

Abstract
OBJECTIVES
To perform a genome-wide linkage analysis in a large atrial fibrillation (AF) kindred using AF and abnormally prolonged signal-averaged (SA) P-wave duration as the phenotype.
BACKGROUND
While inherited forms of AF exist, phenotypic complexity has limited efforts to ascertain mutation carriers and thus identify causal genes. The identification of intermediate or endophenotypes may accelerate this effort.
METHODS
A genome-wide linkage analysis was performed in a 4-generation AF kindred of 27 individuals, 8 with AF documented by ECG. The analysis was performed using AF as the phenotype, and repeated using an abnormally prolonged SA P-wave duration as the phenotype.
RESULTS
Linkage analysis and fine mapping generated a maximum multipoint logarithm of the odds (LOD) score of 3.0 at chromosome 5p15 between markers D5S406 and D5S635. Importantly, eight heterozygous carriers had a prolonged SA P-wave (203±21 msec) compared with 17 non-carriers (116±12 msec, P<0.00001). Using prolonged SA P-wave (conventionally defined as >155 msec) as an endophenotype, a maximum LOD score of 3.6 was obtained in the same region of chromosome 5p15, a span of 5.75 cM.
CONCLUSIONS
In a large AF kindred, we have identified a novel AF locus on chromosome 5p15 and demonstrated that affected individuals with AF and mutation carriers can be identified by a prolonged SA P-wave duration. Importantly, identification of an endophenotype in this kindred not only aided ascertainment of additional family members but also increased the LOD score providing increased support for linkage at this locus. Identification of the causal gene, mapped to chromosome 5p15, will advance our understanding of the molecular basis of AF.
Condensed Abstract
While inherited forms of atrial fibrillation (AF) exist, phenotypic complexity has limited efforts to ascertain mutation carriers and identify causal genes. In a large AF kindred, we have identified a new AF locus on chromosome 5p15 and demonstrated that that affected individuals with AF and mutation carriers can be identified by a prolonged signal-averaged (SA) P-wave duration. Identification of an intermediate or endophenotype not only aided ascertainment of additional family members but also increased the LOD score and will accelerate identification of the causal gene in this kindred.
Keywords: atrial fibrillation, familial, genetics, P-wave duration, endophenotype
Atrial fibrillation (AF), the most common cardiac arrhythmia, affects approximately two million Americans (1) and results in substantial morbidity and mortality (2). AF is also the most common arrhythmia requiring drug therapy and the limited success of drug therapy for AF may in part reflect poor understanding of its diverse molecular underlying pathophysiology. Recent studies have provided evidence of a genetic contribution to AF (3,4). Mutations in potassium-channel genes have been associated with familial AF but account for only a small fraction of all cases of AF (5–9). In addition, a number of loci that confer increased vulnerability to AF have been described (9,10). Although Mendelian forms of AF are not rare (3), large kindreds such as those used to identify disease genes in other inherited arrhythmia syndromes such as congenital long QT syndrome (11,12), are unusual. One possible reason for such complexity is reduced penetrance with less apparent heritability, and thus a smaller genetic contribution to the ultimate clinical phenotype. Another possible mechanism is that the phenotype may be the result of interaction of several genes i.e. polygenic, each with a small overall contribution. Finally, AF may require additional environmental events to reduce ‘AF threshold’ sufficiently to trigger an episode of AF (13).
The paroxysmal nature and variable symptoms in AF, a high prevalence in the general population and a late age of onset in many individuals, all make assignment of the clinical phenotype challenging. A number of strategies can be used to minimize misphenotyping, including “affecteds-only” analysis and “diagnosis by offspring”, all of which reduce the power and maximum logarithm of the odds (LOD) score. This complexity has compelled a search for new more effective methods for investigating the genetics of complex diseases, such as AF (14). One approach is to use the families of affected individuals as an enriched target population for the definition and evaluation of novel phenotypes. The psychiatric field has pioneered systematic approaches to phenotyping in an attempt to discover intermediate or endophenotypes, i.e., subtle or novel phenotypes which are causally related to the poorly penetrant classical clinical syndromes (15). One such example is impaired attention, which has proved useful for genetic studies of schizophrenia (16). In the case of AF, examples of potential endophenotypes include SA P-wave duration, pulmonary venous anatomy as assessed by CT or MRI, and profiles of biomarkers (17,18). If such markers of a reduced threshold for AF can be defined, then not only will they be useful clinically (e.g. to subset patients with AF), but they will also accelerate the identification of causal genes.
Signal-averaged P-wave electrocardiography has been shown to have a potential role in identifying patients at risk of developing paroxysmal AF and those likely to progress from paroxysmal AF to chronic AF (19). The SA P-wave ECG measures delayed potentials indicative of delayed intra- and inter-atrial conduction. As conduction delays are critical for the initiation of reentrant arrhythmias, evidence of a prolonged P-wave duration using signal-averaging techniques provides a means by which patients at risk of developing AF can be identified. In this study, we describe the identification of a novel locus for familial AF that is inherited in an autosomal dominant manner. In addition, we show that affected individuals with AF can be identified by a prolonged SA P-wave, whereas non-carriers have normal P-wave duration supporting a primary defect in atrial activation as the cause of AF in this kindred.
Methods
Clinical Evaluations
All studies were performed with the approval of the Institutional Review Board of Vanderbilt University Medical Center. Each family member had a physical examination and a detailed history to identify past medical conditions, symptoms while in AF, and the medical history of first-degree relatives. Each subject was evaluated by 12-lead ECG, echocardiogram, P-wave SA ECG, and laboratory studies including thyroid-stimulating hormone.
The proband and relatives were clinically classified using a consistently applied set of definitions. AF was defined as replacement of sinus P waves by rapid oscillations or fibrillatory waves that varied in size, shape and timing and were associated with an irregular ventricular response when atrioventricular conduction was intact. Documentation of AF on an ECG, rhythm strip, event or Holter monitor was required. Lone AF was defined as AF in individuals <60 years of age without hypertension or overt structural heart disease by clinical examination, ECG and echocardiography. The upper limits of normal for cardiac chamber dimensions were based on age and body surface area (20). We defined as affected those individuals with ECG documented AF. We defined as unaffected only those individuals >60 years of age with no personal history of AF and no offspring with a history of AF. All other family members were defined as unknown for the purpose of initial genetic analyses.
Signal-averaged P-wave duration
All subjects were in sinus rhythm at the time of the SA P-wave recording. In addition, all cardioactive drugs were withdrawn for at least five half lives before the recordings were performed. The methodology of SA P-wave duration recording and analysis has been described previously (19,21). This was recorded using the MAC 5000 (GE Medical Systems, Milwaukee, WI). In brief, the SA P-wave duration was obtained from a relaxed patient in a supine position in a quiet room, free from electrical interference. It incorporates three bipolar orthogonal leads referred to as the X, Y and Z leads, which correspond to those used for the acquisition of the standard ECG. The signal from each lead was amplified to 5 μV/cm and passed through a low bandpass filter of 300 Hz and a high bandpass filter of 40 Hz and then converted from analog-to-digital data to a 12-bit accuracy at the sampling rate of 1 kHz. A specially filtered P-wave derived from the selected dominant sinus P-wave of lead II served as a reference signal for all processing. After passing through a P-wave recognition program to eliminate ectopic atrial beats, >200 beats were averaged, on a trigger point within the specially filtered P-wave. If the noise level remained >0.5 μV, even after 200 beats averaging, the averaging was continued until the peak noise was reduced to <0.5 μV. The filtered signals for the X, Y and Z leads were combined into a vector magnitude, the root mean square (RMS) value. The filtered P wave duration in this vector was calculated. Previous studies have identified an abnormal SA P-wave filtered duration as >155 msec (22).
Genetic Analyses
Whole blood for genomic DNA extraction was obtained from the proband and all consenting family members. Genetic analyses were performed on all available individuals, regardless of affection status. A genome-wide scan was performed by deCODE (Reykjavik, Iceland) using their 546 marker panel and corresponding genetic map (23,24). These microsatellite markers span the human genome by every ~8 centi-Morgans (cM). Genotypes were ascertained without knowledge of clinical status. Fine mapping was performed at candidate loci using additional markers identified at the Genethon and Center for Medical Genetics, Marshfield Medical Research Foundation, databases.
Statistical Analyses
Two-point and multipoint LOD scores were calculated assuming a disease penetrance of 0.95. Allele frequencies were estimated from the population. Two-point LOD scores were calculated with MLINK program (25). To estimate the most likely location for disease, multipoint LOD scores were calculated using the program SIMLINK (26). The parameters used for multipoint linkage analysis were identical to those for 2-point linkage analysis. The distance (cM) between markers was based on the data from the Center for Medical Genetics, Marshfield Medical Research Foundation database. Data for the SA P-wave duration is presented as the mean value ± SD. Comparison of means was performed using Student’s t-test.
Results
Clinical characteristics of the pedigree
In the four-generation family, AF segregated as autosomal dominant trait (Figure 1). The family originated from the Middle Tennessee region of the US. In total 27 family members (18 males and 9 females; ages from 12 to 70 years) were involved in the study, 26 of whom provided DNA samples. Nine family members had the diagnosis excluded as discussed above. One consanguineous marriage occurred in this family. There were eight individuals with documented AF on the ECG. I:1 was coincidentally diagnosed with paroxysmal AF at the age of 41 when her ECG was recorded for occupational reasons. Although the patient had noticed fatigue and decreased exercise tolerance, these complaints had not prompted her to see a physician. At the present time, I:1 has chronic AF that is managed with rate control therapy. I:2 was diagnosed with symptomatic paroxysmal AF at the age of 45 years. He initially responded to antiarrhythmic drugs, but gradually over the next decade, his AF became drug-resistant and he was placed on rate control therapy for chronic AF. As he died at the age of 56, probably of cardiovascular cause, a DNA sample was not available for this subject. The proband (II:18) developed symptoms of palpitations, lightheadedness, and dizziness at the age of 32 but did not seek medical attention until he was in his 40’s. At the present time, he continues to have symptomatic paroxysmal AF that is adequately controlled with a combination of digoxin and beta-blockers.
Figure 1.

Pedigree and haplotypes for family VAF-1. Black symbols indicate individuals affected with atrial fibrillation (AF); gray, obligate carriers; and diagonal lines, deceased individuals. Proband is indicated by arrow. Underlined numbers indicate unaffected individuals used in linkage analyses. Results of genotypic analysis are shown for markers D5S2088, D5S406, D52054, D5635, D5807, and D5S1486 below each individual. Vertical black line to the left of some series of numbers indicates disease haplotype. One consanguineous marriage is indicated by =.
In affected individuals, the mean age of onset of AF was 32±10 (range 18–45) years. In this kindred, AF began with paroxysmal episodes but in two family members (I:1 and I:2) became chronic over time. The QT interval was normal in all affected individuals. In most affected subjects, there was evidence of mild left atrial dilatation and no other evidence of structural heart disease. However, echocardiography did reveal mild concentric left ventricular hypertrophy in 2 subjects (II:16 and II: 20). However, in both these subjects, AF was diagnosed at least 2 years before treatment for hypertension was initiated. No members of this kindred have undergone invasive electrophysiologic evaluation.
Linkage Analysis
We identified preliminary evidence of linkage with the marker D5S2054 (maximum LOD score = 2.4, θ= 0) on chromosome 5p15. Additional markers including D5S2088, D5S406, D5S635, D5S807 and D5S1486 were used for fine mapping. The peak multipoint LOD score of 3.0 was obtained for a region between markers D5S406 and D5S635. These results indicate that the autosomal dominant AF gene is located between markers D5S406 and D5S635, a region of 5.75 cM in length (Figure 2).
Figure 2.
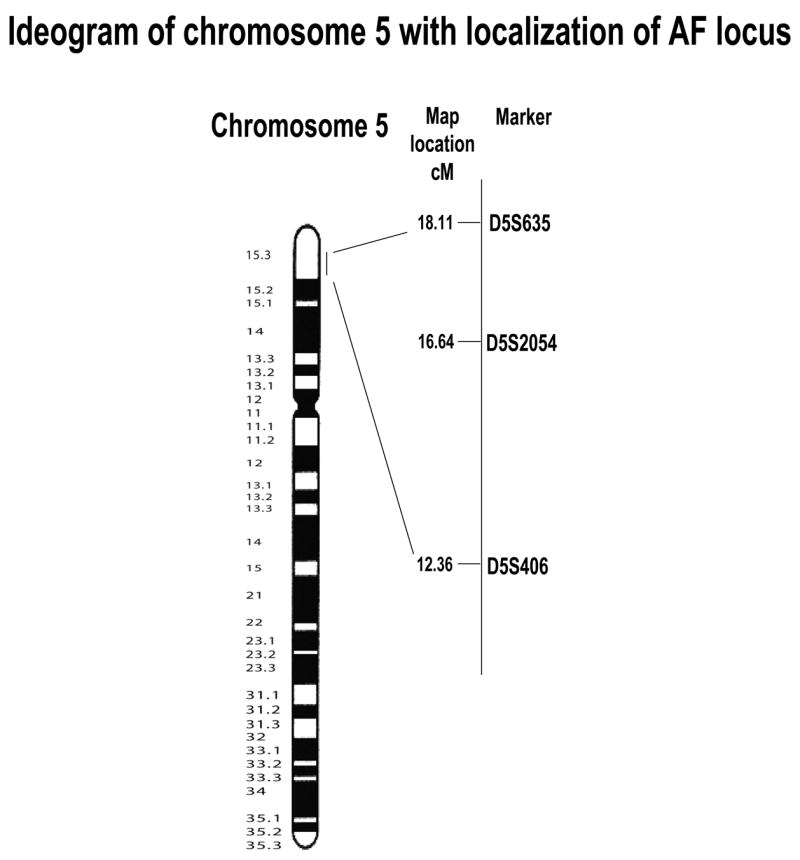
Ideogram of chromosome 5 with Geimsa banding patterns and localization of the AF locus. Genetic map with chromosome 5p15 markers and location of putative AF gene is shown on right.
Signal-averaged P-wave Duration
This was performed in 25 family members including 8 family members with documented AF and 17 individuals in whom the diagnosis was ruled out. As subject I:1 was in chronic AF and I:2 was deceased, no SA P-wave duration was obtained in these individuals. Individuals carrying the AF haplotype and with a documented history of AF, the SA P-wave duration was prolonged (Table 1) (Figure 3). Importantly, eight heterozygous carriers had a prolonged SA P-wave (203±21 msec) compared with 17 non-carriers (116±12 msec, P<0.00001). Using prolonged SA averaged P-wave (conventionally defined as >155 msec (22)) as an endophenotype for AF, genome-wide linkage was performed with 8 individuals classified as affected and 17 unaffected. We identified evidence of linkage with the marker D5S2054 (maximum LOD score = 3.6, θ= 0) in the same region of chromosome 5p15 (Table 2). This LOD score is robust to changes in penetrance or allele frequency, and even analyses of affected individuals only result in 2-point LOD scores of >2.8. Because this marker was fully informative, a multipoint calculation achieved no higher LOD score. Although a prolonged SA P-wave duration allowed us to improve the LOD score for this kindred, it did not narrow the region of interest.
Table 1.
Clinical characteristics of family members.
| Pedigree Number | Age at evaluation (yrs) | Age at onset (yrs) | Rhythm | Symptoms | Echo | Signal averaged P-wave (msec)- | LA size (mm) | EF | Therapy |
|---|---|---|---|---|---|---|---|---|---|
| Affected and haplotype carriers | |||||||||
| I: 1 | 75 | 41 | Chronic lone AF | Yes | Normal | - | 45 | 0.60 | Rate control |
| I: 2 | Deceased | NA | Chronic AF | Yes | NA | - | NA | NA | Rate control |
| II: 10 | 58 | 42 | Paroxysmal AF | Yes | Normal | 190 | 46 | 0.55 | AAD |
| II: 16 | 66 | 32 | Paroxysmal AF | Yes | LVH | 189 | 44 | 0.60 | AAD |
| II: 18 | 68 | 32 | Paroxysmal lone AF | Yes | Normal | 200 | 40 | 0.60 | Rate control |
| II: 20 | 70 | 30 | Paroxysmal AF | Yes | LVH | 180 | 42 | 0.55 | AAD |
| III: 1 | 38 | 18 | Paroxysmal lone AF | Yes | Normal | 210 | NA | 0.65 | Rate control |
| III: 4 | 32 | - | NSR | Yes | Normal | 250 | 38 | 0.65 | NA |
| III: 6 | 34 | 18 | Paroxysmal lone AF | Yes | Normal | 198 | 44 | 0.65 | AAD |
| IV: 1 | 22 | - | NSR | No | Normal | 210 | 39 | 0.65 | NA |
| Unaffected (individuals without a disease haplotype) | |||||||||
| II: 1 | 52 | - | NSR | No | Normal | 112 | 36 | 0.60 | - |
| II: 2 | 54 | - | NSR | No | Normal | 104 | 34 | 0.60 | - |
| II: 3 | 57 | - | NSR | No | Normal | 108 | 38 | 0.55 | - |
| II: 4 | 61 | - | NSR | No | Normal | 107 | 40 | 0.60 | - |
| II: 5 | 64 | - | NSR | No | Normal | 112 | 38 | 0.50 | - |
| II: 6 | 65 | - | NSR | No | Normal | 141 | 36 | 0.55 | - |
| II: 7 | 67 | - | NSR | No | Normal | 105 | 37 | 0.55 | - |
| II: 8 | 72 | - | NSR | No | Normal | 117 | 36 | 0.50 | - |
| II: 9 | 55 | - | NSR | No | Normal | 118 | 38 | 0.60 | - |
| II: 13 | 75 | - | NSR | No | LVH | 134 | 40 | 0.65 | - |
| II: 14 | 72 | - | NSR | No | Normal | 140 | 39 | 0.55 | - |
| II: 15 | 64 | - | NSR | No | Normal | 110 | 36 | 0.65 | - |
| II: 21 | 63 | - | NSR | No | Normal | 125 | 40 | 0.60 | - |
| II: 22 | 62 | - | NSR | No | Normal | 118 | 38 | 0.55 | - |
| III: 2 | 60 | - | NSR | No | Normal | 120 | 36 | 0.60 | - |
| III: 3 | 34 | - | NSR | No | Normal | 105 | 38 | 0.60 | - |
| III: 5 | 36 | - | NSR | No | Normal | 110 | 38 | 0.60 | - |
AF, atrial fibrillation; AAD, antiarrhythmic drug; EF, ejection fraction; LA, left atrial; LVH, left ventricular hypertrophy; NA, not available.
Figure 3.
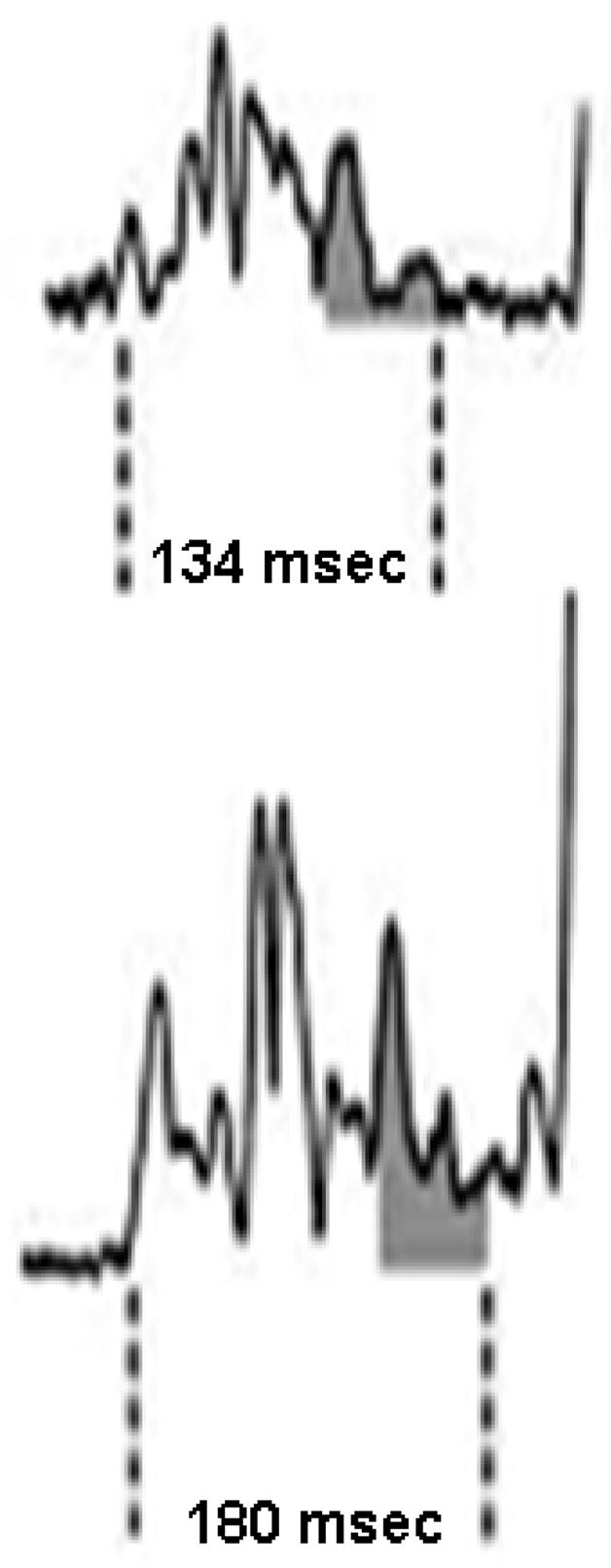
Representative tracings of signal-averaged P-wave duration in a subject (II:13) with no history or symptoms of AF who did not carry the AF haplotype and had a normal P-wave duration (top). The bottom tracing is from a subject (II:20) with AF and the disease haplotype who had prolonged SA P-wave duration.
Table 2.
Linkage in Family VAF-1 to markers at 5p15.
| Recombination Fraction (θ)
|
||||||
|---|---|---|---|---|---|---|
| Marker | 0.00 | 0.05 | 0.10 | 0.20 | 0.30 | 0.40 |
| D5S2088 | 0.68 | 0.42 | 0.12 | 0.28 | 0.49 | 0.40 |
| D5S406 | 0.72 | 0.62 | 0.40 | −0.03 | −0.19 | −0.12 |
| D5S2054 | 3.62* | 3.08 | 2.97 | 2.10 | 1.58 | 0.98 |
| D5S635 | 2.64 | 2.55 | 2.11 | 1.78 | 1.12 | 0.48 |
| D5S807 | 0.26 | 0.08 | 0.00 | 0.29 | 0.39 | 0.58 |
| D5S1486 | 0.08 | −0.26 | 0.24 | 0.28 | 0.32 | 0.16 |
Maximum 2-point LOD score.
Discussion
In this study, we have identified a novel locus for AF that is mapped to chromosome 5p15 and characterized by early onset of AF. In addition, we have also demonstrated that prolongation of the SA P-wave duration cosegregates with AF and appears to be an intermediate marker (endophenotype) for identifying individuals at increased risk for developing AF in this kindred. Importantly, identification of an endophenotype not only aided ascertainment of additional family members but also increased the LOD score providing increased support for linkage at this locus. Identification of the causal gene, mapped to chromosome 5p15, will not only advance our understanding of the molecular basis of AF but may also provide new therapeutic approaches to treat this common and morbid condition.
The locus we have defined on chromosome 5p15 currently extends over 5.75-cM. On the basis of the locations of the recombinant boundaries, and the sequence of the human genome, the region includes five annotated genes. One putative candidate gene is A Disintegrin-like and Metalloprotease with Thrombospondin type 1 motif, 16 (ADAMTS-16)(27), whose functions include collagen processing as procollagen N-proteinase, cleavage of the matrix proteoglycans aggrecan, as well as inhibition of angiogenesis and blood coagulation homeostasis (28). However, we have been unable to identify transcripts of this gene in a human cardiac cDNA library. We have also performed direct DNA sequence analysis on the remaining four genes (LOC340094, FLJ33360, KIAA0947 and MED10) in affected subjects II:10 and II:18 with unaffected II:22 as the control. However, no mutation was identified in the coding and intron splice sequences of these genes.
Several lines of evidence raise the possibility that AF and some forms of dilated cardiomyopathy may be allelic. For example, this region of 5p is close to but does not overlap with a previously reported locus for neonatal AF that is associated with sudden death and variable cardiomyopathy (29). However, in our AF kindred, dilated cardiomyopathy was not feature of the clinical presentation. Support for this has also recently come from identification of mutations in the cardiac sodium channel (SCN5A) that manifest as early onset dilated cardiomyopathy and AF (30,31). The cloning of the causative genes at the 10q22, 6q14-16 and 5p13 loci will further clarify the relationship between AF and cardiomyopathy (9,10,29).
The genetic basis for the majority of patients with AF remains unknown. Although familial AF has been mapped to three other loci, identification of the causative genes has proved elusive. One possible reason relates to phenotypic complexity of AF with the lack of a biological basis for the classification of the disease. Others include reduced penetrance of the gene and polygenic nature of the disease. Consequently, the identification of well-defined endophenotypes that cosegregate with AF have been proposed in order to aid gene mapping (14). An endophenotype should not only cosegregate with the condition but also be present in an individual whether or not AF is present. While it’s possible that as the left atrial size increases secondary to AF, the P-wave duration may lengthen, in our study there was no uniform increase in left atrial size despite prolongation of the SA P-wave duration in individuals carrying the AF haplotype. In fact, the one individual with the AF haplotype (IV:1) but no documented history or symptoms of AF had a normal left atrial size. Therefore, in this study, we showed that not only individuals with AF but also those who do not carry the AF haplotype could be identified by SA electrocardiography.
Abnormalities of the P-wave such as increased duration, right and left atrial hypertrophy, are associated with an increased incidence of AF (32). In our kindred, strikingly abnormal P-wave durations occurred in the absence of other pathology. The routine ECG may however, not identify many other P-wave abnormalities that are risk factors for AF and are seen during intracardiac recording after programmed stimulation. These abnormalities not only reflect structural abnormalities of the atria but also alterations in electrical properties, the most important of which are intra-atrial and inter-atrial conduction abnormalities and abnormalities of refractoriness throughout the atria. As the SA P-wave ECG is able to record low level electrical signals more precise measurements of duration and amplitude of the P-wave can be made. Furthermore, SA P-wave duration has been shown, in retrospective and prospective studies, to be able to discriminate between those at risk and those without risk of developing AF (33,34).
Our study demonstrated that in this kindred SA P-wave duration was able to identify affected individuals and carriers of the AF haplotype. However, validation studies will need to be performed in additional cohorts before it can be concluded that it is a useful endophenotype for genetic studies of AF.
Acknowledgments
This work was supported by NIH grant HL075266 to DD and UO1 HL65962 to DMR
Abbreviation List
- AF
atrial fibrillation
- ECG
electrocardiogram
- LOD
logarithm of the odds
- SA
signal-averaged
Footnotes
No conflicts of interest
References
- 1.Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155:469–73. [PubMed] [Google Scholar]
- 2.Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–52. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- 3.Darbar D, Herron KJ, Ballew JD, et al. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol. 2003;41:2185–92. doi: 10.1016/s0735-1097(03)00465-0. [DOI] [PubMed] [Google Scholar]
- 4.Fox CS, Parise H, D’Agostino RB, Sr, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–5. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 5.Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–4. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y, Xia M, Jin Q, et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75:899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia M, Jin Q, Bendahhou S, et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012–9. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 8.Olson TM, Alekseev AE, Liu XK, et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–91. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 9.Ellinor PT, Shin JT, Moore RK, Yoerger DM, MacRae CA. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation. 2003;107:2880–3. doi: 10.1161/01.CIR.0000077910.80718.49. [DOI] [PubMed] [Google Scholar]
- 10.Brugada R, Tapscott T, Czernuszewicz GZ, et al. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. 1997;336:905–11. doi: 10.1056/NEJM199703273361302. [DOI] [PubMed] [Google Scholar]
- 11.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–80. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 12.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 13.Otway R, Vandenberg JI, Guo G, et al. Stretch-sensitive KCNQ1 mutation A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J Am Coll Cardiol. 2007;49:578–86. doi: 10.1016/j.jacc.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 14.Ellinor PT, Macrae CA. The genetics of atrial fibrillation. J Cardiovasc Electrophysiol. 2003;14:1007–9. doi: 10.1046/j.1540-8167.2003.03307.x. [DOI] [PubMed] [Google Scholar]
- 15.Garver DL, Holcomb JA, Christensen JD. Heterogeneity of response to antipsychotics from multiple disorders in the schizophrenia spectrum. J Clin Psychiatry. 2000;61:964–72. doi: 10.4088/jcp.v61n1213. quiz 973. [DOI] [PubMed] [Google Scholar]
- 16.Cornblatt BA, Malhotra AK. Impaired attention as an endophenotype for molecular genetic studies of schizophrenia. Am J Med Genet. 2001;105:11–5. [PubMed] [Google Scholar]
- 17.Ellinor PT, Low AF, Patton KK, Shea MA, Macrae CA. Discordant atrial natriuretic peptide and brain natriuretic peptide levels in lone atrial fibrillation. J Am Coll Cardiol. 2005;45:82–6. doi: 10.1016/j.jacc.2004.09.045. [DOI] [PubMed] [Google Scholar]
- 18.Ellinor PT, Low A, Patton KK, et al. C-Reactive protein in lone atrial fibrillation Reduced apelin levels in lone atrial fibrillation. Am J Cardiol. 2006;97:1346–50. doi: 10.1016/j.amjcard.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 19.Darbar D, Jahangir A, Hammill SC, Gersh BJ. P wave signal-averaged electrocardiography to identify risk for atrial fibrillation. Pacing Clin Electrophysiol. 2002;25:1447–53. doi: 10.1046/j.1460-9592.2002.01447.x. [DOI] [PubMed] [Google Scholar]
- 20.Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age. Circulation. 1980;62:1054–61. doi: 10.1161/01.cir.62.5.1054. [DOI] [PubMed] [Google Scholar]
- 21.Jordaens L, Tavernier R, Gorgov N, Kindt H, Dimmer C, Clement DL. Signal-averaged P wave: predictor of atrial fibrillation. J Cardiovasc Electrophysiol. 1998;9:S30–4. [PubMed] [Google Scholar]
- 22.Zaman AG, Archbold RA, Helft G, Paul EA, Curzen NP, Mills PG. Atrial fibrillation after coronary artery bypass surgery: a model for preoperative risk stratification. Circulation. 2000;101:1403–8. doi: 10.1161/01.cir.101.12.1403. [DOI] [PubMed] [Google Scholar]
- 23.Hakonarson H, Bjornsdottir US, Halapi E, et al. A major susceptibility gene for asthma maps to chromosome 14q24. Am J Hum Genet. 2002;71:483–91. doi: 10.1086/342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong A, Gudbjartsson DF, Sainz J, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–7. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 25.Ott J. Analysis of Human Genetic Linkage. The John Hopkins University Press; 1991. [Google Scholar]
- 26.Ploughman LM, Boehnke M. Estimating the power of a proposed linkage study for a complex genetic trait. Am J Hum Genet. 1989;44:543–51. [PMC free article] [PubMed] [Google Scholar]
- 27.Cal S, Obaya AJ, Llamazares M, Garabaya C, Quesada V, Lopez-Otin C. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene. 2002;283:49–62. doi: 10.1016/s0378-1119(01)00861-7. [DOI] [PubMed] [Google Scholar]
- 28.Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberti C, Wang L, Li L, et al. Genome-Wide Linkage Scan Identifies a Novel Genetic Locus on Chromosome 5p13 for Neonatal Atrial Fibrillation Associated With Sudden Death and Variable Cardiomyopathy. Circulation. 2004;110:3753–3759. doi: 10.1161/01.CIR.0000150333.87176.C7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNair WP, Ku L, Taylor MR, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–7. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 31.Olson TM, Michels VV, Ballew JD, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–54. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leier CV, Meacham JA, Schaal SF. Prolonged atrial conduction. A major predisposing factor for the development of atrial flutter. Circulation. 1978;57:213–6. doi: 10.1161/01.cir.57.2.213. [DOI] [PubMed] [Google Scholar]
- 33.Yamada T, Fukunami M, Shimonagata T, et al. Dispersion of signal-averaged P wave duration on precordial body surface in patients with paroxysmal atrial fibrillation. Eur Heart J. 1999;20:211–20. doi: 10.1053/euhj.1998.1281. [DOI] [PubMed] [Google Scholar]
- 34.Yamada T, Fukunami M, Shimonagata T, et al. Prediction of paroxysmal atrial fibrillation in patients with congestive heart failure: a prospective study. J Am Coll Cardiol. 2000;35:405–13. doi: 10.1016/s0735-1097(99)00563-x. [DOI] [PubMed] [Google Scholar]