

Abstract
Backbone dynamics of the camphor monoxygenase cytochrome P450cam (CYP101) as a function of oxidation/ligation state of the heme iron were investigated via hydrogen/deuterium exchange (H/D exchange) as monitored by mass spectrometry. Main chain amide NH hydrogens can exchange readily with solvent and the rate of this exchange depends upon, among other things, dynamic fluctuations in local structural elements. A fluxional region of the polypeptide will exchange more quickly with solvent than one that is more constrained. In most regions of the enzyme, exchange rates were similar between oxidized high-spin camphor-bound and reduced camphor-and CO-bound CYP101 (CYP-S and CYP-S-CO, respectively.) However, in regions of the protein that have previously been implicated in substrate access by structural and molecular dynamics investigations, the reduced enzyme shows significantly slower exchange rates than the oxidized CYP-S. This observation corresponds to increased flexibility of the oxidized enzyme relative to the reduced form. Structural features previously found to be perturbed in CYP-S-CO upon binding of the biologically relevant effector and reductant putidaredoxin (Pdx) as determined by nuclear magnetic resonance are also more protected from exchange in the reduced state. To our knowledge, this study represents the first experimental investigation of backbone dynamics within the P450 family using this methodology.
Keywords: redox-dependent dynamics, electron transfer, tandem mass spectrometry, hydrogen/deuterium exchange
1. Introduction
The cytochromes P450 are heme-containing monooxygenases that catalyze selective oxidations in both eukaryotic and prokaryotic organisms. The general reaction catalyzed by P450 enzymes is shown in Eq. 1.
| (Eq. 1) |
In humans, P450 enzymes are important in steroid hormone biosynthesis, xenobiotic and drug metabolism and clearance [1]. Cytochrome P450cam (CYP101) from the soil bacterium Pseudomonas putida catalyzes the 5-exo hydroxylation of camphor via two discrete electron transfer steps from the iron sulfur protein cofactor putidaredoxin (Pdx). CYP101 serves a good model for P450s because it is readily soluble, crystal structures are available for multiple forms of the enzyme and its reaction mechanism has been extensively studied [1-3].
Protein dynamics are critical in determining the catalytic efficiency of an enzyme as they play an important role in the stability, flexibility and interconversion of various conformational states that contribute to enzymatic turnover. However, little is known concerning the dynamics of CYP101 and the role they play in function. Enzyme dynamics are responsive to the presence/absence of substrate, allosteric effectors and co-factors; this is particularly relevant to P450s as they require allosteric effectors for turnover. In addition, redox-active proteins often show different dynamics depending upon oxidation state, and redox-dependent dynamics can have functional consequences. For example, the Fe2S2 ferredoxin putidaredoxin (Pdx) is both the physiological reductant and allosteric effector for CYP101, and shows structural and dynamic changes as a function of oxidation state which regulates its binding to CYP101 [4,5]. We wished to determine whether CYP101 also displays redox-dependent dynamics, and chose to use mass spectrometry (MS) to measure hydrogen/deuterium (H/D) exchange of backbone amide hydrogens as a probe of local backbone dynamics as a function of heme oxidation state. Amide hydrogens of proteins are chemical reporters that can describe the flexibility and energetics of the protein backbone and allow investigation of protein-protein interactions and protein folding [6-9]. Main chain amide hydrogens exchange with solvent at a rate inversely dependent upon the degree of protection afforded by local secondary structural elements [10]. A rapidly fluctuating region will exchange amide hydrogens more quickly with solvent than one that is more constrained. Exchange can be readily measured by allowing amide hydrogens to exchange with deuterons under the appropriate conditions, rapidly quenching the exchange reactions (usually by low temperature and acidic conditions), followed by peptic digest and then conducting mass spectrometry (MS) on the peptic segments to determine the extent of H/D exchange.
2. Materials and Methods
2.1 Production and Purification of CYP101
Growth and purification of CYP101 followed published procedures [11,12]. Briefly, the mutant C334A CYP101 was used in which a surface cysteine has been replaced by alanine in order to prevent dimerization. This mutant has been shown to be spectroscopically and enzymatically identical to the wild-type enzyme, but does not aggregate at high concentrations [13]. C334A CYP101 (plasmid pDNC334A) was expressed in Escherichia coli strain NCM533. The inoculants were grown in M9 medium containing trace metals (M9+) at 37 °C. At an optical density (O.D600) of 1.0, 1 mM IPTG and 5 mM camphor was added to induce the protein expression. The cells were incubated for an additional 12 h and harvested by centrifugation and stored frozen at -80 °C.
The frozen NCM533 cell pellet was sonicated in 50 mM Tris-HCl pH 7.4, 1 mM camphor, 50 mM KCl (L-buffer). Protamine sulfate (20 mg/g cell pellet) and ammonium sulfate (490 mg/ml, 70% saturation) was added to the supernatant. The resulting precipitate was dissolved in 5 ml L-buffer and dialyzed against L-buffer overnight at 4 °C. The protein solution was diluted into 100 ml L-buffer and loaded to a DEAE sepharose 20 mL fast flow column (Amersham Biosciences, Piscataway, NJ) followed by another P100 column (Bio-Rad). The purity of the enzyme was determined by measuring absorption ratios A391/A280 on a Hitachi U-2000 UV/visible spectrophotometer. The fractions with absorption ratio greater than 1.4 were used for the following experiments.
2.2 General proteolysis, segment separation and mass spectrometric procedures [9,14]
Fifty μl of exchanged, quenched, and frozen sample was thawed at 1 °C. The thawed solution was immediately pumped over a column (104 μl bed volume) filled with porcine pepsin (Sigma) immobilized on Poros 20 AL media at 30 mg/ml per the manufacturer’s instructions, with 0.05% TFA (200 μl/min) for three min with contemporaneous collection of proteolytic products by a reverse-phase trap column (4 μl bed volume). Subsequently the peptide segments were eluted from the trap column and separated on a C18 column (Magic C18, Michrom BioResources, Inc, Auburn, CA) with HPLC using a linear gradient of 13% solvent B to 40% solvent B over 23 minutes (solvent A, 0.05% TFA in water; solvent B, 95% acetonitrile, 5% solvent A; flow rate 5 μl/min – 10 μl/min). Mass spectrometric analyses were carried out with a Thermo Finnigan LCQ™ mass spectrometer (Thermo Electron Corporation, San Jose, CA) with capillary temperature at 215 °C and needle voltage at 2.2 kV.
2.3 Optimization of Digestion/Separation Conditions
To ensure the highest sequence coverage of the protein for this analysis, various experimental parameters, such as type of protease column, quenching conditions, and HPLC conditions, were optimized prior to the H/D exchange experiments using aqueous exchange buffer instead of deuterated exchange buffer. To quickly identify pepsin-generated peptides for each condition employed, spectral data was acquired in data-dependent MS/MS mode with dynamic exclusion. SEQUEST software program (Thermo Electron Corporation, Xcorr >0.7 for +1, >1.0 for +2, and >1.5 for +3; database, cytochrome P450cam) was used to identify the sequence of the dynamically selected parent peptide ions. This tentative peptide identification was verified by visual confirmation of the parent ion charge state presumed by the SEQUEST program for each peptide. This set of peptides was then further examined to determine if the quality of the measured isotopic envelope of peptides was sufficient to allow accurate measurement of the geometric centroid of isotopic envelopes on deuterated samples. The deuterium buildup curves of analogous peptides were cross-checked for consistency. The best sequence coverage map generated identified peptides for 99% of the protein sequence (410/414 residues, see supplementary material).
2.4 H/D exchange of CYP101 Oxidized and Reduced Forms
Deuterium exchange was initiated by diluting 4 μl of 2 mg/ml oxidized CYP101 in 50 mM Tris-HCl, 50 mM KCl, pH 7.4, 1 mM camphor in H2O with 16 μl of 50 mM Tris-HCl, 50 mM KCl, pH 7.4, 1 mM camphor in D2O. The reaction mixture was incubated at room temperature for 30, 100, 300, 1000 s, respectively. Isotope exchange was quenched by the addition of 30 μl of an ice-chilled acidic buffer (3.2 M GuHCl, 0.8% formic acid, pH 2.5) and by decreasing the temperature immediately in liquid nitrogen after mixing the exchanged and quenched CYP101 (yielding a 50 μl of frozen sample). Reduced CYP101 was obtained by placing the enzyme sample under a carbon monoxide atmosphere prior to and after the addition of 0.25 M sodium dithionite in 50 mM Tris-HCl, pH 7.4. To ensure the samples remained reduced during transfer and incubation, the CYP-S-CO (reduced CO-and substrate-bound CYP101) complexes were placed inside an anaerobic chamber under N2 atmosphere. The CYP-S (oxidized substrate-bound CYP101) complexes were also prepared under anaerobic conditions for the best comparison with CYP-S-CO samples. These partially deuterated frozen samples were then subjected to H/D exchange analysis as above, along with control samples of non-deuterated (run without deuterated buffers) and fully deuterated CYP101 (incubated in 80 mM TCEP-HCl in 80% D2O at 60 °C for two hours).
The centroids of probe peptide isotopic envelopes were measured using a program developed in-house in collaboration with Sierra Analytics. The corrections for back-exchange were made employing methods described previously [8]. Percent deuteration and fractional deuterium incorporation were calculated using Eqs. 2 and 3:
| (Eq. 2) |
| (Eq. 3) |
where m(P), m(N), and m(F) are the centroid value of partially deuterated peptide, non-deuterated peptide, and fully deuterated peptide, respectively. MaxD is the maximum deuterium incorporation calculated by subtracting the number of prolines in the third or later amino acid and two from the number of amino acids in the peptide of interest (assuming the first two amino acids cannot retain deuterons [15]). For CYP101, the deuterium recovery of a fully deuterated sample ((m(F) – m(N)) / MaxD) was an average of 64%.
3. Results and Discussion
3.1 Background on H/D exchange MS
CYP101 was prepared in the oxidized substrate-bound (CYP-S) and reduced substrate-and CO-bound (CYP-S-CO) forms, as previously reported [11,12]. To briefly describe the H/D exchange MS procedure, samples were incubated in D2O for appropriate times under native conditions; exchange was quenched with cold 3.2 M GuHCl, 0.8% formic acid, pH 2.5, followed by passage over an activated pepsin column and HPLC separation of peptic segments prior to MS analysis. The identity of the segments was established by tandem MS-MS methods, yielding better than 99% coverage of the 414-residue CYP101 sequence. Each peptic segment quickly loses the deuterons attached to the first two residues [15]. The N-terminal amide becomes an amino group and loses the deuteron instantaneously when exposed to an aqueous environment, such as, during HPLC separation. While the second amide remains intact, the deuteron on the second amide exchanges on average 25 times faster than the rest of the amides due to the induction effect from the positively charged amino group [15]. Therefore, the information obtained from the peptide segment 1-24 is in fact the deuteration level for the segment 3-24. This accounts for the many gaps seen in Figure 1.
Figure 1.
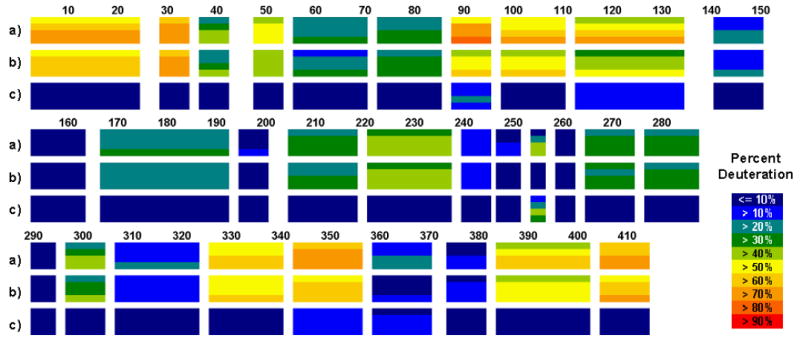
H/D-ex of CYP101 at 25 °C. From top, deuteration level of a) oxidized CYP-S, b) reduced CYP-S-CO and c) difference between CYP-S and CYP-S-CO. Each block represents a peptide fragment that was analyzed and contains four time points, 30, 100, 300, and 1,000 s from top to bottom. The deuteration level and difference in deuteration levels at each time point at each peptide is color-coded as shown at right.
CYP101 showed a well–folded structure; no region of the protein is fully exchanged within 30 s. However, a comparison of CYP-S (oxidized) and CYP-S-CO (reduced) shows well-defined regions with obvious differences in H/D exchange rates as a function of oxidation state (Figure 1 and Table 1).
Table 1.
The average deuteration levels and the average difference in deuteration levels between the oxidized and reduced forms of CYP101a. Structure column indicates to standard helix designations for P450 structures.10
| segment | structure | oxidized | reduced | difference |
|---|---|---|---|---|
| 3–24 | - | 68% | 63% | 5% |
| 29–34 | - | 71% | 72% | 0% |
| 37–42 | A helix | 34% | 31% | 3% |
| 48–53 | - | 49% | 45% | 4% |
| 56–70 | B helix | 27% | 24% | 3% |
| 73–85 | - | 31% | 31% | 0% |
| 88–95 | B’ helix | 72% | 54% | 18% |
| 98–110 | C helix | 63% | 55% | 9% |
| 113–134 | C helix | 59% | 45% | 14% |
| 141–150 | - | 18% | 15% | 4% |
| 153–163 | E helix | -2% | -2% | 0% |
| 167–192 | F helix | 28% | 25% | 3% |
| 195–200 | - | 8% | 5% | 3% |
| 205–218 | H helix | 32% | 29% | 3% |
| 221–237 | - | 42% | 40% | 2% |
| 240–245 | I helix | 15% | 11% | 3% |
| 247–251 | I helix | 10% | 4% | 6% |
| 254–256 | I helix | 29% | -2% | 31% |
| 259–262 | I helix | 2% | 1% | 0% |
| 265–274 | J helix | 33% | 30% | 2% |
| 277–287 | K helix | 33% | 31% | 2% |
| 290–294 | - | 4% | 4% | 1% |
| 297–304 | - | 38% | 33% | 5% |
| 307–323 | - | 18% | 16% | 2% |
| 326–340 | - | 59% | 56% | 3% |
| 343–356 | - | 76% | 59% | 17% |
| 359–370 | L helix | 20% | 9% | 11% |
| 374–381 | L helix | 13% | 12% | 1% |
| 384–402 | H helix | 57% | 52% | 5% |
| 405–414 | - | 67% | 65% | 2% |
Values are based on average deuteration levels taken at 4 time points of 30, 100, 300, and 1000 s.
The rate of exchange of amide hydrogens depends upon the extent of protection afforded by hydrogen bonding in regular secondary structure. For exchange to occur, local fluctuations must separate the H-bond donor-acceptor pair by 5 Å or more [16]. The measured rate of exchange can be used to provide an estimate of the equilibrium constant for opening of the structure. Figure 2 shows representative data for two segments; one represents a large increase in deuteration over time while the other undergoes little exchange. The deuterium buildup curves would appear distorted if there were significant differences between the exchange rates. Alternatively, results presented here and elsewhere are shown as a percent deuteration after a specified time [9,14].
Figure 2.
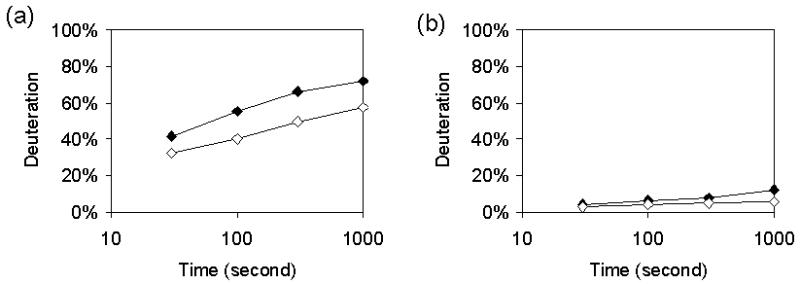
Representative data from fragments 113-134 containing residues in the C and D helices (left) and 195-200 (right). Graphs represent deuterium content of oxidized CYP-S (black) and reduced (white) CYP-S-CO as a function of time measured at 30, 100, 300, and 1000 s. Times are plotted on a log scale. Fragment 113-134 illustrates a significant difference in percent deuteration between oxidation states.
The level of deuteration is determined by MS from the average molecular mass of a segment, which depends on both the masses and relative intensities of the isotopic peaks, with an uncertainty in the average molecular mass of the peptides of 0.02-0.2 Da [17]. In all segments for which differences are observed, the extent of H/D exchange is greater for the oxidized CYP-S than for the reduced CYP-S-CO. This indicates that the oxidized form exchanges more readily with solvent, implying a more dynamic structure than the reduced form.
3.2 Reduced CYP-S-CO is less dynamic than the oxidized CYP-S
Segments corresponding to residues 88–95, 113–134, 254–256, 343–356, and 359–370 show between 11% and 31% higher deuteration levels in CYP-S than in CYP-S-CO, and segment 98–110 shows 9% greater deuteration in CYP-S (Figures 1 and 3). These correspond to portions of the B’ and C helices, both of which are perturbed upon binding of effector (Pdx) as determined by nuclear magnetic resonance (NMR) methods, as well as portions of the I and L helices and the loop of polypeptide containing the axial heme cysteinyl ligand, Cys 357 [12]. The side chains of residues on the B’ and I helices interact directly with substrate in the CYP101 active site, and movements of the B’ helix have been implicated in substrate access and egress from the active site by crystallographic studies and dynamic simulations [18,19].
Figure 3.
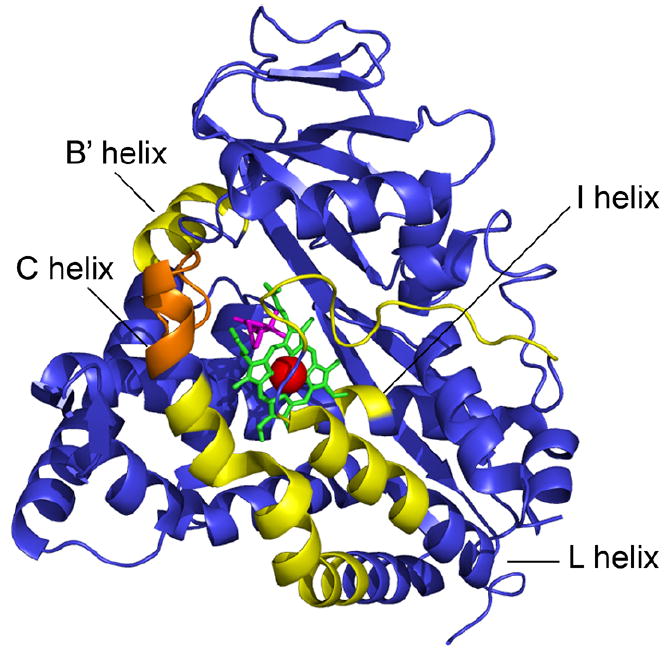
CYP101 structure (PDB entry 3CPP) colored to reveal differences in deuteration levels between the oxidized CYP-S and reduced CYP-S-CO states. Yellow represents a greater than 10% change in deuteration levels between the two states; orange represents a 9% difference in the levels of deuteration. Heme is colored green, iron red, and camphor magenta. Decreased areas of deuteration correspond to a change in flexibility of the backbone. Yellow and orange areas are more flexible in oxidized CYP-S than in the reduced CYP-S-CO, indicating a less dynamic reduced form.
The C helix and the loop containing the axial heme ligand Cys 357 form part of the effector (Pdx) binding site on the proximal surface of CYP101, and NMR resonances corresponding to these features are strongly perturbed in CYP-S-CO upon binding of reduced Pdx [11,12,20]. Arg 109 and Arg 112 on the C helix (residues 106–120) are proposed to interact with negatively charged residues on the surface of Pdx, proximal to the heme binding site on CYP101. Segments 98-110 and 113–134, which are also in the Pdx binding region, show a 9% and 14% difference in deuteration between the oxidized and reduced forms, respectively. Also showing slower H/D exchange in CYP-S-CO are segments corresponding to the Cys 357 loop and nearby L helix, with the segment from 343-356 showing a 17% decrease in H/D exchange and that from 359-370 showing an 11% decrease upon reduction. These findings indicate that CYP-S-CO has more restricted conformational fluctuations than CYP-S, particularly in secondary structure adjacent to the heme. Also, the decreased flexibility of CYP-S-CO in the Pdx binding site relative to CYP-S complements a corresponding decrease in dynamics observed upon reduction of Pdx, and supports the proposal that the binding interactions between the two redox partners are largely modulated by redox-dependent dynamics [4].
3.3 Differences in dynamics between the two oxidation states of CYP101 may account for differences in long-range perturbations due to Pdx binding
We have previously observed long-range perturbations in the distal region of CYP-S-CO adjacent to the active site upon binding of Pdx by NMR, and have suggested a possible route for mechanical transmission of those perturbations due to Pdx binding to CYP-S-CO at the proximal face [11]. This transmission could occur via contacts between the C helix and the N-terminal region of the I helix, and through the loop regions connecting the B’ and C helices, the C helix being adjacent to the Cys 357 loop. Such mechanical transmission is likely more efficient in the less mobile CYP-S-CO, and indeed, we see no corresponding Pdx-induced perturbations in CYP-S by NMR. From current results, residues in the distal B’ helix (90-96) and portions of the I helix are less dynamic in the reduced enzyme, with a segment (88-95) corresponding to the B’ helix showing an 18% decrease in H/D exchange in CYP-S-CO and segment 254–256, representing the middle portion of the I helix, showing a 31% difference in deuteration levels between the CYP-S and CYP-S-CO. The majority of the I helix is within 14 Å of the heme and poses significant challenges to traditional NMR methods for oxidized CYP-S, as 1H resonances are broadened into the noise due to paramagnetic relaxation. The H/D exchange MS unambiguously reveals a change in dynamics of the I helix between the oxidized and reduced forms. The H/D exchange MS of the I helix indicates that the I helix is conformationally more restricted near the active site in the reduced enzyme, which could help constrain substrate in the correct orientation for oxidation.
3.4 Correlation between B-factors and protection factors
Amide hydrogen exchange within native protein structures occurs with a low energy of activation, so that local structural fluctuations and H/D exchange rates can be correlated. This allows interpretation of exchange rate as an indicator of flexibility and dynamics. A correlation between the temperature factor (B-factor) of a crystal structure and the exchange rate of amide hydrogens has been observed [9,21].
The H/D exchange protection factor for each segment was calculated for CYP-S-CO. Protection factors are defined as the ratio of the exchange rate of a given solvent-exposed backbone amide hydrogen from model peptides to the observed exchange rate of the exchangeable amide hydrogen within a folded protein [22]. Protection factors were calculated based on the following analysis [9]. First, the intrinsic exchange rate for each amino acid of CYP-S-CO was calculated using the program HXPEP (http://hx2.med.upenn.edu/download.html). Next, the deuterium buildup curve for each segment was calculated from the intrinsic rates and a new set of exchange rates was obtained by dividing the intrinsic exchange rate by a single protection factor for each segment. A number of fits were iterated to optimize exchange rates and protection factors. In the present case, this analysis provides only a rough estimate of the protection factors due to the limited number of time points. Average B-factor values for each segment were also calculated. B-factor values from the crystal structure are given for each amino acid. On the other hand, the resolution H/D exchange data of this protein was on average 14 amino acids (414 residues divided into 30 segments). For comparison, the B-factors for each segment used in the MS analysis were averaged. The R-square value of the protection factor and the average B-factor for each segment was calculated.
A can be seen from Figures 4 and 5, the protection factors for CYP-S-CO correlate reasonably well with B-factors for main-chain atoms from the crystal structure of oxidized camphor bound CYP101 (1DZ4) [23]. This suggests to us that the oxidized enzyme in the crystal lattice has a restricted range of conformations available to it relative to those observed for the oxidized enzyme in solution, and conversely, that similar conformational restrictions take place upon reduction and CO binding as in the conformational selection that must occur during the crystallization process.
Figure 4.
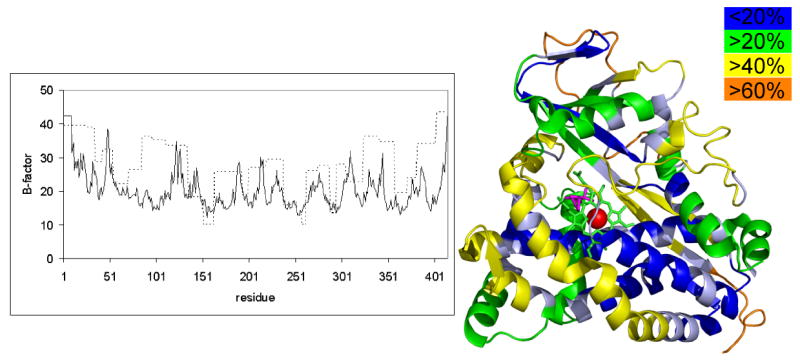
(Left) B- factor, solid line, from (PDB entry 1DZ4) and H/D exchange protection factor, dotted line, for reduced cytochrome P450cam. (Right) CYP101 structure colored to reveal deuteration levels in the reduced CYP-S-CO state. Blue areas are less than 20% deuterated in the reduced state, green areas are greater than 20% deuterated, yellow is greater than 40% deuterated, and orange is greater than 60% deuterated. Areas of high protection factor correspond to areas of low deuteration, shown here in blue and green.
Figure 5.
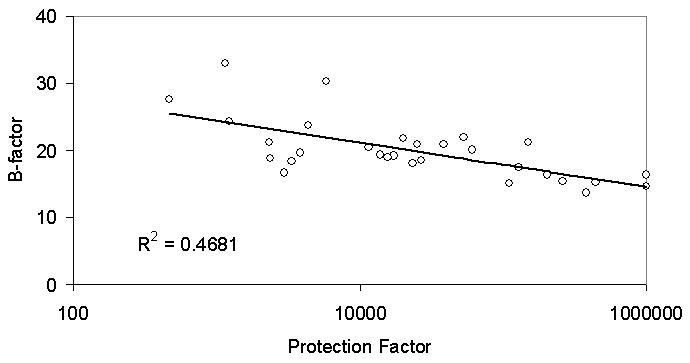
Correlation between B-factor (1DZ4) and logarithm of H/D exchange protection factor for reduced cytochrome P450cam.
4. Conclusions
Overall, CYP101 shows a well-ordered structure, and none of the peptide segments are completely exchanged before the first time point of 30 s. Five segments exchanged significantly faster in the oxidized CYP-S than in the reduced CYP-S-CO, corresponding to the B’, C, I and L helices, and the loop containing Cys 357, the axial heme iron ligand. Our current hypothesis is that reduction of the enzyme decreases the number of accessible conformational substates. The effect of such conformational restriction on the distal side (the B’, I and L helices) helix is to render loss of substrate less likely (by preventing the opening of transient channels to the surface) and increase a particular orientational preference for substrate in the active site. The effect on the proximal side (the C helix and the Cys 357 loop) would be to decrease the entropy price to be paid upon binding of effector (Pdx) [11].
The dynamic changes in the distal region around the active site, while inferred from previous NMR results, could not be completely supported previously, as key residues in the active site cannot be assigned due to paramagnetic broadening in the oxidized enzyme. However, the MS data clearly demonstrates that the I helix, which is a central structural feature of CYP101 and provides a primary contacts with substrate in the active site, exhibits a decrease in dynamics upon reduction. CYP101 oxidizes camphor with nearly perfect efficiency and is typically a much less efficient enzyme for oxidation of other hydrocarbons. However, many P450 enzymes, especially those involved in drug and xenobiotic mtabolism in eukaryotes, are more promiscuous and their active sites can often accommodate multiple substrate molecules simultaneously. All of the P450 enzymes for which structures have been determined feature a helix backbone similar to the I helix in CYP101, and mutagenesis studies combined with NMR and MS dynamic investigations could provide crucial clues as to whether the specificity of the P450 substrate binding is dependent upon dynamics in this helix.
The mechanism by which the redox-dependent modulation of dynamics occurs in CYP101 remains unknown, but an analogy with the situation in Pdx might provide some insight. Decrease in the global dynamics of Pdx upon reduction appear to be related to the increased negative charge on the Fe-S cluster in that protein after reduction, which in turn causes a shortening of hydrogen bonds between the backbone amides of the Fe-S cluster binding loop and concomitant puckering of that loop. This puckering draws the ends of the loop closer to the metal center, and decreases the conformational space available for fluctuations in the adjacent regions of the protein [24]. In the case of CYP101, reduction of the heme may require protonation of nearby functionality to compensate for the decreased positive charge on the iron. One obvious candidate for protonation is the heme propionate, which is closely associated with the imidazole of His 355. In turn, His 355 is located in and between secondary structural features that show redox dependent dynamics, and as such is a likely candidate for transmitting redox-dependent charge effects to the affected secondary structures. We note that a conserved His residue serves such the role of transmitting redox-dependent dynamic changes in vertebrate-type ferredoxins [25].
The combination of NMR and MS is proving to be an invaluable combination for detecting global motions that are mechanistically important in this enzyme. To our knowledge, while this is not the first example of the application of segment-specific MS analysis for a P450 enzyme (REF), it is the first to analyze the conformational dynamics in a cytochrome P450 via H/D exchange MS methods. While we have used this methodology to examine redox-dependent dynamics, it is clear that this method is applicable to the characterization of other types of structural perturbations, such as substrate and effector binding, and we will describe such experiments in future publications.
Supplementary Material
Sequence coverage of the proteolytic digest of CYP101 as well as H/D exchange curves for all segments of both CYP-S and CYP-S-CO are available as supplementary material.
Acknowledgments
This work was supported by US PHS grant RO1-GM44191 (TCP).
Abbreviations used
- CYP101
cytochrome P450cam
- CYP-S
oxidized substrate-bound cytochrome P450cam
- CYP-S-CO
reduced CO- and substrate-bound cytochrome P450cam
- H/D exchange
hydrogen/deuterium exchange
- GuHCl
guanidinium hydrochloride
- HPLC
high pressure liquid chromatography
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- Pdx
putidaredoxin
- TCEP-HCl
tris(2-carboxyethyl)phosphine hydrochloride
- TFA
trifluoroacetic acid
- Tris-HCl
tris(hydroxymethyl)aminomethane hydrochloride
References
- 1.Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Plenum Press; New York: 1995. [Google Scholar]
- 2.Guengerich FP. J Biol Chem. 1991;266:10019–10022. [PubMed] [Google Scholar]
- 3.Poulos TL, Finzel BC, Howard AJ. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 4.Lyons TA, Ratnaswamy G, Pochapsky TC. Protein Sci. 1996;5:627–639. doi: 10.1002/pro.5560050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sevrioukova IF. J Mol Biol. 2005;347:607–621. doi: 10.1016/j.jmb.2005.01.047. [DOI] [PubMed] [Google Scholar]
- 6.Baerga-Ortiz A, Hughes CA, Mandell JG, Komives EA. Protein Sci. 2002;11:1300–1308. doi: 10.1110/ps.4670102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horn J, Kraybill B, Petro EJ, Coales SJ, Morrow JA, Hamuro Y, Kossiakoff A. Biochemistry. 2006;45:8488–8498. doi: 10.1021/bi0604328. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Smith DL. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Englander SW, Kallenbach NR. Q Rev Biophys. 1984;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 11.Rui L, Pochapsky SS, Pochapsky TC. Biochemistry. 2006;45:3887–3897. doi: 10.1021/bi052318f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pochapsky SS, Pochapsky TC, Wei JY. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- 13.Nickerson DP, Wong LL. Protein Eng. 1997;10:1357–1361. doi: 10.1093/protein/10.12.1357. [DOI] [PubMed] [Google Scholar]
- 14.Hamuro Y, Coales SJ, Southern MR, Nemeth-Cawley JF, Stranz DD. J Biomol Tech. 2003;14:171–182. [PMC free article] [PubMed] [Google Scholar]
- 15.Bai YW, Milne JS, Mayne LC, Englander SW. Proteins Struct Funct Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Englander SW. Annu Rev Biomol Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 17.Smith DL, Deng Y, Zhang Z. J Mass Spectrom. 1997;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 18.Poulos TL, Finzel BC, Howard AJ. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
- 19.Ludemann SK, Lounnas V, Wade RC. J Mol Biol. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- 20.Westlake ACG, Harford-Cross CF, Donovan J, Wong LL. Eur J Biochem. 1999;265:929–935. doi: 10.1046/j.1432-1327.1999.00793.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Post CB, Smith DL. Biochemistry. 1996;35:779–791. doi: 10.1021/bi952227q. [DOI] [PubMed] [Google Scholar]
- 22.Houry WA, Sauder JM, Roder H, Scheraga H. Proc Natl Acad Sci USA. 1998;95:4299–4302. doi: 10.1073/pnas.95.8.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 24.Pochapsky TC, Kostic M, Jain N, Pejchal R. Biochemistry. 2001;40:5602–5614. doi: 10.1021/bi0028845. [DOI] [PubMed] [Google Scholar]
- 25.Kostic M, Bernhardt R, Pochapsky TC. Biochemistry. 2003;42:8171–8182. doi: 10.1021/bi034500r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence coverage of the proteolytic digest of CYP101 as well as H/D exchange curves for all segments of both CYP-S and CYP-S-CO are available as supplementary material.