

Abstract
Neuronal activity in the central nervous system evokes localized changes in blood flow, a response termed neurovascular coupling or functional hyperaemia. Modern functional imaging methods, such as functional magnetic resonance imaging (fMRI), measure signals related to functional hyperaemia in order to determine localization of brain function and to diagnose disease. The cellular mechanisms that underlie functional hyperaemia, however, are not well understood. Glial cells have been hypothesized to be intermediaries between neurons and blood vessels in the control of neurovascular coupling, owing to their ability to release vasoactive factors in response to neuronal activity. Using an in vitro preparation of the isolated, intact rodent retina, we have investigated two likely mechanisms of glial control of the vasculature: glial K+ siphoning and glial induction of vasoactive arachidonic acid metabolites. Potassium siphoning is a process by which a K+ current flowing through glial cells transfers K+ released from active neurons to blood vessels. Since slight increases in extracellular K+ can cause vasodilatation, this mechanism was hypothesized to contribute to neurovascular coupling. Our data, however, suggest that glial K+ siphoning does not contribute significantly to neurovascular coupling in the retina. Instead, we suggest that glial cells mediate neurovascular coupling by inducing the production of two types of arachidonic acid metabolites, epoxyeicosatrienoic acids (EETs) and 20-hydroxyeicosatetraenoic acid (20-HETE), which dilate and constrict vessels, respectively. We show that both light flashes and direct glial stimulation produce vasodilatation or vasoconstriction mediated by EETs and 20-HETE, respectively. Further, we show that the type of vasomotor response observed (dilatation or constriction) depends on retinal levels of nitric oxide. Our data also demonstrate that glial cells are necessary intermediaries for signalling from neurons to blood vessels, since functional hyperaemia does not occur when neuron-to-glia communication is interrupted. These results indicate that glial cells play an important role in mediating functional hyperaemia and suggest that the regulation of blood flow may involve both vasodilating and vasoconstricting components.
The central nervous system (CNS) must receive a continuous supply of blood to match the local metabolic needs of activated neurones. Neuronal activity within a localized brain region evokes increases in blood flow, a response termed functional hyperaemia. This response was first described over a century ago by Roy & Sherrington (1890). Functional hyperaemia is controlled by complex mechanisms involving a co-ordinated interaction between neurones, glial cells and cells of the vessel wall. Owing to the close relationship between these cells, they are called collectively ‘the neurovascular unit’ (Iadecola, 2004). The tone of vascular smooth muscle cells sets the diameter of blood vessels and is the most important factor influencing changes in blood flow. According to Poiseuille’s equation, small changes in the diameter of a vessel can have dramatic effects on blood flow, since flow is proportional to the fourth power of the vessel radius (Badeer, 2001). Vascular tone and diameter are influenced by a variety of factors released from neurones and glia during synaptic transmission (Hamel, 2006; Girouard & Iadecola, 2006).
Here we review recent findings from our laboratory on the role of glial cells in neurovascular coupling in the retina. The retina is the most accessible part of the CNS and is an ideal preparation for studying neurovascular coupling, since retinal neurones can be activated by their natural stimulus, light. In addition, the in vitro retina preserves a relatively intact vascular network, owing to a planar geometry of the blood vessels. Using an isolated retina with preserved vasoactivity to light, our laboratory has investigated two likely mechanisms of glial control of the vasculature: K+ siphoning and glial induction of vasoactive arachidonic acid metabolites.
Although electrically inexcitable (they cannot fire action potentials), glial cells respond to transmitters released from neurones with raised intracellular Ca2+ levels and, in turn, initiate physiological responses through the release of transmitters (Nedergaard, 1994; Schipke & Kettenmann, 2004; Newman, 2005a,b). This special form of Ca2+-based excitability present in glia is correlated with vasomotor responses. Depending on the CNS region studied and the type of preparation used, neuronal activity produces Ca2+ increases in glial cells, followed by vasoconstriction (Mulligan & MacVicar, 2004; Metea & Newman, 2006) or vasodilatation (Zonta et al. 2003; Takano et al. 2006; Metea & Newman, 2006) of adjacent arterioles. Although many vasoactive factors can be produced following activation of glial cells (ions, products of cellular metabolism and transmitters), arachidonic acid metabolites such as prostaglandins, epoxyeicosatrienoic acids (EETs) and 20-hydroxyeicosatetraenoic acid (20-HETE) have been shown to play a key role in glial control of blood flow (see Fig. 3K; Harder et al. 2000; Medhora et al. 2001).
Figure 3. Light stimulation evokes vasodilatation and vasoconstriction, an in vitro model of neuro-vascular coupling in the retina.

A–F, IR-DIC images of arterioles at the vitreal surface of the retina. Shown are vessels before (A) and during light-evoked vasodilatation (B); before (C) and during light-evoked constriction (D); and before (E) and during light-evoked sphincter-like constriction (F). Scale bars represent 10 μm. G, time course of light-evoked vasodilatation in 6 trials. H, time course of vasodilatation can be very fast (latency < 500 ms). I, time course of vasoconstriction in 5 trials. J, the concentration of NO determines the type of vascular response to light stimulation. Raising NO levels increases the percentage of vessels which constrict to light. Below 70 nM NO, light stimulation evokes vasodilatation in all vessels (n = 12). As NO is raised, a greater percentage of vessels constrict. K, schematic diagram of vasoactive metabolites of arachidonic acid. See Metea & Newman (2006) for details.
A second mechanism hypothesized to underlie glial-mediated neurovascular coupling is a specialized form of K+ spatial buffering termed K+ siphoning, through which glial cells control extracellular K+ ([K+]o; Kofuji & Newman, 2004). Potassium siphoning is a process by which an increase in [K+]o generated by active neurons results in K+ influx into glial cells, followed by K+ efflux at distant sites. During K+ siphoning, K+ influx depolarizes glial cells and drives out an equal amount of K+ from distant glial endfeet, where K+ channel density is high (Newman, 1984; Brew et al. 1986). These glial endfeet are directly apposed to blood vessels. Moderate increases in [K+]o around blood vessels produce vasodilatation by hyperpolarizing the smooth muscle cells (Haddy et al. 2006). Thus, K+ siphoning by glia has been considered a likely mechanism of neurovascular coupling (Edvinsson et al. 1993). In the retina, inwardly rectifying Kir4.1 K+ channels carry most of the K+ siphoning current in glial cells and are expressed at high density on the endfeet of Müller cells apposed to blood vessels (Kofuji et al. 2000; Butt & Kamada, 2006).
We have addressed both K+ siphoning-mediated and arachidonic acid metabolite-mediated mechanisms of glial control of vasomotor responses in our experiments. Our data indicate that, contrary to a previous hypothesis (Paulson & Newman, 1987), glial K+ siphoning in the retina does not contribute significantly to neurovascular coupling. Instead, we demonstrate that glial cells induce the production of two types of arachidonic acid metabolites, EETs and 20-HETE, which have vasodilating and vasoconstricting effects, respectively. Further, we suggest that nitric oxide (NO) levels determine whether vasodilatation or vasoconstriction will dominate. Nitric oxide acts as a switch to control the type of vascular response observed following neuronal activity. Our results indicate that the regulation of blood flow may involve co-ordinated vasodilating and vasoconstricting components mediated by glial cells. Our data also show that glial cells are necessary intermediaries for signalling from neurones to blood vessels in the retina, since functional hyperaemia does not occur when neurone-to-glia communication is interrupted.
Potassium siphoning and neurovascular coupling
Müller cells, the principal glia of the retina, are highly permeable to K+. This permeability is based on the expression of Kir4.1 K+ channels (Kofuji et al. 2000, 2002; Kofuji & Connors, 2004). The demonstration of glial K+ siphoning in the retina (Newman et al. 1984; Karwoski et al. 1989; Kofuji et al. 2002), as well as evidence for K+-induced vasodilatation (Chrissobolis et al. 2000; Chrissobolis & Sobey, 2003; Haddy et al. 2006) supports the hypothesis that glial cells dilate vessels by release of K+ (Paulson & Newman, 1987). Vasodilatation in response to increased K+ occurs by activation of vascular smooth muscle Na+,K+-ATPases as well as Kir channels (Haddy et al. 2006). However, it has never been directly demonstrated that glial K+ siphoning mediates neurovascular coupling.
Our laboratory performed two tests to determine whether K+ efflux from glial cell endfeet can initiate vasodilatation (Metea et al. 2007): first, the diameter of retinal arterioles was monitored as K+ efflux from glial cell endfeet was evoked by cell depolarization; and second, vascular responses elicited by flickering light were assessed in Kir4.1 knockout mice, in which K+ efflux from glial cells is substantially reduced (Kofuji et al. 2000).
Potassium efflux, elicited by patch-pipette depolarization of single glial cells (which spreads to many cells in the glial network), failed to generate vascular responses (Fig. 1), suggesting that K+ efflux from glia is not sufficient to initiate smooth muscle cell responses. A second test of the K+ siphoning hypothesis was performed using knockout mice lacking Kir4.1 channels. If K+ siphoning contributes to neurovascular coupling, then light-evoked vasodilatation should be substantially reduced in Kir4.1 knockout animals because these channels are essential for carrying K+ siphoning currents (Kofuji et al. 2000). However, light-evoked vasomotor responses were the same in wild-type and knockout animals (Fig. 2). We conclude that in the retina, K+ siphoning by glia is, at most, a secondary mechanism in neurovascular coupling and is not involved in the initiation of vasodilatation. It is possible that K+ siphoning plays a more subtle, modulatory role in neurovascular coupling, such as influencing the basal vascular tone. However, the in vitro preparation is not well suited to investigate such a mechanism.
Figure 1. Light stimulation, but not glial cell depolarization, evokes vasodilatation.
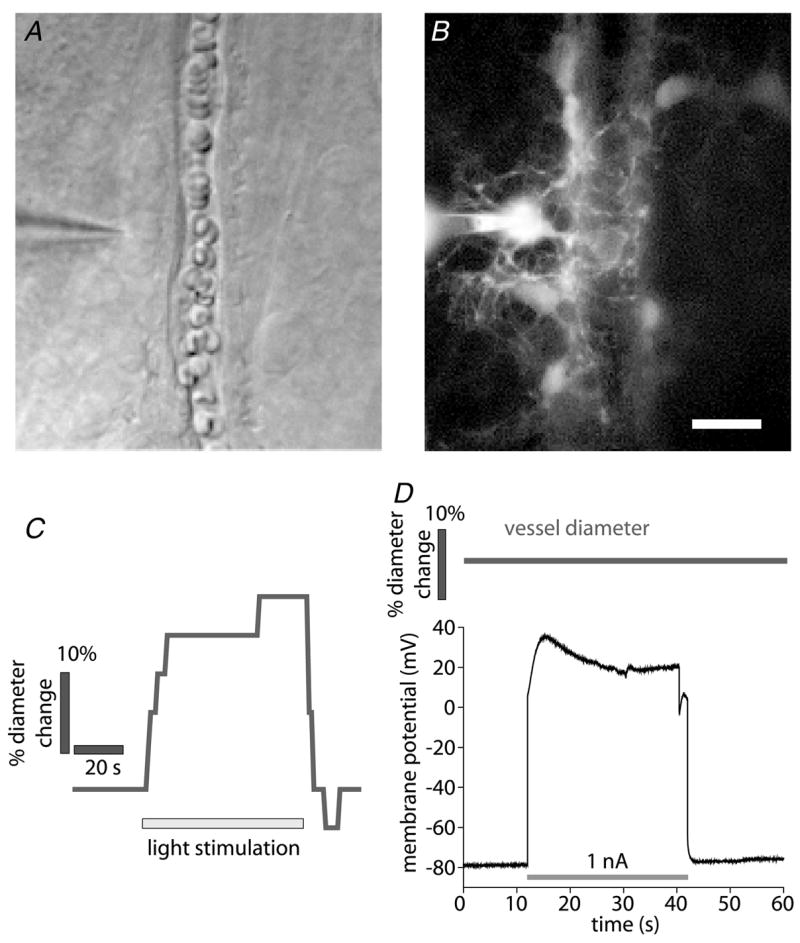
A and B, micrographs showing a whole cell-patched astrocyte contacting an arteriole. The patch pipette is seen at the left. A, infrared-differential interference contrast (IR-DIC) image. B, fluorescence image showing the Lucifer Yellow-filled astrocyte contacting the arteriole. Additional astrocytes coupled to the patched cell are also seen. Scale bar for A and B represents 10 μm. C, time course of vessel diameter change. Flickering light stimulation evokes vasodilatation, demonstrating neurovascular coupling in the retina. D, the astrocyte shown in A and B is depolarized by injection of 1 nA current. Arteriole diameter does not change during the depolarization, indicating that K+ siphoning is not sufficient to initiate vasodilatation. In a series of experiments, glial cells were depolarized to membrane potentials ranging from −40 to +120 mV, sufficient to produce K+ siphoning in many glial cells coupled together. Each cell was depolarized multiple times. No change in arteriole diameter was observed during glial depolarization.
Figure 2. Light-evoked vasodilatation is not reduced in Kir4.1 knockout (KO) mice despite the absence of K+ currents in retinal glial cells.

A, flickering light stimulation evokes vasodilatation of similar amplitude in both wild-type (WT) and Kir4.1 knockout mice.
B, Ba2+-sensitive current–voltage relations of Müller cells from Kir4.1 WT and KO mice. Ba2+-sensitive inward current is completely absent in the KO cell.
Arachidonic acid metabolites and neurovascular coupling
We also investigated the role that vasoactive arachidonic acid metabolites play in neurovascular coupling (Metea & Newman, 2006). Using the light-responsive rat retina preparation, we found that flickering light-induced neuronal activity can elicit either vasodilatation or vasoconstriction of retinal arterioles, depending on the concentration of NO present in the preparation (Fig. 3). Both responses were sensitive to drugs that interfered with arachidonic acid metabolism. EETs acids were responsible for vasodilatation to light and 20-HETE mediated vasoconstriction (Metea & Newman, 2006). Notably, responses of the same vessel changed from vasodilatation to vasoconstriction when the concentration of NO was raised (Fig. 3J), indicating that NO acts as a modulator of these vasomotor responses. The role of NO in determining the type of vasomotricity observed may explain previous contradictory studies concerning the involvement of arachidonic acid metabolites in neurovascular coupling in the brain. Either vasodilatation or vasoconstriction induced by glial cell stimulation has been observed in these studies (Zonta et al. 2003; Mulligan & MacVicar, 2004).
We also investigated whether glial cells mediate vasodilatation and vasoconstriction and whether these responses are correlated with glial cell Ca2+ excitability. Our laboratory has previously shown that following neuronal activity, glial Ca2+ increases are evoked owing to purinergic receptor activation (Newman, 2005a,b). We found that when retinal purinergic signalling is interrupted by an antagonist, neurovascular responses are inhibited. In the presence of purinergic antagonists, experimentally evoked increases in glial Ca2+ still produce vasoactive responses similar to those elicited by light (Fig. 4) (Metea & Newman, 2006). This indicates that glial cells are necessary mediators of neurovascular coupling in the retina. In addition, Ca2+ increases propagated as Ca2+ waves from an initiation site to distant (> 100 μm) blood vessels evoked vasomotor responses (Fig. 5), suggesting that the glial network can mediate neurovascular coupling over large distances.
Figure 4. Activation of glial cells produces the same types of vascular responses as light stimulation.

Glial cells were stimulated by UV photolysis of caged-Ca2+ within the cells. Photolysis evoked propagated Ca2+ increases in networks of glial cells apposed to blood vessels. These Ca2+ increases were correlated with vessel dilatation (A), constriction (B) or sphincter-like constriction (C). These glial-evoked vasomotor responses occurred even when transmitter release from neurones was blocked by tetanus toxin.
Figure 5. Propagated glial Ca2+ waves evoke vasodilatation in distant arterioles.
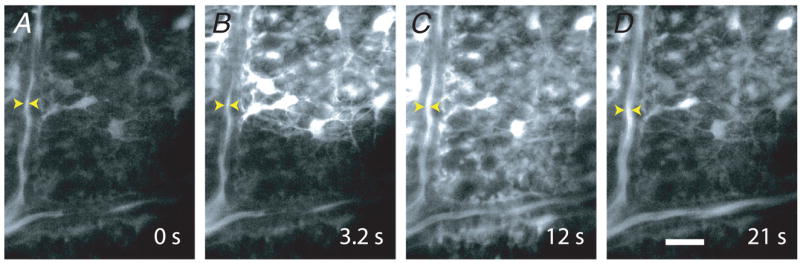
A–D, a Ca2+ wave produced by ejection of ATP propagates through glial cells at the surface of the retina. The Ca2+ wave dilates an arteriole when it reaches the vessel. The site of focal ATP ejection, used to initiate the Ca2+ wave, was just beyond the upper right corner of the images. Scale bar represents 20 μm.
Discussion
Several recent studies indicate that glial cells can act as intermediaries in signalling from neurones to blood vessels within the neurovascular unit (Koehler et al. 2006). In the retina, we have found that glial cells are necessary for neurovascular coupling. When neurone-to-glia purinergic signalling is interrupted, neuronal-induced but not glial-induced vasomotricity is abolished (Metea & Newman, 2006). The precise role of glial cells in mediating neurovascular coupling is difficult to investigate, in part because glial cells, in addition to directly controlling vascular responses, are also key elements in neurometabolic coupling (Magistretti & Pellerin, 1999; Paemeleire, 2002). Although glia play key roles in regulating cerebral blood flow, we presently do not have a clear understanding of all of the complex interactions that occur within the neurovascular unit.
The fact that activated glia are able to produce either vasodilatation or vasoconstriction raises important questions regarding the mechanisms controlling blood flow in the CNS. It is possible that both vasodilatation and vasoconstriction are necessary in order to direct blood precisely to areas of need. We have found that glial cells can produce either dilatation or constriction evoked by the same stimulus but under different NO concentrations. Glial cells sense synaptic activity and respond with Ca2+ increases (Newman, 2005b). These increases may influence NO production, since nitric oxide synthase is Ca2+ sensitive. By controlling NO levels, glial Ca2+ increases could also influence the production of arachidonic acid metabolites, since the synthetic enzymes for EETs and 20-HETE are inhibited by NO. Our data indicate that glial-induced, vasodilating epoxygenase metabolites of arachidonic acid are active at low concentrations of NO, while vasoconstricting 20-HETE is favoured at higher concentrations.
These data suggest a model whereby a heterogeneous spatial activation of glial cells could serve as a mechanism for precise control over the vasculature, redistributing blood flow from constricted to relaxed vessels. In support of this hypothesis, we have also observed sphincter-like vasoconstrictions localized close to capillary branch points and sometimes occurring in a vessel displaying a general vasodilatation (Fig. 3E and F). Sphincter-like vasoconstrictions have also been observed in retinal capillaries (Peppiatt et al. 2006) and in brain vessels (Hamel, 2006), and might underlie a controlled closing of vascular collateral branches in order to maximize blood flow to other areas. It is also possible that blood flow can be directed towards or deflected from entire capillary beds as a result of strategic sphincter precapillary contractions. These active vessel constrictions might account for the generation of complex functional magnetic resonance imaging (fMRI) signals observed following activation of restricted brain regions (Harel et al. 2002).
A fuller understanding of glial function within the neurovascular unit will be necessary in order to develop an accurate model of the regulation of cerebral blood flow and may have an impact on the interpretation of brain function studies. It may also lead to new treatments for types of brain pathology where cerebral circulation is compromised.
Acknowledgments
This work was supported by NIH grants EY004077 to E.A.N. and an NSF predoctoral fellowship to M.R.M.
References
- Badeer HS. Hemodynamics for medical students. Adv Physiol Educ. 2001;25:44–52. doi: 10.1152/advances.2001.25.1.44. [DOI] [PubMed] [Google Scholar]
- Brew H, Gray PTA, Mobbs P, Attwell D. Endfeet of retinal glial cells have higher densities of ion channels that mediate K+ buffering. Nature. 1986;324:466–468. doi: 10.1038/324466a0. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kamada T. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis S, Sobey CG. Inwardly rectifying potassium channels in the regulation of vascular tone. Curr Drug Targets. 2003;4:281–289. doi: 10.2174/1389450033491046. [DOI] [PubMed] [Google Scholar]
- Chrissobolis S, Ziogas J, Chu Y, Faraci FM, Sobey CG. Role of inwardly rectifying K+ channels in K+-induced cerebral vasodilatation in vivo. Am J Physiol Heart Circ Physiol. 2000;279:H2704–H2712. doi: 10.1152/ajpheart.2000.279.6.H2704. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, MacKenzie ET, McCulloch J. Cerebral Blood Flow and Metabolism. Raven Press; New York: 1993. [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Haddy FJ, Vanhoutte PM, Feletou M. Role of potassium in regulating blood flow and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2006;290:R546–R552. doi: 10.1152/ajpregu.00491.2005. [DOI] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059–1064. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Harder DR, Roman RJ, Gebremedhin D. Molecular mechanisms controlling nutritive blood flow: role of cytochrome P450 enzymes. Acta Physiol Scand. 2000;168:543–549. doi: 10.1046/j.1365-201x.2000.00707.x. [DOI] [PubMed] [Google Scholar]
- Harel N, Lee SP, Nagaoka T, Kim DS, Kim SG. Origin of negative blood oxygenation level-dependent fMRI signals. J Cerebr Blood Flow Metab. 2002;22:908–917. doi: 10.1097/00004647-200208000-00002. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Muller (glial) cells. Science. 1989;244:578–580. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. 2006;100:307–317. doi: 10.1152/japplphysiol.00938.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Biedermann B, Siddharthan V, Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A, Reichenbach A. Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia. 2002;39:292–303. doi: 10.1002/glia.10112. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Connors NC. Molecular substrates of potassium spatial buffering in glial cells. Mol Neurobiol. 2004;28:195–208. doi: 10.1385/MN:28:2:195. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci. 1999;354:1155–1163. doi: 10.1098/rstb.1999.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhora M, Narayanan J, Harder D. Dual regulation of the cerebral microvasculature by epoxyeicosatrienoic acids. Trends Cardiovasc Med. 2001;11:38–42. doi: 10.1016/s1050-1738(01)00082-2. [DOI] [PubMed] [Google Scholar]
- Metea MR, Kofuji P, Newman EA. Neurovascular coupling is not mediated by potassium siphoning from glial cells. J Neurosci. 2007;27:2468–2471. doi: 10.1523/JNEUROSCI.3204-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. 2006;26:2862–2870. doi: 10.1523/JNEUROSCI.4048-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- Newman EA. Regional specialization of retinal glial cell membrane. Nature. 1984;309:155–157. doi: 10.1038/309155a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J Neurosci. 2005a;25:5502–5510. doi: 10.1523/JNEUROSCI.1354-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glia and synaptic transmission. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford University Press; Oxford: 2005b. pp. 355–366. [Google Scholar]
- Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paemeleire K. The cellular basis of neurovascular metabolic coupling. Acta Neurol Belg. 2002;102:153–157. [PubMed] [Google Scholar]
- Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237:896–898. doi: 10.1126/science.3616619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy CS, Sherrington CS. On the regulation of the blood-supply of the brain. J Physiol. 1890;11:85–108. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipke CG, Kettenmann H. Astrocyte responses to neuronal activity. Glia. 2004;47:226–232. doi: 10.1002/glia.20029. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]