

Abstract
An important focus in cell biology is understanding how different feedback mechanisms regulate G protein–coupled receptor systems. Toward this end we investigated the regulation of endogenous β2 adrenergic receptors (β2ARs) and phosphodiesterases (PDEs) by measuring cAMP signals in single HEK-293 cells. We monitored cAMP signals using genetically encoded cyclic nucleotide-gated (CNG) channels. This high resolution approach allowed us to make several observations. (a) Exposure of cells to 1 μM isoproterenol triggered transient increases in cAMP levels near the plasma membrane. Pretreatment of cells with 10 μM rolipram, a PDE4 inhibitor, prevented the decline in the isoproterenol-induced cAMP signals. (b) 1 μM isoproterenol triggered a sustained, twofold increase in phosphodiesterase type 4 (PDE4) activity. (c) The decline in isoproterenol-dependent cAMP levels was not significantly altered by including 20 nM PKI, a PKA inhibitor, or 3 μM 59-74E, a GRK inhibitor, in the pipette solution; however, the decline in the cAMP levels was prevented when both PKI and 59-74E were included in the pipette solution. (d) After an initial 5-min stimulation with isoproterenol and a 5-min washout, little or no recovery of the signal was observed during a second 5-min stimulation with isoproterenol. (e) The amplitude of the signal in response to the second isoproterenol stimulation was not altered when PKI was included in the pipette solution, but was significantly increased when 59-74E was included. Taken together, these data indicate that either GRK-mediated desensitization of β2ARs or PKA-mediated stimulation of PDE4 activity is sufficient to cause declines in cAMP signals. In addition, the data indicate that GRK-mediated desensitization is primarily responsible for a sustained suppression of β2AR signaling. To better understand the interplay between receptor desensitization and PDE4 activity in controlling cAMP signals, we developed a mathematical model of this system. Simulations of cAMP signals using this model are consistent with the experimental data and demonstrate the importance of receptor levels, receptor desensitization, basal adenylyl cyclase activity, and regulation of PDE activity in controlling cAMP signals, and hence, on the overall sensitivity of the system.
INTRODUCTION
G protein–coupled receptors (GPCRs) are critical links in relaying information from the extracellular space to the intracellular environment. Elucidating the regulation of GPCRs is an essential step in understanding cellular physiology (Clark, 1986; Palczewski and Benovic, 1991; Burns and Baylor, 2001; Kohout and Lefkowitz, 2003). To date, the coordination of GPCR desensitization and cyclic nucleotide phosphodiesterase (PDE) activity in controlling cyclic nucleotide signals is best understood in sensory neurons (Detlev and Restrepo, 1998; Burns and Baylor, 2001; Fain et al., 2001). In particular, the unique structure of retinal rod outer segments, high concentrations of signaling proteins such as rhodopsin (∼3 mM), and endogenous cyclic nucleotide-gated channels have allowed integration of data obtained both in vitro and in vivo, and provided unique insight into this signaling system (Stryer, 1991; Lagnado and Baylor, 1992; Pugh and Lamb, 1993; Yarfitz and Hurley, 1994; Yau, 1994; Polans et al., 1996; Pugh et al., 1997; Molday, 1998).
Other GPCR-mediated signaling systems are not as well understood. This is especially true of β2 adrenergic receptor (β2AR)–mediated signaling pathways. The low cellular concentrations of these receptors, the existence of multiple receptor subtypes in a single cell, the lack of specificity of protein kinase inhibitors, and, until recently, the inability to measure cAMP signals in single cells, have significantly hindered efforts to unravel the relative contributions of β2AR desensitization and PDE activity in shaping cAMP signals (Clark and Rich, 2003). For these reasons, investigators have relied on the overexpression or knockdown of receptors, G protein–coupled receptor kinases (GRKs), arrestin, or PDEs to elucidate the molecular mechanisms that regulate β2AR signaling (Clark, 1986; Devic et al., 2001; Friedman et al., 2002; Kohout and Lefkowitz, 2003; Xiang et al., 2005; Violin et al., 2006a,b). While these approaches have yielded tremendous amounts of information about what may be happening in endogenous signaling systems, it is well documented that cells have a remarkable ability to adapt to the overexpression, knockout, or knockdown of specific proteins (Krumins and Gilman, 2006; Violin et al., 2006b). Thus, it is critical to complement these approaches with studies of unperturbed signaling systems using real-time readouts of intracellular signals.
We previously examined the molecular mechanisms underlying transient, PGE1-induced cAMP signals in human embryonic kidney (HEK)-293 cells. We found that the rate of prostaglandin-induced cAMP synthesis does not significantly decay in these cells, at least in the time-frame of our experiments, and that in response to PGE1, PDE4 activity increased two- to threefold in a PKA-dependent manner (Rich et al., 2001a; Rich et al., 2007). We next monitored cAMP concentration in single cells by measuring cyclic nucleotide-gated (CNG) channel activity in the whole cell patch clamp configuration. This approach allowed us to selectively disrupt PKA-mediated signaling using either PKI, a highly selective peptide inhibitor of PKA (Cheng et al., 1986), or Ht31, a peptide that disrupts A-kinase anchoring protein (AKAP)–PKA interactions (Carr et al., 1991). We observed that including either PKI or Ht31 in the pipette solution significantly inhibited the decline in the cAMP response. These observations, in conjunction with biochemical and molecular approaches, demonstrated that PKA-mediated stimulation of PDE4 activity was primarily responsible for the decline in transient, PGE1-induced cAMP signals (Willoughby et al., 2006; Rich et al., 2007). As part of our study we developed a quantitative model of PGE1-induced cAMP signals, and used this model to ask a simple question: what would happen to the time course of the observed cAMP signals if the receptors did desensitize? Surprisingly, simulations of the model indicated that in the presence of PKA-mediated stimulation of PDE4 activity, receptor desensitization would only have small effects on the time course of cAMP signals.
In the present work we test this prediction by studying the endogenous β2AR signaling system in the same HEK-293 cell line. Our results indicate that exposure of cells to isoproterenol, a β-adrenergic agonist, triggers a sustained, twofold increase in PDE4 activity, and that, by itself, this feedback mechanism is capable of causing decays in isoproterenol-induced cAMP transients. However, in contrast to the observations described above, we demonstrate that isoproterenol-induced cAMP signals remain transient when PKA-dependent stimulation of PDE4 activity is pharmacologically inhibited. Our results demonstrate that GRK-mediated desensitization of β2ARs is also capable of causing decays in isoproterenol-induced cAMP transients. In addition, we present evidence that GRK-mediated desensitization, but not PKA-dependent stimulation of PDE4 activity, mediates the suppression of β2AR signaling over a 5-min wash period. On the basis of these data we developed a mathematical model of the β2-adrenergic signaling system. Simulations using this model reproduced the major aspects of the observed cAMP signals and provide a framework with which we can interpret the concerted actions of receptor levels, GRK-mediated receptor desensitization, and PDE4 activity in regulating cAMP signals. Abstracts describing parts of this work have already appeared (Rich, T.C., S.J. Vayttaden, R.B. Clark, and W.K. Xin. 2007a. FASEB J. 21:A791; Rich, T.C., W.K. Xin, A.L. Britain, and J.W. Karpen. 2007c. FASEB J. 21:A792).
MATERIALS AND METHODS
Cell Culture and Channel Expression
Culture and adenovirus infection of HEK-293 cells were performed as described previously (Rich et al., 2000). In brief, HEK-293 cells were maintained in 10 ml of minimal essential medium (MEM; Life Technologies Inc.) containing 10% vol/vol FBS (Gemini), and grown in 100-mm culture dishes at 37°C in a humidified atmosphere of 95% air, 5% CO2. Cells were plated at ∼60% confluence in 100-mm dishes for infection with an adenovirus encoding the C460W/E583M CNG channel at a multiplicity of infection of ∼10 pfu/cell (Rich et al., 2001b). 2 h post-infection, hydroxyurea was added to the cell media at a final concentration of 1 mM to inhibit viral replication. 24 h post-infection, cells were detached with PBS containing 0.03% EDTA, resuspended in serum-containing media, and assayed within 12 h. All experiments were conducted at room temperature, 20–22°C. Unless otherwise stated, all reagents were purchased from Sigma-Aldrich.
Measurement of PDE activity
Cyclic AMP PDE activity was measured according to the method of Thompson and Appleman (1971) as detailed previously (Hansen et al., 2000; Richter and Conti, 2002; Rich et al., 2007). In brief, after incubation for various times with or without 1 μM isoproterenol, cells were harvested and homogenized in ice-cold hypotonic buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 0.2 mM EGTA, 100 mM Na2PO4, 1.4 mM β-mercaptoethanol, 0.5% NP-40, 1 μM microcystin-LR, and a “complete protease inhibitor” tablet (Roche Diagnostics). Cells were homogenized using an all-glass homogenizer. Aliquots of the homogenates were assayed for PDE activity using 1 μM cAMP as the substrate. PDE4 activity was defined as the fraction of cAMP PDE activity inhibited by 10 μM rolipram. Protein concentrations were determined using the Bio-Rad Laboratories, Inc. protein assay using BSA as a standard. Experiments were repeated at least three times.
Measurement of Total Cellular cAMP Levels
HEK-293 cells were plated at 33% confluence in 100-mm dishes and assayed 24–48 h later. Cells were washed and assayed in a solution containing (in mM): 145 NaCl, 4 KCl, 10 HEPES, 10 d-glucose, 1 MgCl2, 1 CaCl2, pH 7.4. Additions were made from 10× solutions. Reactions were terminated by addition of 1 N HCl (0.1 N HCl final) and plates were incubated on ice for 15 min, after which the cells were scraped from the dish. Cellular cAMP levels were measured using an enzyme immunoassay (Cayman Chemical). Sample cAMP concentrations were calculated from standard curves. Data are presented as mean ± SEM, performed in triplicate.
Single Cell Measurement of cAMP Signals
The whole cell patch clamp technique was used to measure cAMP in single cells, as described previously (Rich et al., 2001a, 2007). In brief, recordings were made by an HEKA EPC10 patch clamp amplifier system. To ensure adequate voltage control, pipette resistance was limited to 4 MΩ and averaged 2.2 ± 0.1 MΩ (n = 115). Voltage offsets were zeroed with the pipette in the bath solution; no additional corrections were made for the liquid junction potential difference. Experiments with a series resistance-induced error in excess of 5 mV were discarded. After achieving whole cell configuration, the preparation was allowed to equilibrate for at least 10 min to ensure sufficient time for dialysis of compounds from the patch pipette into the cell. Current records were typically sampled at 10 kHz and filtered at 2 kHz and stored on a PC. Currents were recorded during 400-ms steps to a membrane potential of +20 mV from a holding potential of 0 mV. The pipette solution contained (in mM) 140 KCl, 0.5 MgCl2, 10 HEPES, 5 Na2ATP, 0.5 Na2GTP, pH 7.4; the bath solution contained (in mM) 140 NaCl, 4 KCl, 10 d-glucose, 10 HEPES, and either 0.1 or 10 MgCl2, pH 7.4. PGE1 and rolipram (Calbiochem) were added to control solutions from concentrated DMSO stocks (final DMSO concentrations ≤0.2%). Isoproterenol was dissolved in a solution of 0.1 mM ascorbate, 1 mM thiourea, pH 7.0. PKI (Calbiochem), St-Ht31 (Promega), and the 59-74E peptide (Sigma-Genosys) were aliquoted as 1000× stock solutions and stored at −20°C. Extracellular solutions were applied using a SF-77B solution switcher (Warner Instruments) with a mechanical switch time of 1–2 ms. The time to exchange the extracellular solution was measured by applying a 140 mM KCl solution to a depolarized cell (+50 mV) and monitoring changes in current through endogenous voltage-gated K+ channels; for each experiment, it was <100 ms. The bulk solution within the bath chamber was changed within 20 s using a custom-built, gravity-driven perfusion system. In some experiments, cells were treated with 100 ng/ml of pertussis toxin (PTX) for 18–24 h before experiments to test for possible influence of PTX-sensitive G proteins in β2AR signaling. To ensure that PTX was active in our system, we demonstrated that PTX could inhibit sphingosine-1-phosphate–mediated Ca2+ release from intracellular stores (not depicted), as described previously (van Koppen et al., 1996). The baseline current (non-CNG channel current) was monitored by measuring the residual current in the presence of 10 mM Mg2+ (a CNG channel blocker), approximately once per minute. No Mg2+-blockable currents were observed in control cells (cells which do not express CNG channels). All data were analyzed using custom scripts written in the MATLAB programming environment (v 7.4, MathWorks), and statistical analysis was performed using Student's t test. Simulations were conducted using the fourth order Runga Kutta method as implemented within the MATLAB programming environment. Electrophysiological data were converted to formats compatible with MATLAB software using a custom script provided by Bruxton Corporation.
Mathematical Equations Describing the β2 Adrenergic Signaling Pathway
To investigate the relative contributions of receptor desensitization and PDE activity in regulating cAMP signals, we developed a kinetic model of cAMP turnover near the plasma membrane of HEK-293 cells. This model is based on a recently published model of prostaglandin-induced cAMP signals in the same cells (Rich et al., 2007). Interestingly, in HEK-293 cells, prostaglandin-induced signals do not significantly desensitize at the level of cAMP synthesis (Rich et al., 2001a, 2007). Here we have expanded the model such that it can now be used to examine the effects of β2AR expression levels, as well as receptor desensitization, on cAMP signals. In this model, GRK activity triggers the desensitization of β2ARs, whereas elevated PKA activity stimulates PDE4 activity. PKA-mediated regulation of β2ARs is not considered. Initial conditions were obtained by setting the basal adenylyl cyclase activity and letting the system run to equilibrium (without stimulating receptor activity). The equations used to describe this model are given below. Parameters and initial conditions are given in Table I.
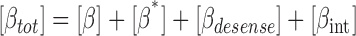 |
(1) |
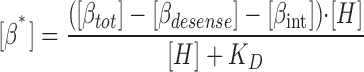 |
(2) |
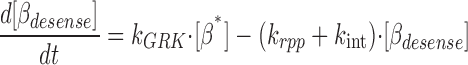 |
(3) |
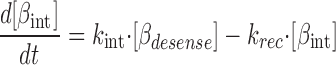 |
(4) |
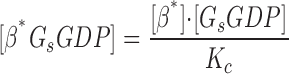 |
(5) |
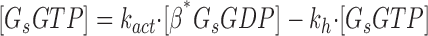 |
(6) |
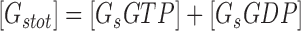 |
(7) |
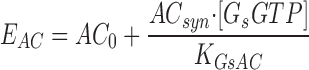 |
(8) |
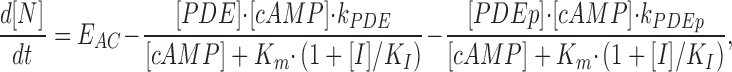 |
(9) |
where [βtot] is the total concentration of receptors, [β*] is the concentration of activated receptor at the plasma membrane, K D is the dissociation constant of isoproterenol, [βdesense] is the concentration of desensitized β2ARs, [βint] is the concentration of internalized β2ARs, k GRK is the rate constant of receptor phosphorylation by GRK, kr pp is the rate constant of receptor dephosphorylation, [H] is the hormone concentration, [Gs] is the total Gs concentration, [GsGTP] is the activated Gs concentration, ACsyn is the maximal cAMP synthesis rate, KGsAC is the equilibrium constant between activated G protein and AC, kact is the rate constant of Gs activation by β*, and kh is the rate constant of GTP hydrolysis. We assumed the rate of cAMP synthesis was not dependent on [ATP], because, in our experiments, the intracellular levels of ATP were clamped at high concentrations compared with the K m of AC for ATP (5 and 0.315 mM, respectively). [N] is the total cAMP concentration, [cAMP] is the concentration of free cAMP, E AC is the cAMP synthesis rate, [PDE] is the concentration of unphosphorylated PDE4, k PDE is the cAMP hydrolysis rate constant for unphosphorylated PDE4, [PDEp] is the concentration of phosphorylated PDE4, k PDEp is the cAMP hydrolysis rate constant for phosphorylated PDE4, K m is the Michaelis constant for PDE4, [I] is the concentration of a competitive inhibitor such as rolipram, and K I is the inhibition constant. The effects of 59-74E (a peptide inhibitor of GRK) were modeled as an 80% reduction in k grk. The phosphorylation of PDE4 is described by the equations below.
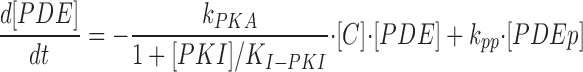 |
(10) |
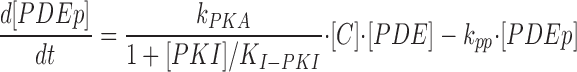 |
(11) |
where k PKA is the rate constant of PKA-mediated phosphorylation, K I-PKI is the inhibition constant for the noncompetitive inhibitor PKI, k pp is the rate constant of phosphatase-mediated dephosphorylation, and [C] is the concentration of free catalytic PKA subunits. The equations describing PKA activity are given below.
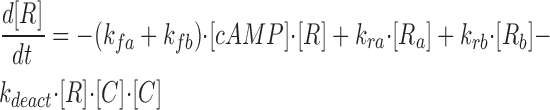 |
(12) |
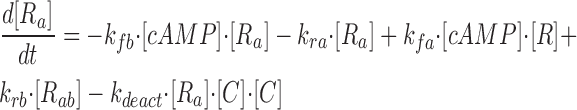 |
(13) |
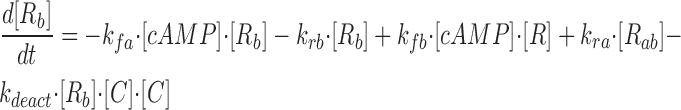 |
(14) |
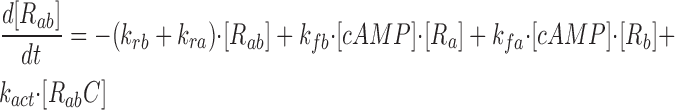 |
(15) |
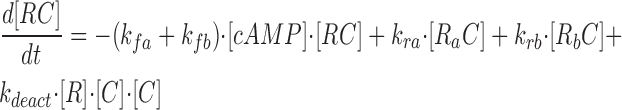 |
(16) |
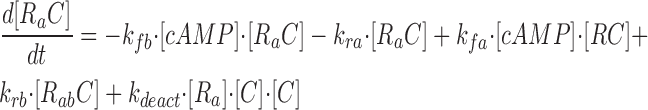 |
(17) |
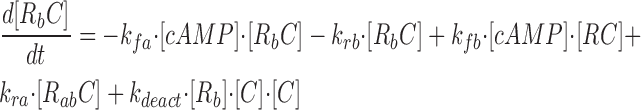 |
(18) |
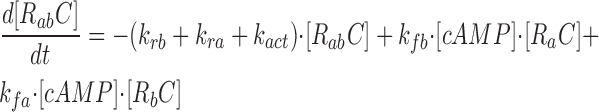 |
(19) |
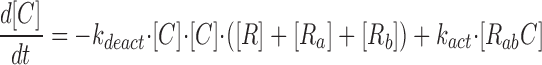 |
(20) |
where R represents the regulatory subunit of PKA with two cAMP binding sites, a and b. [RC] is the concentration of unliganded regulatory subunits bound to catalytic subunits, [R a C] is the concentration with cAMP bound to site a, [R b C] is the concentration with cAMP bound to site b, and [R ab C] is the concentration with cAMP bound to sites a and b. The rate constants for cAMP binding and unbinding to sites a and b are k fa, k fb, and k ra, k rb. The rate constants for the dissociation and association of the R and C subunits are k act and k deact. Note that the dissociation and association of the R and C subunits are considered irreversible reactions. The concentration of free cAMP and activation of CNG channels are described as follows:
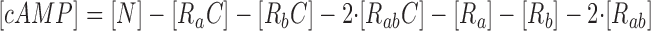 |
(21) |
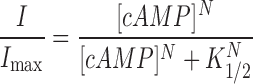 |
(22) |
where I/I max is the fractional current through CNG channels, K 1/2 is the concentration of cAMP that elicits half-maximal current, and N is the Hill coefficient.
TABLE I.
Parameters Used to Simulate Transient cAMP Signals Near the Plasma Membrane of HEK-293 Cells
| Parameter | Parameter definition | Value | Initial condition | Reference |
|---|---|---|---|---|
| [βtot] | Total β2AR concentration | 1 μM | ** | |
| [β] | Inactive receptor concentration | 0.2 μM | ||
| [β*] | Activated receptor concentration | 0.8 μM | ||
| [βdesense] | Desensitized receptor concentration | 0 μM | ||
| [βint] | Internalized receptor concentration | 0 μM | ||
| kGRK | Rate constant of receptor phosphorylation | 0.005 s−1 | Tran et al., 2004 | |
| krpp | Rate constant of receptor dephosphorylation | 0.0005 s−1 | Iyer et al., 2006; Tran et al., 2007a | |
| kint | Rate constant of receptor internalization | 0.01 s−1 | ||
| krec | Rate constant of receptor recycling | 0.005 s−1 | ||
| kact | Rate constant of Gs activation of β2AR | 15 s−1 | Frace et al., 1993 | |
| kh | Rate constant of GTP hydrolysis | 0.8 s−1 | Frace et al., 1993 | |
| KD | Dissociation constant of isoproterenol | 250 nM | Whaley et al., 1994 | |
| Gstot | Total concentration of Gs | 4 μM | ** with initial estimate from Post et al., 1995 | |
| Kc | Equilibrium constant between activated receptor and G protein | 15 μM | ||
| KGsAC | Equilibrium constant between activated G protein and AC | 315 μM | ||
| ACsyn | cAMP synthesis rate | 10 μM·s−1 | ** | |
| AC0 | Basal cAMP synthesis rate | 0.005 μM·s−1 | ** | |
| [N] | cAMP concentration | 0.5 μM | Beavo et al., 1974 | |
| V max-PDE | Maximal hydrolysis rate of unphosphorylated PDE | 0.15 μM⋅s−1 | Reeves et al., 1987; Rich et al., 2001a, 2007 | |
| V max-PDEp | Maximal hydrolysis rate of phosphorylated PDE | 2.5⋅ V max-PDE | Rich et al., 2007 | |
| K m | Michaelis constant for PDE | 1 μM | Houslay et al., 1998 | |
| K I | Inhibition constant of rolipram | 0.1 μM | Houslay et al., 1998; Richter and Conti, 2004 | |
| K 1/2 | cAMP concentration that half maximally activates CNG channels | 1.1 μM | Rich et al., 2001a,b | |
| N | Hill coefficient of cAMP binding to CNG channels | 2.1 | Dhallan et al., 1990; Rich et al., 2001b | |
| k PKA | Rate constant of PKA-mediated phosphorylation | 0.015 μM−1·s−1 | ** | |
| k pp | Rate constant of PDE dephosphorylation | 0.005 s−1 | ** | |
| [PKA] | PKA concentration | 1 μM | Beavo et al., 1974; Hofmann et al., 1977; Rich and Karpen, 2002 | |
| k fa | Rate constant of cAMP binding to site a of the R subunit | 5 μM−1·s−1 | Doskeland and Ogreid, 1984; Rich and Karpen, 2002 | |
| k fb | Rate constant of cAMP binding to site b | 0.4 μM−1·s−1 | Doskeland and Ogreid, 1984; Rich and Karpen, 2002 | |
| k ra | Rate constant of cAMP dissociation from site a | 1 s−1 | Doskeland and Ogreid, 1984; Houge et al., 1990; Rich and Karpen, 2002 | |
| k rb | Rate constant of cAMP dissociation from site b | 0.2 s−1 | Doskeland and Ogreid, 1984; Houge et al., 1990; Rich and Karpen, 2002 | |
| kPKA act | Rate constant of R and C subunit dissociation | 70 s−1 | Smith et al., 1981; Houge et al., 1990; Huang and Taylor, 1998; Rich and Karpen, 2002 | |
| kPKA deact | Rate constant of R and C subunit association | 0.75 μM−2·s−1 | Smith et al., 1981; Houge et al., 1990; Huang and Taylor, 1998; Rich and Karpen, 2002 | |
| K I-PKI | Inhibition constant of PKI | 2.3 nM | Cheng et al., 1986 |
Initial conditions were estimated based on steady-state parameter values in the presence of basal adenylyl cyclase activity. ** denotes values that were fit to the time course of cAMP signals as described in Rich et al. (2007). Concentrations of enzymes should be considered approximate, whether estimated or experimentally derived, because, with the exceptions of data provided from our work, experimentally derived data were obtained from other cellular systems, either overexpression systems or other cell types. In addition, there have been few studies estimating the localized concentrations of enzymes within signaling complexes.
RESULTS
We and others recently reported that PKA-mediated activation of PDE4 is an important mechanism for shaping GPCR-mediated signals (Terrin et al., 2006; Willoughby et al., 2006; Rich et al., 2007). To better understand the interplay between PDE activation and GPCR desensitization in regulating cAMP signals we examined signaling via the endogenous β2ARs in HEK-293 cells. Cyclic AMP signals were monitored in single cells using CNG channels as real-time biosensors. CNG channels with the C460W and E583M mutations (C460W/E583M channels) were expressed in HEK-293 cells via adenovirus infection. These mutations increase both the specificity and the sensitivity of the channels to cAMP (Rich et al., 2001b). CNG channel activity was measured using the whole cell voltage clamp technique, as described in the Materials and methods. The activation of C460W/E583M channels in response to 20, 50, and 1000 nM isoproterenol is shown in Fig. 1. CNG channel activation in response to 20 nM isoproterenol reached a steady plateau over a 7-min stimulation, indicating a sustained increase in cAMP levels. Conversely, channel activation in response to 50 and 1000 nM isoproterenol was transient, indicating an initial increase and subsequent decrease in cAMP levels near the plasma membrane. We next sought to determine the cellular mechanisms responsible for the transient cAMP signals observed in response to high concentrations of isoproterenol.
Figure 1.

Measurement of isoproterenol-induced cAMP signals near the plasma membrane of HEK-293 cells. HEK-293 cells were infected with CNG-channels that transduce cAMP signals to electrical current. Responses were elicited by exposure of each cell to the indicated concentrations of isoproterenol. Shown are cAMP signals in response to 20 nM (A), 50 nM (B), and 1 μM (C) isoproterenol. All traces are representative of the range of responses from at least five experiments.
We previously reported that PKA-mediated stimulation of PDE4 activity is primarily responsible for the decline in PGE1-induced cAMP signals in HEK-293 cells; that 5-min stimulation with PGE1, forskolin, or isoproterenol triggers a PKA-dependent, two- to threefold increase in PDE4 activity; and that this stimulation of PDE4 is responsible for the decline in PGE1-induced cAMP signals (Rich et al., 2001b, 2007). By analogy, it seemed likely that PKA-mediated stimulation of PDE4 activity also contributes to the decay of cAMP signals triggered by β-adrenergic agonists. To ensure that the time course of PDE4 activation was consistent with this hypothesis, we measured PDE4 activity in response to isoproterenol as described in the Materials and methods. Stimulation of HEK-293 cells with 1 μM isoproterenol triggered a threefold increase in PDE4 activity within 1 min, followed by a slight reduction in activity to a sustained level of twofold over basal (Fig. 2 A). PDE4 activation was inhibited by H89, a PKA inhibitor, in a dose-dependent manner (Rich et al., 2007). H89 did not affect the non-PDE4 activity, which contributes ∼50% of total PDE activity in these cells (unpublished data). Taken together, these data suggest that β-adrenergic agonists trigger PKA-mediated stimulation of PDE4 activity, which may contribute to the decay of transient cAMP signals.
Figure 2.

Effects of PKA on isoproterenol (1 μM)-induced cAMP synthesis and hydrolysis in HEK-293 cells. (A) Time course of isoproterenol-induced stimulation of PDE4 activity in HEK-293 cell extracts. Cells were incubated with 1 μM isoproterenol for the indicated times. At the end of the incubation, cells were harvested and homogenates subjected to PDE activity assays using 1 μM cAMP as substrate. PDE activity was measured in the absence or presence of 10 μM rolipram. The rolipram-inhibited PDE4 activity is reported. It is clear that isoproterenol induced a two- to threefold increase in PDE4 activity. The increase in PDE4 activity was due to PKA-dependent phosphorylation of PDE4 and was prevented by pretreatment with PKA inhibitors (not depicted). (B) cAMP accumulation measured in HEK-293 cells using an enzyme immunoassay. Cells were stimulated with 10 μM isoproterenol for 5 min following pretreatment with vehicle (DMSO, 10 min), 10 μM H89 (10 min), 10 μM rolipram (5 min), or 10 μM H89 (10 min) and 10 μM rolipram (5 min). H89 had little or no effect on isoproterenol-induced cAMP accumulation in the presence of rolipram, indicating that PKA does not substantially stimulate receptor desensitization or inhibit the rate of cAMP synthesis following stimulation with saturating concentrations of isoproterenol. Data are the mean ± SEM of three (A) or four (B) separate experiments, each performed in triplicate.
It has been proposed that PKA-mediated stimulation of GRK activity may have contributed to the time course of cAMP signals (Cong et al., 2001). More recently, several groups presented data consistent with an alternate proposal, that at high agonist concentrations PKA has little effect on β2AR activity (Krasel et al., 2004, 2005; Tran et al., 2004; Violin et al., 2006b). In addition, it is possible that PKA-mediated inhibition of adenylyl cyclase activity could slow the rate of cAMP accumulation. To determine if PKA did indeed inhibit the rate of cAMP synthesis, either by inhibition of adenylyl cyclase activity or by stimulation of GRK activity, we measured the effect of H89 (10 μM) on isoproterenol-induced (10 μM) cAMP accumulation in both the presence and absence of rolipram (a specific inhibitor of PDE4) using enzyme immunoassays (Fig. 2 B). In these experiments we used a higher isoproterenol concentration than in other experiments because H89 is a competitive antagonist of β2ARs (Penn et al., 1999). The data demonstrate that H89 has little or no effect on isoproterenol-induced cAMP accumulation in the presence of rolipram. Thus, PKA does not inhibit cAMP synthesis triggered by saturating concentrations of isoproterenol in this cellular system. These data are consistent with our previous work demonstrating that H89 had little or no effect on forskolin- or PGE1-induced cAMP accumulation in the presence of rolipram (Rich et al., 2007).
To further elucidate the molecular mechanisms underlying near-membrane cAMP signals, we monitored isoproterenol-induced C460W/E583M channel activity in the presence of rolipram, PKI (a peptide inhibitor of PKA), PTX (an inhibitor of Gi), St-Ht31 (a peptide that disrupts AKAP-PKA complexes), and 59-74E (a peptide inhibitor of GRK characterized by Winstel et al., 2005). Inhibitors were included in the pipette solution to ensure that we exposed the intracellular space to known inhibitor concentrations. Representative traces of cAMP signals in the presence of these compounds are shown in Fig. 3 (A–G). The percentage of current remaining 3 min after the peak current was used to quantify the extent to which the different compounds altered the decay in cAMP signals (Fig. 3 H). Including rolipram (10 μM) in the pipette solution abolished the decline in the isoproterenol-induced cAMP signal, demonstrating the significance of PDE4 activity in shaping near-membrane cAMP signals. It is important to note that rolipram will inhibit both basal and stimulated PDE4 activity (∼99% of all PDE4 activity at 10 μM rolipram and 1 μM cAMP); whereas PKI will only inhibit PKA-mediated processes such as the stimulation of PDE4 activity, leaving basal PDE4 activity intact. When either PKI (20 nM) or St-Ht31 (10 μM) were included in the pipette solution, no significant effects on isoproterenol-induced signals were observed (Fig. 3, B and C). These results were in stark contrast to our previous observations that both PKI and St-Ht31 prevented the decline in PGE1-induced cAMP signals in the same cells (Rich et al., 2007). Thus, it was apparent that mechanisms other than stimulation of PDE4 activity contribute to shaping the time course of isoproterenol-induced cAMP signals.
Figure 3.

Pharmacological profiling of the molecular mechanisms underlying transient isoproterenol-induced cAMP signals. Responses were elicited by exposure of CNG channel–expressing cells to 1 μM isoproterenol. (A) The decline in the cAMP transients was abolished when 10 μM rolipram (a PDE4 inhibitor) was included in the patch pipette. The decline in the response remained largely intact when 20 nM PKI (a PKA inhibitor, B) or 10 μM St-Ht31 (an AKAP-PKA disruptor, C) were included in the patch pipette, indicating the PKA was not solely responsible for the decline in the signal. (D) An 18–24-h pretreatment with PTX (an inhibitor of Gi) had little effect on the overall kinetics of the signal. (E) 3 μM 59-74E (a GRK inhibitor) did not prevent the decline in the cAMP transients. However, the decline in the response was largely eliminated when both PKI and 59-74E (F) or St-Ht31 and 59-74E (G) were included in the patch pipette. (H) Data were quantified by measuring the percentage of current remaining 3 min after the peak current.
One potential mechanism responsible for the decline in cAMP levels is Gi-mediated inhibition of adenylyl cyclase activity. Several studies have demonstrated that purified β2AR or a synthetic peptide corresponding to the third intracellular loop of the β2AR can activate Gi in reconstituted systems, albeit with a significantly lower coupling efficiency relative to Gs (Cerione et al., 1985; Rubenstein et al., 1991). To test this possibility, we treated cells with 100 ng/ml PTX for 18–24 h before measuring CNG channel activity, as described in the Materials and methods. We observed that some PTX-treated cells displayed higher basal cAMP levels and slightly slower rates of decline in the transient cAMP signal (Fig. 3 D); however, these results were not statistically significant (Fig. 3 H). These data indicate that potential β2AR-mediated activation of Gi, and subsequent inhibition of adenylyl cyclase activity, had little or no effect on the time course of cAMP signals in this cellular system.
Another possible mechanism that may underlie the transient responses is GRK-mediated receptor desensitization and the subsequent reduction in adenylyl cyclase activity. To test this possibility, we included the GRK inhibitor 59-74E (3 μM) in the pipette solution. 59-74E inhibits phosphorylation of rhodopsin by purified GRK subtypes 2, 3, and 5 with IC50s of 0.6, 1.5, and 1.6 μM, respectively (Winstel et al., 2005). Again, although small changes in the kinetics of the cAMP signal were observed in some cells (Fig. 3 E), overall, the decay of the signal was not significantly altered (Fig. 3 H). Thus, individually, inhibition of PKA-mediated stimulation of PDE4 activity, preventing Gi activity, or inhibition of GRK activity did not prevent the decline in isoproterenol-induced cAMP signals.
We next sought to determine if multiple signaling pathways worked in concert to control the decline of cAMP signals. To test this possibility we included both PKI (20 nM) and 59-74E (3 μM) in the patch pipette solution. Under these conditions, the decay in isoproterenol-induced cAMP responses was significantly blunted (Fig. 3, F and H). Similarly, when St-Ht31 and 59-74E were included in the patch pipette, the decline of the cAMP transient was blunted (Fig. 3, G and H). Treatment with PTX had no additional effects on cells exposed to either 59-74E or PKI and 59-74E (Fig. 3 H). Based upon these data, we propose that both GRK-mediated desensitization (in the presence of basal PDE activity) and PKA-mediated stimulation of PDE4 activity are each sufficient to dampen cAMP signals.
We then tested the effects of these pharmacological agents on signals elicited during a two-pulse stimulation with isoproterenol. During this protocol, cells were exposed to 1 μM isoproterenol for 5 min, washed for 5 min, and then reexposed to 1 μM isoproterenol (Fig. 4). There was little or no response to the second stimulation with isoproterenol when either vehicle or 20 nM PKI was included in the pipette solution (Fig. 4, A, B, and E). In contrast, there was a substantial response to the second pulse of isoproterenol when 59-74E was included in the pipette solution (Fig. 4, C and E). Interestingly, when both PKI and 59-74E were included in the pipette solution, there was substantial response to the second pulse of isoproterenol (Fig. 4 D), but the relative amplitude of the second response was not significantly greater than with 59-74E alone (Fig. 4 E). The incomplete recovery may be due to incomplete inhibition of GRK activity with 59-74E, or to the presence of a GRK(s) that is not significantly inhibited by 59-74E. Based upon these observations, we propose that GRK-mediated desensitization is primarily responsible for the sustained suppression of cAMP signals during the 5-min wash period.
Figure 4.

GRKs mediate the sustained suppression of isoproterenol-induced cAMP signals. HEK-293 cells were exposed to 1 μM isoproterenol, followed by a 5-min washout, and a rechallenge with isoproterenol at the times indicated. Shown are representative traces of cells exposed to control intracellular solution (A) or cells exposed to intracellular solution containing PKI (B), 59-74E (C), or PKI + 59-74E (D). (E) Ratio of the peak currents elicited by exposure to isoproterenol (second exposure/first exposure).
To gain further insight into the molecular mechanisms underlying the cAMP signals described above, we developed a mathematical model of the system. The model is summarized in the schematic depicted in Fig. 5. Aspects of the model (e.g., PKA-mediated regulation of PDE4 activity) have been published previously (Rich et al., 2007). According to this model, β2ARs couple to Gs which in turn leads to activation of adenylyl cyclase (AC). Conversely, endogenous β2ARs do not couple to Gi in HEK-293 cells, or, if they do, activation of Gi has no significant effect on cAMP signals (Fig. 3). Isoproterenol-induced increases in cAMP concentration subsequently lead to activation of PKA, which exerts its effects on cAMP signals through phosphorylation and stimulation of PDE4 activity. We have excluded other potential mechanisms of PKA-mediated feedback, such as phosphorylation of adenylyl cyclase or β2ARs, because we observed little or no PKA-mediated inhibition of forskolin-induced cAMP synthesis in HEK-293 cells (Rich et al., 2007). This indicates that PKA-dependent inhibition of adenylyl cyclase activity does not play a significant role in shutting down cAMP signals in HEK-293 cells. In addition, we have assumed that at saturating isoproterenol concentrations there is little PKA-mediated desensitization of β2AR because PKA inhibitors (10 μM H89) have no additional effects on isoproterenol-induced cAMP accumulation in the presence of rolipram (Fig. 2 B). This assumption is only valid at high agonist concentration where GRK-mediated desensitization of β2ARs is predominant (Krasel et al., 2004, 2005; Tran et al., 2004; Violin et al., 2006b). For a considered discussion of PKA-mediated desensitization of β2ARs, see Whaley et al. (1994). The equations are detailed in the Materials and methods.
Figure 5.

Schematic presentation of the feedback mechanism primarily responsible for the regulation of β2 adrenergic signaling in HEK-293 cells. Ligand binding to β2ARs leads to activation of adenylyl cyclase and cAMP production. cAMP activates PKA, which in turn phosphorylates and activates PDE4. GRK-mediated phosphorylation of the receptor, and subsequent arrestin binding, is also sufficient to trigger a decay in the near-membrane cAMP signal.
The model reproduces the experimental data presented (compare Figs. 3 and 6). In the presence of either PKI or 59-74E, the transient nature of the signal is maintained; however, in the presence of both PKI and 59-74E the decline in the signal is blunted. Fig. 7 depicts simulations of the two pulse protocol. Panels A, C, E, and G depict currents through CNG channels. Panels B, D, F, and H depict active receptor levels. In these simulations, active receptors are receptors in the surface membrane that have not been phosphorylated. While it is likely that the binding of arrestin, and not receptor phosphorylation, is responsible for desensitizing the receptor (by blocking receptor–G protein interactions), the binding of arrestin to phosphorylated receptors is fast compared with the rate of GRK-mediated phosphorylation (Krasel et al., 2004, 2005), and therefore is not considered in the model.
Figure 6.

Mathematical simulations of the system described by the schematic in Fig. 5. Panels on left depict the signal being measured, panels on right depict the underlying cAMP signals. (A and B) Simulations of normalized current through CNG channels (I/I max) due to isoproterenol-induced cAMP signals under control conditions. (C and D) Simulations of isoproterenol-induced currents and cAMP signals in the presence of 20 nM PKI (included in the pipette solution). Including PKI in the pipette solution inhibits PKA-mediated stimulation of PDE activity, allowing increased cAMP levels, but the signal is still transient due to basal PDE4 activity and GRK-mediated receptor desensitization. (E and F) Simulations describing the effect of GRK inhibition (3 μM 59-74E) on isoproterenol-induced signals. The signals are still transient due to PKA-mediated stimulation of PDE4 activity. (G and H) Simulations of currents and cAMP signals when both PKA and GRK activity are inhibited (with PKI and 59-74E). Inhibiting receptor desensitization and the stimulation of PDE4 activity prevents the decay of isoproterenol-induced signals.
Figure 7.

Simulations of the recovery of isoproterenol-induced signals following a 5-min stimulation and a 5-min washout (the two-pulse protocol). The simulations depict the response of the system to an initial 5-min exposure to 1 μM isoproterenol, followed by a 5-min wash, and then a second exposure to 1 μM isoproterenol. Under control conditions (A), and in the presence of 20 nM PKI (C), only small currents are induced by a second exposure to isoproterenol. (B and D) This was primarily due to a reduction in the number of active receptors. (E and F) Inhibition of GRK activity with 3 μM 59-74E allows a second isoproterenol response that is substantially larger (∼50% of the peak response to the initial pulse) due to an increased number of receptors at the start of the second pulse. (G and H) In the presence of both PKI and 59-74E, the cAMP response was further increased, due to the increased number of receptors at the start of the second pulse, and the loss of PKA-mediated regulation of PDE activity. These simulations are consistent with the data presented in Fig. 4.
Simulations describing the system under control conditions or in the presence of 20 nM PKI show small responses to the second pulse of isoproterenol, consistent with the experimental observations (Fig. 7, A and B). Under these conditions, active receptor levels are still low at the end of the wash period (Fig. 7, B and D), thus there are few receptors (∼16%) available to respond to a second pulse of isoproterenol. However, simulations describing the system in the presence of 59-74E show a marked increase in the number of receptors present (Fig. 7 F), and a substantial response to the second pulse of isoproterenol (Fig. 7 E). Interestingly, simulations depicting both PKI and 59-74E in the patch pipette (Fig. 7, G and H) show a slightly higher second response than experimental observations (compare Fig. 6 D with Fig. 7 G), consistent with the possibility that we are not completely inhibiting GRK activity. Taken together, the data presented in Figs. 1–4 and the simulations presented in Figs. 6 and 7 strongly suggest that either GRK-mediated desensitization of β2ARs or PKA-mediated stimulation of PDE4 activity are sufficient to dampen cAMP signals during a sustained exposure to saturating concentrations of isoproterenol, and that GRK-mediated desensitization is primarily responsible for the sustained suppression of β2AR-mediated signals.
We next used this model to examine the effects of increasing basal adenylyl cyclase activity on the responsiveness of the system (Fig. 8). We let the system reach equilibrium at each level of basal adenylyl cyclase activity (in the absence of receptor activation) to determine the initial conditions (e.g., initial cAMP levels, PKA and PDE activities, etc.). Interestingly, in the presence of small (twofold) increases in basal adenylyl cyclase activity, receptor-mediated cAMP signals were markedly slower with smaller amplitudes. These effects were due to the increased basal cAMP levels activating PKA, and, in turn, PKA stimulating PDE4 activity. In essence, increased basal adenylyl cyclase activity primed PDE-mediated feedback on subsequent cAMP signals. 10-fold increases in basal adenylyl cyclase activity triggered even further reductions in the peak cAMP levels. The higher basal adenylyl cyclase activity also largely eliminated the transient nature of the response, again due to increased PKA and PDE activity.
Figure 8.

Simulated effects of increased basal AC activity on β2AR-mediated cAMP signaling. (A and B)Increasing basal adenylyl cyclase activity from control levels of 0.005 μM/s to 0.05 μM/s increased both the basal PKA and PDE activities, in essence priming the negative feedback loop. The effects of this priming phenomena were (B) smaller cAMP signals leading to (A) decreased peak currents through CNG channels. In addition, the overall kinetics of the signal were slowed with increased basal AC activity, and, at the highest basal AC activity shown, were no longer transient.
Lastly, we used this model to investigate the interactions between receptor levels, GRK-mediated receptor desensitization, and PKA-mediated stimulation of PDE4 activity in regulating isoproterenol-induced cAMP signals by comparing peak cAMP levels elicited during the first and second stimulation of the two pulse protocol (Fig. 9). The black circles depict peak cAMP levels in response to different isoproterenol concentrations measured during the first isoproterenol pulse. The open circles depict the cAMP levels in response to different isoproterenol concentrations measured at the end of the second isoproterenol pulse (following a 5-min pulse to 1 μM isoproterenol and 5-min wash). It should be noted that, as stated above, the effects of PKA-mediated desensitization of β2ARs were not modeled; thus, cAMP levels may be overestimated at low ligand concentrations.
Figure 9.

Simulations demonstrate the effects of receptor levels on [cAMP] in the absence and presence of PDE4 activity. Solid circles represent normalized peak cAMP levels (from model simulations) in response to a 5-min stimulation with isoproterenol. Open circles represent cAMP levels measured at the end of the second stimulation with isoproterenol (following a 5-min stimulation with 1 μM isoproterenol followed by a 5-min wash). (A–F) The effects of receptor desensitization in the following scenarios: (A) high receptor levels (100 μM) and PDE4 activity inhibited (10 μM rolipram); (B) low receptor levels (1 μM) and PDE activity inhibited; (C) high receptor levels with basal PDE activity (20 nM PKI to inhibit PKA activity); (D) low receptor levels with basal PDE activity; (E) high receptor levels and PKA-mediated regulation of PDE activity intact; (F) low receptor levels and PKA-mediated regulation of PDE activity intact.
Simulations were used to investigate the effects of receptor levels and PDE activity on cAMP signals. In the simulations depicted in Fig. 9 (A and B), PDE activity was inhibited with 10 μM rolipram. When receptor levels were high, receptor desensitization primarily caused a loss of sensitivity (∼threefold right shift in the dose–response curve, Fig. 9 A); whereas, when receptor levels were low, receptor desensitization caused a loss of efficacy (∼50% reduction in cAMP levels, Fig. 9 B).
In the simulations depicted in Fig. 9 (C and D), PKA-mediated stimulation of PDE4 activity was inhibited with 20 nM PKI. This allowed us to determine the effects of basal PDE activity and receptor desensitization on the responsiveness of the system. At high receptor levels, receptor desensitization caused a similar right shift in the isoproterenol dose–response (Fig. 9, compare A and C), as well as a reduction in the efficacy. The reduction in efficacy was more pronounced at higher agonist concentrations (at which cAMP levels more rapidly reached concentrations higher than the K m of the PDE (∼1 μM)). At low receptor levels, basal PDE activity caused a further reduction in efficacy (Fig. 9, compare B and D).
In the simulations depicted in Fig. 9 (E and F), PKA-mediated stimulation of PDE4 activity was left intact. This allowed us to examine the effects of PKA-mediated stimulation of PDE activity in conjunction with receptor desensitization. The simulations demonstrate that when receptor levels were high, stimulation of PDE activity caused an additional loss of sensitivity (an additional right shift in the dose–response curve, Fig. 9 E). When the receptor levels were low, PDE activity further blunted cAMP accumulation during the second isoproterenol pulse (Fig. 9 F). These simulations reveal the extent to which receptor desensitization as well as basal and stimulated PDE activities suppress cAMP signals.
DISCUSSION
Early studies quantitatively examined the complex interplay between receptor desensitization and PDE activity in controlling cAMP signals (Barber et al., 1984, 1992; Barber, 1986). Since that time, a wide range of work has identified the molecular components of β2AR desensitization and the regulation of PDE activity (for reviews see Houslay et al., 1998; Clark et al., 1999; Gainetdinov et al., 2004; Wang et al., 2006; Conti and Beavo, 2007; DeWire et al., 2007). However, relatively few studies have further examined the interplay between desensitization and PDE activity in regulating cAMP levels, and a majority of these studies have relied on either the overexpression or knockdown of β2ARs. In the present work we have taken a slightly different approach. We investigated an endogenous signaling system by monitoring isoproterenol-induced cAMP signals in HEK-293 cells using CNG channels as real-time cAMP sensors. We observed that activation of endogenous β2ARs with isoproterenol (1 μM) triggers transient cAMP signals. The time course of the decline of these signals is similar to the time course of PDE4 activation (Fig. 2), as well as GRK-mediated β2AR phosphorylation and subsequent receptor desensitization (Seibold et al., 2000; Krasel et al., 2004, 2005; Tran et al., 2004). Interestingly, inhibition of either GRK or PKA activity does not significantly alter the time course of the response; whereas inhibition of both GRK and PKA activities significantly reduces the decline in the signal. These data strongly support the hypothesis that both receptor desensitization and PKA-mediated stimulation of PDE4 activity are individually capable of generating a decline in the transient response, and that both processes occur over a similar time frame. When we examined the effects of GRK and PKA inhibitors during a second pulse of isoproterenol, we observed that GRK-mediated desensitization is primarily responsible for a more sustained suppression of β2AR-mediated signaling. In addition, the high resolution cAMP measurements presented here have allowed us to develop a kinetic model of this system (discussed below).
Local PDE4 Activity Is Sufficient to Ensure Transient cAMP Signals
Several recent studies have identified other mechanisms that may contribute to the regulation of cAMP signals in HEK-293 cells. For example, it has been proposed that PDE4 binds to arrestin and is subsequently recruited to the receptor (Perry et al., 2002; Houslay and Baillie, 2005). This process would allow PDE4 to regulate PKA-mediated phosphorylation of β2ARs (Houslay and Baillie, 2005). In addition, it has been suggested that PKA-mediated phosphorylation triggers a switch in the coupling of β2ARs from Gs to Gi (Daaka et al., 1997; Houslay and Baillie, 2005; Lynch et al., 2005); however, the existence of this switch remains controversial and may be cell type specific (Friedman et al., 2002; Schmitt and Stork, 2002). Although we did not directly test for arrestin recruitment of PDE4, previous work (Rich et al., 2001a, 2007; Willoughby et al., 2006) and the results presented here suggest that PKA-mediated stimulation of resident PDE4 activity is sufficient to dampen cAMP signals, even in the presence of sustained adenylyl cyclase activity. In addition, the data presented here demonstrate that treatment with PTX alone, or in conjunction with the GRK inhibitor 59-74E, does not significantly alter isoproterenol-induced cAMP signals mediated by endogenous β2ARs. As such, it is improbable that in this system PKA-mediated phosphorylation switches the coupling of an appreciable fraction of β2ARs from Gs to Gi. Recently, two other groups have come to similar conclusions regarding the coupling of endogenous β2ARs to Gi in HEK-293 cells (Willoughby et al., 2007; Violin et al., 2008).
One likely explanation for the differences in the results of these studies is that much of the work investigating molecular mechanisms of β2 adrenergic signaling has relied on overexpression systems. HEK-293 cells are often used in these studies because they express low levels of endogenous β2ARs. High levels of overexpressed receptors are likely to facilitate low occurrence molecular interactions, such as β2AR coupling to Gi. In addition, we have found that stable overexpression of β2ARs triggers a substantial up-regulation of PDE types 1, 3, and 4 (unpublished data). Expression levels of other relevant proteins, such as GRKs and arrestins, also may be affected. As such, molecular interactions identified in studies using overexpressed β2ARs may be more consistent with cell types expressing high local receptor levels, such as cardiac myocytes.
More recently, siRNA techniques were used to identify specific proteins that regulate cAMP signals in HEK-293 cells. Willoughby et al. (2007) presented data indicating that β-arrestin 2 and specific PDE4D subtypes regulate cAMP signals near the plasma membrane of these cells. They did not examine the effects of siRNA-mediated knockdown of these proteins on other components of the cAMP signaling pathway, such as receptor levels, adenylyl cyclase activity, or basal cAMP levels. The results presented here demonstrate that small changes in basal cAMP levels have dramatic effects on agonist-induced signals. Hence, great care must be taken in interpreting the effects of siRNA-mediated knockdown of proteins on agonist-induced cAMP signals. Regardless, the data presented by these authors clearly implicate the involvement of β-arrestin 2, PDE4D3, and PDE4D5 in the regulation of cAMP signals in HEK-293 cells.
The roles of specific GRKs are less well established. Early studies suggested that GRK2 was primarily responsible for GRK-mediated phosphorylation of β2ARs (Krupnick and Benovic, 1998). More recently, two groups presented data indicating that GRK5 and GRK6 may play important roles in the phosphorylation of β2ARs (Tran et al., 2007b; Violin et al., 2008). Clearly, further research using a variety of techniques will be required to clarify the potential roles of different GRK subtypes in the regulation of β2AR signaling.
Mathematical Modeling of Second Messenger Systems
Recently investigators have begun to use a variety of mathematical modeling techniques to describe signaling systems (Bhalla and Iyengar, 1999; Rich et al., 2000; Saucerman et al., 2003, 2006; Suh et al., 2004; Iancu et al., 2007). These approaches, when used in conjunction with carefully controlled experimental data, have given insight into both the possible and actual signaling paradigms. For example, early experimental work provided strong experimental evidence for the subcellular compartmentalization of cAMP signals (Manganiello and Vaughan, 1973; Corbin and Keely, 1977; Brunton et al., 1981; Jurevicius and Fischmeister, 1996). Further evidence was provided by the discovery of the ubiquitous scaffolding proteins, AKAPs (Sarkar et al., 1984; Bregman et al., 1989; Wong and Scott, 2004). More recently, a series of quantitative studies used modeling in conjunction with experimental approaches to estimate the effective diffusion coefficient for cAMP (Rich et al., 2000), predict effects of cAMP and cGMP compartmentalization (Rich et al., 2001a; Piggott et al., 2006), and examine signals triggered by different GPCR agonists as well as subcellular gradients of cAMP (Saucerman et al., 2006; Iancu et al., 2007; Warrier et al., 2007).
Here we used mathematical models to unravel the complex interactions between receptor levels, receptor desensitization, and regulation of PDE activity in controlling the sensitivity and efficacy of this system. The simulations clearly demonstrate that both GRK-mediated desensitization of β2ARs and PKA-mediated stimulation of resident PDE4 activity can generate declines in agonist-induced cAMP signals, consistent with our interpretation of the data presented here. The simulations also demonstrate the critical importance of basal adenylyl cyclase activity. Small increases in adenylyl cyclase activity trigger modest increases in PKA activity, which, in turn, stimulate basal PDE activity. The increased basal PDE activity dramatically affects the kinetics of subsequent, agonist-induced cAMP signals, slowing the kinetics of the signals and reducing peak cAMP levels. Our model predicts that at higher basal adenylyl cyclase activities, cAMP signals would no longer be transient. These results are consistent with previous studies of photoreceptors in which basal PDE activity had profound effects on the kinetics of the light response (Nikonov et al., 1998; Nikonov et al., 2000). Both the results of Nikonov et al. and those presented here demonstrate that slight increases in cyclase or PDE activity may have dramatic effects on the amplitude and kinetics of agonist-induced responses.
The simulations also reveal that in the absence of PDE activity, receptor desensitization caused a loss of sensitivity in cells with high receptor levels and a loss of efficacy in cells with low receptor levels (Fig. 9). Thus, these simulations are consistent with earlier studies that quantitatively examined β2AR desensitization (Barber et al., 1984; Barber, 1986; Whaley et al., 1994; Clark et al., 1999). The models presented here allowed us to examine the effects of receptor desensitization in the presence of PDE activity. In this case, simulations revealed that, together, these processes were responsible for reductions in the sensitivity and efficacy of the system. We believe this overall approach is critical for understanding the potential roles of a variety of feedback mechanisms in regulating second messenger systems.
Our experimental data and mathematical simulations demonstrate important differences between isoproterenol- and PGE1-induced cAMP signals. Both GPCR agonists trigger transient cAMP responses, yet the mechanisms underlying these responses are different. As described here, both GRK-mediated receptor desensitization and PKA-mediated stimulation of PDE4 activity contribute to the decay in isoproterenol-induced cAMP signals; whereas, PKA-mediated stimulation of PDE4 activity is primarily responsible for the decay in PGE1-induced signals. This important difference is critical to how each system responds to subsequent regulation. For example, our data and simulations clearly demonstrate that prolonged exposure to isoproterenol dramatically reduces the rate of cAMP synthesis. Thus, a subsequent reduction in PDE activity (e.g., by ERK-mediated phosphorylation of PDE4) or an increase in adenylyl cyclase activity (e.g., by Ca2+-calmodulin–mediated stimulation of adenylyl cyclase type 8) would lead to attenuated changes in cAMP levels. Conversely, following prolonged exposure to PGE1, adenylyl cyclase activity is still high, as is PDE4 activity (Rich et al., 2001a, 2007). Subsequent inhibition of PDE activity or stimulation of adenylyl cyclase activity will lead to rapid increases in cAMP levels. Thus, the two GPCR systems are inherently different. In other words, GRK-mediated desensitization not only desensitizes initial GPCR signals, it also, in effect, limits subsequent cAMP production. In systems in which GRK-mediated desensitization occurs more slowly (or in cellular systems with spare receptors stimulated by high concentrations of agonist), sustained receptor activation leads to increased rates of cAMP production and hydrolysis; therefore small changes in PDE or AC activity lead to rapid changes in cAMP levels.
In conclusion, we have resolved the kinetics of β2AR-triggered cAMP signals in HEK-293 cells. In addition, we have used experimental and mathematical modeling approaches to estimate the relative contributions of GRK-mediated desensitization of β2ARs and PKA-mediated stimulation of PDE4 activity in suppressing cAMP signals. The use of mathematical modeling has also allowed us to probe the effects of receptor levels on this signaling system, making it possible to extrapolate from the results presented here to other cellular environments. As such, this work represents an initial step toward the quantitative understanding of cAMP signaling systems. What remains to be determined are the local concentrations of relevant enzymes, including receptors, G proteins, adenylyl cyclase, and phosphodiesterase. Revealing the subcellular concentrations and kinetics of these key signaling enzymes in different cellular settings is the next critical step in understanding how signaling specificity is achieved.
Acknowledgments
We would like to thank Drs. Marco Conti and Jeffrey Karpen for valuable discussions about this research, Drs. Mark Gillespie and Troy Stevens for helpful comments on the manuscript, and Ms. Andrea Britain for technical assistance.
This work was supported by National Institutes of Health (NIH) grants R01-HL074278 and K02-HL081463 (T.C. Rich), and the State of Texas Advanced Technology Program Award 011618–0106-2003 and NIH GM031208 (R.B. Clark).
Olaf S. Andersen served as editor. served as editor.
Abbreviations used in this paper: AC, adenylyl cyclase; AKAP, A-kinase anchoring protein; β2AR, β2 adrenergic receptor; CNG, cyclic nucleotide-gated; GPCR, G protein–coupled receptor; GRK, G protein–coupled receptor kinase; HEK, human embryonic kidney; PDE, phosphodiesterase; PKI, a peptide inhibitor of PKA; PTX, pertussis toxin.
References
- Barber, R. 1986. Discrimination between intact cell desensitization and agonist affinity changes. Mol. Cell. Endocrinol. 46:263–270. [DOI] [PubMed] [Google Scholar]
- Barber, R., T.J. Goka, and R.W. Butcher. 1984. Effects of desensitization on the responses of WI-38 and S49 cells to hormones. Mol. Cell. Endocrinol. 36:29–35. [DOI] [PubMed]
- Barber, R., T.J. Goka, and R.W. Butcher. 1992. Cyclic AMP turnover in intact tissue: role of cyclic nucleotide phosphodiesterases. Adv. Second Messenger Phosphoprotein Res. 25:1–11. [PubMed] [Google Scholar]
- Beavo, J.A., P.J. Bechtel, and E.G. Krebs. 1974. Activation of protein kinase by physiological concentrations of cyclic AMP. Proc. Natl. Acad. Sci. USA. 71:3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla, U.S., and R. Iyengar. 1999. Emergent properties of networks of biological pathways. Science. 283:381–387. [DOI] [PubMed] [Google Scholar]
- Bregman, D.B., N. Bhattacharyya, and C.S. Rubin. 1989. High affinity binding protein for the regulatory subunit of cAMP-dependent protein kinase II-B. Cloning, characterization, and expression of cDNAs for rat brain P150. J. Biol. Chem. 264:4648–4656. [PubMed] [Google Scholar]
- Brunton, L.L., J.S. Hayes, and S.E. Mayer. 1981. Functional compartmentation of cAMP and protein kinase in heart. Adv. Cyclic Nucleotide Res. 14:391–397. [PubMed] [Google Scholar]
- Burns, M.E., and D.A. Baylor. 2001. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu. Rev. Neurosci. 24:779–805. [DOI] [PubMed] [Google Scholar]
- Carr, D.W., R.E. Stofko-Hahn, I.D. Fraser, S.M. Bishop, T.S. Acott, R.G. Brennan, and J.D. Scott. 1991. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 266:14188–14192. [PubMed] [Google Scholar]
- Cerione, R.A., C. Staniszewski, J.L. Benovic, R.J. Lefkowitz, M.G. Caron, P. Gierschik, R. Somers, A.M. Spiegel, J. Codina, and L. Birnbaumer. 1985. Specificity of the functional interactions of the β-adrenergic receptor and rhodopsin with guanine nucleotide regulatory proteins reconstituted in phospholipid vesicles. J. Biol. Chem. 260:1493–1500. [PubMed] [Google Scholar]
- Cheng, H.C., B.E. Kemp, R.B. Pearson, A.J. Smith, L. Misconi, S.M. Van Patten, and D.A. Walsh. 1986. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J. Biol. Chem. 261:989–992. [PubMed] [Google Scholar]
- Clark, R.B. 1986. Desensitization of hormonal stimuli coupled to regulation of cyclic AMP levels. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 20:151–209. [PubMed] [Google Scholar]
- Clark, R.B., and T.C. Rich. 2003. Probing the roles of protein kinases in G-protein-coupled receptor desensitization. Mol. Pharmacol. 64:1015–1017. [DOI] [PubMed] [Google Scholar]
- Clark, R.B., B.J. Knoll, and R. Barber. 1999. Partial agonists and G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 20:279–286. [DOI] [PubMed] [Google Scholar]
- Cong, M., S.J. Perry, F.T. Lin, I.D. Fraser, L.A. Hu, W. Chen, J.A. Pitcher, J.D. Scott, and R.J. Lefkowitz. 2001. Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J. Biol. Chem. 276:15192–15199. [DOI] [PubMed] [Google Scholar]
- Conti, M., and J. Beavo. 2007. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 76:481–551. [DOI] [PubMed] [Google Scholar]
- Corbin, J.D., and S.L. Keely. 1977. Characterization and regulation of heart adenosine 3′:5′-monophosphate-dependent protein kinase isozymes. J. Biol. Chem. 252:910–918. [PubMed] [Google Scholar]
- Daaka, Y., L.M. Luttrell, and R.J. Lefkowitz. 1997. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 390:88–91. [DOI] [PubMed] [Google Scholar]
- Detlev, S., and D. Restrepo. 1998. Transduction mechanisms in vertebrate olfactory receptor cells. Physiol. Rev. 78:429–466. [DOI] [PubMed] [Google Scholar]
- Devic, E., Y. Xiang, D. Gould, and B. Kobilka. 2001. β-Adrenergic receptor subtype-specific signaling in cardiac myocytes from β1 and β2 adrenoceptor knockout mice. Mol. Pharmacol. 60:577–583. [PubMed] [Google Scholar]
- DeWire, S.M., S. Ahn, R.J. Lefkowitz, and S.K. Shenoy. 2007. β arrestins and cell signaling. Annu. Rev. Physiol. 69:483–510. [DOI] [PubMed] [Google Scholar]
- Dhallan, R.S., K.-W. Yau, K.A. Schrader, and R.R. Reed. 1990. Primary structure and functional expression of a cyclic nucleotide-activated channel from olfactory neurons. Nature. 347:184–187. [DOI] [PubMed] [Google Scholar]
- Doskeland, S.O., and D. Ogreid. 1984. Characterization of the interchain and intrachain interactions between the binding sites of the free regulatory moiety of protein kinase I. J. Biol. Chem. 259:2291–2301. [PubMed] [Google Scholar]
- Fain, G.L., H.R. Matthews, M.C. Cornwall, and Y. Koutalos. 2001. Adaptation in vertebrate photoreceptors. Physiol. Rev. 81:117–151. [DOI] [PubMed] [Google Scholar]
- Frace, A.M., P.-F. Mery, R. Fischmeister, and H.C. Hartzell. 1993. Rate-limiting steps in β-adrenergic stimulation of cardiac calcium current. J. Gen. Physiol. 101:337–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, J., B. Babu, and R.B. Clark. 2002. β2-Adrenergic receptor lacking the cyclic AMP-dependent protein kinase consensus sites fully activates extracellular signal-regulated kinase 1/2 in human embryonic kidney 293 cells: lack of evidence for Gs/Gi switching. Mol. Pharmacol. 62:1094–1102. [DOI] [PubMed] [Google Scholar]
- Gainetdinov, R.R., R.T. Premont, L.M. Bohn, R.J. Lefkowitz, and M.G. Caron. 2004. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27:107–144. [DOI] [PubMed] [Google Scholar]
- Hansen, G., S. Jin, D.T. Umetsu, and M. Conti. 2000. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc. Natl. Acad. Sci. USA. 97:6751–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, F., P.J. Bechtel, and E.G. Krebs. 1977. Concentrations of cyclic AMP-dependent protein kinase subunits in various tissues. J. Biol. Chem. 252:1441–1447. [PubMed] [Google Scholar]
- Houge, G., R.A. Steinberg, D. Ogreid, and S.O. Doskeland. 1990. The rate of recombination of the subunits (RI and C) of cAMP-dependent protein kinase depends on whether one or two cAMP molecules are bound per RI monomer. J. Biol. Chem. 265:19507–19516. [PubMed] [Google Scholar]
- Houslay, M.D., and G.S. Baillie. 2005. β-Arrestin–recruited phosphodiesterase-4 desensitizes the AKAP79/PKA-mediated switching of β2-adrenoceptor signalling to activation of ERK. Biochem. Soc. Trans. 33:1333–1336. [DOI] [PubMed] [Google Scholar]
- Houslay, M.D., M. Sullivan, and G.B. Bolger. 1998. The multienzyme PDE4 cyclic adenosine monophosphate-specific phosphodiesterase family: intracellular targeting, regulation, and selective inhibition by compounds exerting anti-inflammatory and antidepressant actions. Adv. Pharmacol. 44:225–342. [DOI] [PubMed] [Google Scholar]
- Huang, L.J., and S.S. Taylor. 1998. Dissecting cAMP binding domain A in the RIα subunit of cAMP-dependent protein kinase. Distinct subsites for recognition of cAMP and the catalytic subunit. J. Biol. Chem. 273:26739–26746. [DOI] [PubMed] [Google Scholar]
- Iancu, R.V., S.W. Jones, and R.D. Harvey. 2007. Compartmentation of cAMP signaling in cardiac myocytes: a computational study. Biophys. J. 92:3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, V., T.M. Tran, E. Foster, W. Dai, R.B. Clark, and B.J. Knoll. 2006. Differential phosphorylation and dephosphorylation of β2-adrenoceptor sites Ser262 and Ser355,356. Br. J. Pharmacol. 147:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius, J., and R. Fischmeister. 1996. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proc. Natl. Acad. Sci. USA. 93:295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout, T.A., and R.J. Lefkowitz. 2003. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 63:9–18. [DOI] [PubMed] [Google Scholar]
- Krasel, C., J.P. Vilardaga, M. Bunemann, and M.J. Lohse. 2004. Kinetics of G-protein-coupled receptor signalling and desensitization. Biochem. Soc. Trans. 32:1029–1031. [DOI] [PubMed] [Google Scholar]
- Krasel, C., M. Bunemann, K. Lorenz, and M.J. Lohse. 2005. β-Arrestin binding to the β2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J. Biol. Chem. 280:9528–9535. [DOI] [PubMed] [Google Scholar]
- Krumins, A.M., and A.G. Gilman. 2006. Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J. Biol. Chem. 281:10250–10262. [DOI] [PubMed] [Google Scholar]
- Krupnick, J.G., and J.L. Benovic. 1998. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 38:289–319. [DOI] [PubMed] [Google Scholar]
- Lagnado, L., and D.A. Baylor. 1992. Signal flow in visual transduction. Neuron. 8:995–1002. [DOI] [PubMed] [Google Scholar]
- Lynch, M.J., G.S. Baillie, A. Mohamed, X. Li, C. Maisonneuve, E. Klussmann, G. van Heeke, and M.D. Houslay. 2005. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with β-arrestin to control the protein kinase A/AKAP79-mediated switching of the β2-adrenergic receptor to activation of ERK in HEK293B2 cells. J. Biol. Chem. 280:33178–33189. [DOI] [PubMed] [Google Scholar]
- Manganiello, V., and M. Vaughan. 1973. An effect of insulin on cyclic adenosine 3′:5′-monophosphate phosphodiesterase activity in fat cells. J. Biol. Chem. 248:7164–7170. [PubMed] [Google Scholar]
- Molday, R.S. 1998. Photoreceptor membrane proteins, phototransduction, and retinal degenerative diseases: The Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 39:2491–2513. [PubMed] [Google Scholar]
- Nikonov, S., N. Engheta, and E.N. Pugh Jr. 1998. Kinetics of recovery of the dark-adapted salamander rod photoresponse. J. Gen. Physiol. 111:7–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov, S., T.D. Lamb, and E.N. Pugh Jr. 2000. The role of steady phosphodiesterase activity in the kinetics of the light-adapted salamander rod photoresponse. J. Gen. Physiol. 116:795–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski, K., and J.L. Benovic. 1991. G-protein-coupled receptor kinases. Trends Biochem. Sci. 16:387–391. [DOI] [PubMed] [Google Scholar]
- Penn, R.B., J.L. Parent, A.N. Pronin, R.A. Panettieri, and J.L. Benovic. 1999. Pharmacological inhibition of protein kinases in intact cells: antagonism of β adrenergic receptor ligand binding by H-89 reveals limitations of usefulness. J. Pharmacol. Exp. Ther. 288:428–437. [PubMed] [Google Scholar]
- Perry, S.J., G.S. Baillie, T.A. Kohout, I. McPhee, M.M. Magiera, K.L. Ang, W.E. Miller, A.J. McLean, M. Conti, M.D. Houslay, and R.J. Lefkowitz. 2002. Targeting of cyclic AMP degradation to β2-adrenergic receptors by β arrestins. Science. 298:834–836. [DOI] [PubMed] [Google Scholar]
- Piggott, L.A., K.A. Hassell, Z. Berkova, A.P. Morris, M. Silberbach, and T.C. Rich. 2006. Natriuretic peptides and nitric oxide stimulate cGMP synthesis in different cellular compartments. J. Gen. Physiol. 128:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polans, A., W. Baehr, and K. Palczewski. 1996. Turned on by Ca2+! The physiology and pathology of Ca2+-binding proteins in the retina. Trends Neurosci. 19:547–554. [DOI] [PubMed] [Google Scholar]
- Post, S.R., R. Hilal-Dandan, K. Urasawa, L.L. Brunton, and P.A. Insel. 1995. Quantification of signalling components and amplification in the β-adrenergic-receptor-adenylate cyclase pathway in isolated adult rat ventricular myocytes. Biochem. J. 311:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh, E.N. Jr., and T.D. Lamb. 1993. Amplification and kinetics of the activation steps in phototransduction. Biochim. Biophys. Acta. 1141:111–149. [DOI] [PubMed] [Google Scholar]
- Pugh, E.N. Jr., T. Duda, A. Sitaramayya, and R.K. Sharma. 1997. Photoreceptor guanylate cyclases: a review. Biosci. Rep. 17:429–473. [DOI] [PubMed] [Google Scholar]
- Reeves, M.L., B.K. Leigh, and P.J. England. 1987. The identification of a new cyclic nucleotide phosphodiesterase activity in human and guinea-pig cardiac ventricle. Implications for the mechanism of action of selective phosphodiesterase inhibitors. Biochem. J. 241:535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich, T.C., and J.W. Karpen. 2002. Cyclic AMP sensors in living cells: what signals can they actually measure? Ann. Biomed. Eng. 30:1088–1099. [DOI] [PubMed] [Google Scholar]
- Rich, T.C., K.A. Fagan, H. Nakata, J. Schaack, D.M.F. Cooper, and J.W. Karpen. 2000. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 116:147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich, T.C., K.A. Fagan, T.E. Tse, J. Schaack, D.M.F. Cooper, and J.W. Karpen. 2001. a. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. USA. 98:13049–13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich, T.C., T.E. Tse, J.G. Rohan, J. Schaack, and J.W. Karpen. 2001. b. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J. Gen. Physiol. 118:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich, T.C., W. Xin, C. Mehats, K.A. Hassell, L.A. Piggott, X. Le, J.W. Karpen, and M. Conti. 2007. Cellular mechanisms underlying prostaglandin-induced transient cAMP signals near the plasma membrane of HEK-293 cells. Am. J. Physiol. Cell Physiol. 292:C319–C331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter, W., and M. Conti. 2002. Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs). J. Biol. Chem. 277:40212–40221. [DOI] [PubMed] [Google Scholar]
- Richter, W., and M. Conti. 2004. The oligomerization state determines regulatory properties and inhibitor sensitivity of type 4 cAMP-specific phosphodiesterases. J. Biol. Chem. 279:30338–30348. [DOI] [PubMed] [Google Scholar]
- Rubenstein, R.C., M.E. Linder, and E.M. Ross. 1991. Selectivity of the β-adrenergic receptor among Gs, Gi's and Go: assay using recombinant α subunits in reconstituted phospholipid vesicles. Biochemistry. 30:10769–10777. [DOI] [PubMed] [Google Scholar]
- Sarkar, D., J. Erlichman, and C.S. Rubin. 1984. Identification of a calmodulin-binding protein that co-purifies with the regulatory subunit of brain protein kinase II. J. Biol. Chem. 259:9840–9846. [PubMed] [Google Scholar]
- Saucerman, J.J., L.L. Brunton, A.P. Michailova, and A.D. McCulloch. 2003. Modeling β-adrenergic control of cardiac myocyte contractility in silico. J. Biol. Chem. 278:47997–48003. [DOI] [PubMed] [Google Scholar]
- Saucerman, J.J., J. Zhang, J.C. Martin, L.X. Peng, A.E. Stenbit, R.Y. Tsien, and A.D. McCulloch. 2006. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc. Natl. Acad. Sci. USA. 103:12923–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, J.M., and P.J. Stork. 2002. PKA phosphorylation of Src mediates cAMP's inhibition of cell growth via Rap1. Mol. Cell. 9:85–94. [DOI] [PubMed] [Google Scholar]
- Seibold, A., B. Williams, Z.F. Huang, J. Friedman, R.H. Moore, B.J. Knoll, and R.B. Clark. 2000. Localization of the sites mediating desensitization of the β2-adrenergic receptor by the GRK pathway. Mol. Pharmacol. 58:1162–1173. [DOI] [PubMed] [Google Scholar]
- Smith, S.B., H.D. White, J.B. Siegel, and E.G. Krebs. 1981. Cyclic AMP-dependent protein kinase I: cyclic nucleotide binding, structural changes, and release of the catalytic subunits. Proc. Natl. Acad. Sci. USA. 78:1591–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer, L. 1991. Visual excitation and recovery. J. Biol. Chem. 266:10711–10714. [PubMed] [Google Scholar]
- Suh, B.C., L.F. Horowitz, W. Hirdes, K. Mackie, and B. Hille. 2004. Regulation of KCNQ2/KCNQ3 current by G protein cycling: the kinetics of receptor-mediated signaling by Gq. J. Gen. Physiol. 123:663–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrin, A., G. Di Benedetto, V. Pertegato, Y.F. Cheung, G. Baillie, M.J. Lynch, N. Elvassore, A. Prinz, F.W. Herberg, M.D. Houslay, and M. Zaccolo. 2006. PGE1 stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J. Cell Biol. 175:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, W.J., and M.M. Appleman. 1971. Multiple cyclic nucleotide phosphodiesterase activities from rat brain. Biochemistry. 10:311–316. [PubMed] [Google Scholar]
- Tran, T., J. Friedman, E. Qunabi, F. Baameur, R.H. Moore, and R.B. Clark. 2004. Full and partial agonist-induced PKA and GRK site phosphorylation of the β2-adrenergic receptor using phospho-site specific antibodies: relationship to desensitization, internalization, and ERK activation. Mol. Pharmacol. 65:196–206. [DOI] [PubMed] [Google Scholar]
- Tran, T.M., J. Friedman, F. Baameur, B.J. Knoll, R.H. Moore, and R.B. Clark. 2007. a. Characterization of β2-adrenergic receptor dephosphorylation: comparison with the rate of resensitization. Mol. Pharmacol. 71:47–60. [DOI] [PubMed] [Google Scholar]
- Tran, T.M., R. Jorgensen, and R.B. Clark. 2007. b. Phosphorylation of the β2-adrenergic receptor in plasma membranes by intrinsic GRK5. Biochemistry. 46:14438–14449. [DOI] [PubMed] [Google Scholar]
- van Koppen, C., M. Meyer zu Heringdorf, K.T. Laser, C. Zhang, K.H. Jakobs, M. Bunemann, and L. Pott. 1996. Activation of a high affinity Gi protein-coupled plasma membrane receptor by sphingosine-1-phosphate. J. Biol. Chem. 271:2082–2087. [DOI] [PubMed] [Google Scholar]
- Violin, J.D., S.M. Dewire, W.G. Barnes, and R.J. Lefkowitz. 2006. a. G protein-coupled receptor kinase and β-arrestin-mediated desensitization of the angiotensin II type 1A receptor elucidated by diacylglycerol dynamics. J. Biol. Chem. 281:36411–36419. [DOI] [PubMed] [Google Scholar]
- Violin, J.D., X.R. Ren, and R.J. Lefkowitz. 2006. b. G-protein-coupled receptor kinase specificity for β-arrestin recruitment to the β2-adrenergic receptor revealed by fluorescence resonance energy transfer. J. Biol. Chem. 281:20577–20588. [DOI] [PubMed] [Google Scholar]
- Violin, J.D., L.M. Dipilato, N. Yildirim, T.C. Elston, J. Zhang, and R.J. Lefkowitz. 2008. β2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J. Biol. Chem. 283:2949–2961. [DOI] [PubMed] [Google Scholar]
- Wang, H.Y., J. Tao, E. Shumay, and C.C. Malbon. 2006. G-protein-coupled receptor-associated A-kinase anchoring proteins: AKAP79 and AKAP250 (gravin). Eur. J. Cell Biol. 85:643–650. [DOI] [PubMed] [Google Scholar]
- Warrier, S., G. Ramamurthy, R.L. Eckert, V.O. Nikolaev, M.J. Lohse, and R.D. Harvey. 2007. cAMP microdomains and L-type Ca2+ channel regulation in guinea-pig ventricular myocytes. J. Physiol. 580:765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whaley, B.S., N. Yuan, L. Birnbaumer, R.B. Clark, and R. Barber. 1994. Differential expression of the β-adrenergic receptor modifies agonist stimulation of adenylyl cyclase: a quantitative evaluation. Mol. Pharmacol. 45:481–489. [PubMed] [Google Scholar]
- Willoughby, D., W. Wong, J. Schaack, J.D. Scott, and D.M.F. Cooper. 2006. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J. 25:2051–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby, D., G.S. Baillie, M.J. Lynch, A. Ciruela, M.D. Houslay, and D.M. Cooper. 2007. Dynamic regulation, desensitization, and cross-talk in discrete subcellular microdomains during β2-adrenoceptor and prostanoid receptor cAMP signaling. J. Biol. Chem. 282:34235–34249. [DOI] [PubMed] [Google Scholar]
- Winstel, R., H.G. Ihlenfeldt, G. Jung, C. Krasel, and M.J. Lohse. 2005. Peptide inhibitors of G protein-coupled receptor kinases. Biochem. Pharmacol. 70:1001–1008. [DOI] [PubMed] [Google Scholar]
- Wong, W., and J.D. Scott. 2004. AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 5:959–970. [DOI] [PubMed] [Google Scholar]
- Xiang, Y., F. Naro, M. Zoudilova, S.L. Jin, M. Conti, and B. Kobilka. 2005. Phosphodiesterase 4D is required for β2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc. Natl. Acad. Sci. USA. 102:909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarfitz, S., and J.B. Hurley. 1994. Transduction mechanisms of vertebrate and invertebrate photoreceptors. J. Biol. Chem. 269:14329–14332. [PubMed] [Google Scholar]
- Yau, K.-W. 1994. Phototransduction mechanism in retinal rods and cones: The Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 35:9–32. [PubMed] [Google Scholar]