamel knots as signaling centers linking tooth morphogenesis and odontoblast differentiation. Adv Dent Res. 2001;15:14–8. doi: 10.1177/08959374010150010401. [DOI] [PubMed] [Google Scholar]
- 48.Kollar EJ, Baird GR. The influence of the dental papilla on the development of tooth shape in embryonic mouse tooth germs. J Embryol Exp Morphol. 1969;21:131–48. [PubMed] [Google Scholar]
- 49.Kollar EJ, Baird GR. Tissue interactions in embryonic mouse tooth germs. II. The inductive role of the dental papilla. J Embryol Exp Morphol. 1970;24:173–86. [PubMed] [Google Scholar]
- 50.Lesot H, Lisi S, Peterkova R, Peterka M, Mitolo V, Ruch JV. Epigenetic signals during odontoblast differentiation. Adv Dent Res. 2001;15:8–13. doi: 10.1177/08959374010150012001. [DOI] [PubMed] [Google Scholar]
- 51.Miletich I, Sharpe PT. Neural crest contribution to mammalian tooth formation. Birth Defects Res C Embryo Today. 2004;72:200–12. doi: 10.1002/bdrc.20012. [DOI] [PubMed] [Google Scholar]
- 52.Lesot H, Osman M, Ruch JV. Immunofluorescent localization of collagens, fibronectin, and laminin during terminal differentiation of odontoblasts. Dev Biol. 1981;82:371–81. doi: 10.1016/0012-1606(81)90460-7. [DOI] [PubMed] [Google Scholar]
- 53.Thesleff I, Barrach HJ, Foidart JM, Vaheri A, Pratt RM, Martin GR. Changes in the distribution of type IV collagen, laminin, proteoglycan, and fibronectin during mouse tooth development. Dev Biol. 1981;81:182–92. doi: 10.1016/0012-1606(81)90361-4. [DOI] [PubMed] [Google Scholar]
- 54.Bienze M. Beta-catenin: a pivot between cell adhesion and Wnt signalling. Curr Biol. 2005;15:R64–7. doi: 10.1016/j.cub.2004.12.058. [DOI] [PubMed] [Google Scholar]
- 55.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 56.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–7. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jarvinen E, Salazar-Ciudad I, Birchmeier W, Taketo MM, Jernvall J, Thesleff I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signalling. Proc Natl Acad Sci USA. 2006;143:18627–32. doi: 10.1073/pnas.0607289103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamashiro T, Zheng L, Shitaku Y, Saito M, Tsubakimoto T, Takada K, et al. Wnt10a regulates dentin sialophosphoprotein mRNA expression and possibly links odontoblast differentiation and tooth morphogenesis. Differentiation. 2007;75:452–62. doi: 10.1111/j.1432-0436.2006.00150.x. [DOI] [PubMed] [Google Scholar]
- 59.Melnick M, Chen H, Min Zhou Y, Jaskoll T. The functional genomic response of developing embryonic submandibular glands to NF-kappa B inhibition. BMC Dev Biol. 2001;1:15–33. doi: 10.1186/1471-213X-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 62.Gugasyan R, Grumont R, Grossmann M, Nakamura Y, Pohl T, Nesic D, et al. Rel/NF-kappaB transcription factors: key mediators of B-cell activation. Immunol Rev. 2000;176:134–40. doi: 10.1034/j.1600-065x.2000.00615.x. [DOI] [PubMed] [Google Scholar]
- 63.Murayama T, Mukaida N, Sadanari H, Yamaguchi N, Khabar KS, Tanaka J, et al. The immediate early gene 1 product of human cytomegalovirus is sufficient for up-regulation of interleukin-8 gene expression. Biochem Biophys Res Commun. 2000;279:98–304. doi: 10.1006/bbrc.2000.3923. [DOI] [PubMed] [Google Scholar]
- 64.He B, Weber GF. Synergistic activation of the CMV promoter by NF-kappaB p50 and PKG. Biochem Biophys Res Commun. 2004;321:13–20. doi: 10.1016/j.bbrc.2004.06.101. [DOI] [PubMed] [Google Scholar]
- 65.Malm G, Engman ML. Congenital cytomegalovirus infections. Semin Fetal Neonatal Med. 2007;12:154–9. doi: 10.1016/j.siny.2007.01.012. [DOI] [PubMed] [Google Scholar]