

Abstract
There is growing interest in β-catenin and its role in various human cancers. We recently reported that 2-amino-3-methylimidazo[4,5-f]quinoline (IQ)- and 1,2-dimethylhydrazine (DMH)-induced colon tumors in the rat contain mutations in Ctnnb1, the gene for β-catenin, but the mutation spectrum was influenced by postinitiation exposure to chlorophyllin (CHL) and indole-3-carbinol (I3C) [Blum et al., Carcinogenesis 2001;22:315–320]. The present paper describes a follow-up study in which all of the target organs for IQ- and DMH-induced tumorigenesis were screened; Ctnnb1 mutations were found in 44 of 119 DMH-induced colon tumors, six of 13 IQ-induced colon tumors, 28 of 81 DMH-induced small intestine tumors, none of five IQ-induced small intestine tumors, four of 106 IQ-induced liver tumors, none of 14 DMH-induced Zymbal’s gland tumors, none of 24 IQ-induced Zymbal’s gland tumors, and none of 29 IQ-induced skin tumors. In tumors from rats given carcinogen alone, or carcinogen plus CHL or I3C, Ctnnb1 mutations frequently substituted amino acids adjacent to Ser33, a critical Ser/Thr residue in the glycogen synthase kinase-3β regulatory domain of β-catenin. However, substitution of critical Ser/Thr residues themselves was detected in only three of 24 (12.5%) of the tumors from rats given carcinogen alone, compared with 23 of 58 (40%) of the tumors from rats given carcinogen and treated postinitiation with I3C or CHL (P <0.02). More than 50 of the colon tumors with wild-type β-catenin were examined further for their Apc status; the overall frequency of Apc mutations was <10%, and these genetic changes occurred exclusively in the ‘Mutation Cluster Region’ of Apc. A subset of colon tumors also was examined for expression of β-catenin and c-jun; these proteins were overexpressed in all tumors containing Ctnnb1 mutations, but the expression was highest in tumors with Ctnnb1 mutations affecting Thr41 and Ser45 residues in the glycogen synthase kinase-3β region of β-catenin. Thus, Ctnnb1 mutations occurred more frequently than Apc mutations in colon and small intestine tumors of the rat, and certain mutations upregulated β-catenin/T-cell factor target genes more effectively than others, perhaps influencing the response to phytochemicals administered postinitiation.
Keywords: CTNNB1, APC, Wnt signaling, TCF/LEF target genes, chlorophyllin, indole-3-carbinol
INTRODUCTION
There has been a surge of interest recently in the Wnt signaling pathway and its role in human cancer development [1–4]. Much of this interest evolved from studies of the molecular changes in oncogenes and tumor suppressor genes in colorectal and other human cancers [5]. In human colorectal cancers, the APC tumor suppressor gene is a common target for mutation, but colon tumors with wild-type APC typically have genetic changes in the human β-catenin gene (CTNNB1) [6–8]. The product of the latter gene, β-catenin, is a cadherin-binding protein involved in cell-cell adhesion [9–11], but also functions as a transcriptional activator when complexed in the nucleus with members of the T-cell factor (TCF)/lymphocyte enhancer factor (LEF) family of proteins [1]. Control of the cytosolic levels of β-catenin occurs via a multi-protein complex, in which APC, axin/conductin, and glycogen synthase kinase-3β (GSK-3β) negatively regulate β-catenin expression [1]. In this complex, GSK-3β phosphorylates key Ser/Thr residues in the N-terminal region of β-catenin and the protein is targeted for ubiquitination and proteosomal degradation [12]. In primary human colon tumors and colorectal cancer cell lines, mutations in CTNNB1 substitute critical Ser/Thr residues in the GSK-3β region and stabilize β-catenin, leading to accumulation of β-catenin/TCF complexes in the nucleus [6–8], and activation of target genes, such c-MYC, c-JUN, and CYCLIN D1 [13–15].
Mutations in β-catenin also have been detected in colon tumors of animals treated with chemical carcinogens, such as 2-amino-3-methylimidazo[4,5-f]quinoline (IQ), 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), 1,2-dimethylhydrazine (DMH), azoxymethane, and methylazoxymethanol acetate plus 1-hydroxyanthraquinone [16–19]. Indeed, there are several similarities between human and rat colon tumors with respect to the β-catenin/Apc pathway. First, as in the human situation, colon tumors in the rat contain mutations in Apc or Ctnnb1, but not in both of these genes [17]. Second, β-catenin/TCF target genes frequently are overexpressed, including c-myc, c-jun, and cyclin D1 [16]. Third, genetic changes in human CTNNB1 or murine Ctnnb1 typically substitute amino acids within the N-terminal GSK-3β regulatory domain of β-catenin. However, one interesting species difference is in the spectrum of genetic changes in β-catenin. Thus, whereas the vast majority of β-catenin mutations in human colon cancers substitute critical Ser/Thr residues directly [6–8], in rat colon tumors a high frequency of the mutations substitute amino acids adjacent to key Ser/Thr residues [16–19]. For example, the combined data from studies in which rats were treated with azoxymethane, PhIP, IQ, or 1-hydroxyanthraquinone plus methylazoxymethanol acetate revealed only six of 35 tumors (17%) with mutations that substituted critical Ser/Thr residues directly, whereas 28 of 35 (80%) had genetic changes affecting residues immediately adjacent to Ser33 in β-catenin [19]. Moreover, these genetic changes were localized to two CTGGA sequences in close proximity to one another, suggesting the presence of possible mutational ‘hotspots’ in Ctnnb1.
To complicate matters further, the spectrum and frequency of β-catenin mutations can be influenced by other factors, including exposure to dietary phytochemicals. This was demonstrated in a recent study in which rats were given IQ or DMH and treated postinitiation with chlorophyllin (CHL) or indole-3-carbinol (I3C); in rats given carcinogen alone, virtually all of the mutations resulted in substitutions adjacent to Ser33, whereas in rats given carcinogen followed by CHL or I3C treatment, almost half of the mutations substituted critical Ser/Thr residues directly [16]. The latter report focused almost exclusively on the colon tumors, and suggested the need for further investigation of the other target organs, namely the small intestine, liver, skin, and Zymbal’s gland [20]. Therefore, the present study sought to provide a comprehensive analysis of β-catenin mutations in the major target organs for DMH- and IQ-induced tumorigenesis in the rat. In addition, a subset of colon tumors containing wild-type β-catenin was examined further in order to determine the mutation status of Apc. Finally, the relative expression of β-catenin and β-catenin/TCF target genes was assessed as a function of each specific codon affected (i.e., amino acid substituted) within the GSK-3β regulatory region of β-catenin. The hypothesis was that certain Ctnnb1 mutations might activate β-catenin/TCF target genes more effectively than others, thus influencing the response to phytochemicals administered postinitiation.
MATERIALS AND METHODS
Source of Tumors
Tumors were from a 1-yr study in which male F344 rats were initiated with DMH or IQ during the first 5 wk of the experiment, and 1 wk later the animals were treated with CHL or I3C until termination. Further details, including histopathological findings, were presented in the original report [21]. For information on doses of the test compounds and their routes of administration, see Table 1.
Table 1.
Summary of β-Catenin Mutations in DMH and IQ Tumors (All Target Organs*)
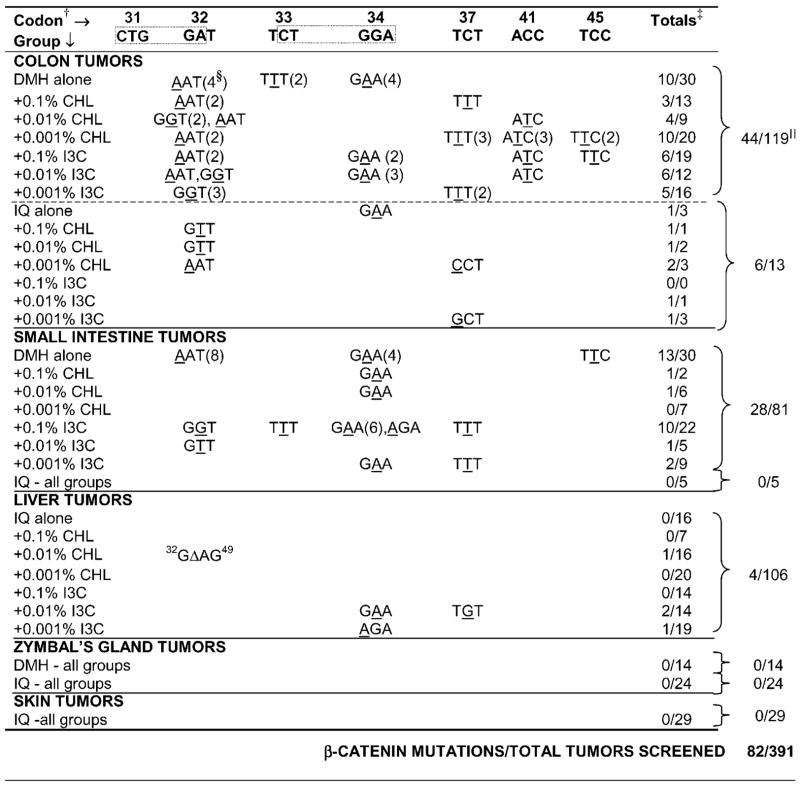
|
Rats were initiated with carcinogen for 5 wk; IQ was given in the diet (AIN-93) at a concentration of 0.03%, whereas DMH was injected s.c. once/wk (20 mg/kg body wt). Starting one wk after the last carcinogen treatment until the end of the study at 1 yr, rats were given CHL in the drinking water or I3C in the diet, at concentrations shown in the table. For details on the pathology of each tumor type, see [21].
Only codons affected by mutation are shown (except codon 31, in gray, to highlight the two CTGGA sequences commonly affected, dotted boxes).
Number of mutations confirmed by sequencing/total tumors successfully amplified.
Total number of tumors with a mutation in the codon indicated.
All groups combined for each carcinogen within the target organ shown. Colon tumor data were from a previous report [16].
Mutation Screening and Sequencing
Tissue samples were extracted with DNAzol genomic isolation reagent (Molecular Research Center, Inc., Cincinnati, OH), and initially a 150-bp region corresponding to the GSK-3β region of β-catenin was amplified by the polymerase chain reaction (PCR), as described before [16,17]. Previous studies of carcinogen-induced rat colon tumors established that mutations in β-catenin were localized exclusively to the GSK-3β region, and no genetic changes were detected in the armadillo region or C-terminal domain [16–19,22]. Nonetheless, for tumors that lacked a mutation in the GSK-3β region we performed comprehensive screening of all 14 exons and the intron-exon boundaries of the entire Ctnnb1 gene, with the PCR primers and conditions reported elsewhere [16,17,22]. In addition, colon tumors with wild-type β-catenin were screened for mutations in Apc, as detailed previously [17,20,23]. In brief, single-strand conformation polymorphism (SSCP) analysis was conducted by means of the GenePhor electrophoresis system (Amersham Pharmacia Biotech, Piscataway, NJ). Six ng of DNA were loaded per well, run at 15°C, 600 V, 25 mAMP, and 15 W for 1.5 h, using the optimal conditions for detecting β-catenin or Apc mutations, based on preliminary studies at different temperatures and in the presence and absence of glycerol [16,22,23]. The gel was silver stained, and bands with altered migration were cut from the gel, reamplified, and cleaned using Wizard PCR Preps (Promega, Madison, WI). All of the samples reported here to contain mutations were confirmed by sequencing in both directions on an ABI Prizm™ 3.3 sequencer.
Reverse Transcriptase-PCR (RT-PCR)
The Micro-FastTrack 2.0 kit (Invitrogen, Carlsbad, CA) was used to isolate mRNA from freshly thawed tissues, and cDNA was synthesized via the Promega RT system. Primers and PCR conditions for the genes of interest, namely cyclin D1, c-myc, and c-jun, were as reported previously [16].
Western Blotting
Tissues were homogenized at room temperature in RIPA lysis buffer (Qiagen, Valencia, CA) and centrifuged at 14,000 g for 2 min through the QIAshredder (Qiagen). Fifteen to twenty micrograms of protein were run on a 4–12% Bis-Tris Gel (Invitrogen) and transferred onto nitrocellulose membrane. After overnight blocking, the membrane was incubated for 1 h with primary antibody, washed, incubated with secondary antibody conjugated with horse-radish peroxidase (BioRad), and developed using ECL reagents 1 and 2 (Amersham Corp.). Equal loading and transfer was confirmed by amido black staining and by comparison with glyceraldehyde-3-phosphate dehydrogenase. Sources and dilutions of the antibodies used, as well as other experimental details, were reported previously [16,24]. Imaging and quantification of the blots was performed on an AlphaInnotech photodocumentation system (AlphaInnotech Corp., San Leandro, CA).
Statistical Analyses
Comparisons were made among treatment groups, and for specific ‘hotspots’ within β-catenin, by means of the Fisher’s Exact Test and ANOVA followed by Fisher’s PLSD, respectively.
RESULTS
DNA was successfully amplified from a total of 391 tumors, and Ctnnb1 mutations were screened by SSCP analysis and confirmed by direct sequencing (Figure 1 and Table 1). All of the mutations were localized within the GSK-3β region of β-catenin, coded by exon 2 of rat Ctnnb1, which corresponds to exon 3 of human CTNNB1 [22].
Figure 1.
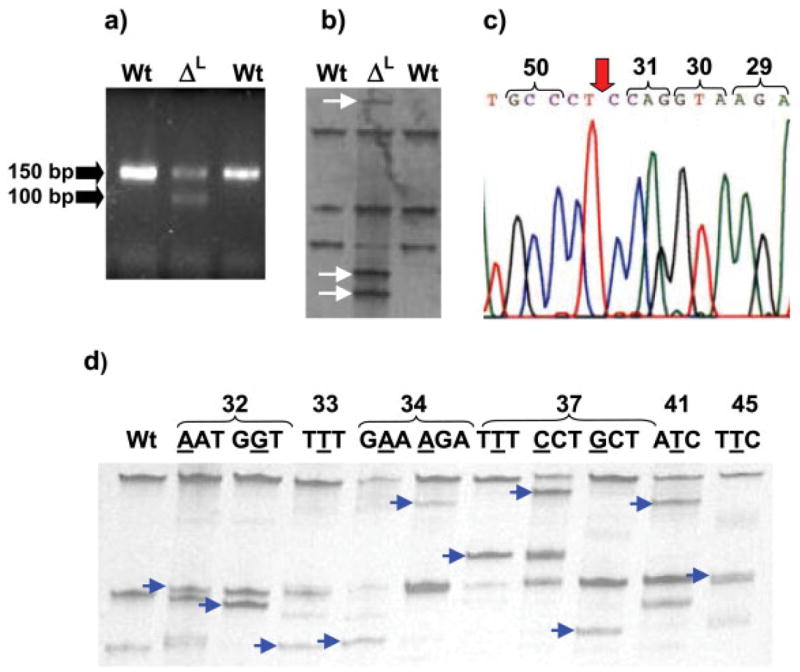
Single-strand conformation polymorphism (SSCP) screening and sequencing of exon 2 of rat Ctnnb1, the region that corresponds to exon 3 of human CTNNB1, and codes for the GSK-3β domain of β-catenin [22]. (a) Ethidium bromide–stained agarose gel and (b) SSCP gel, showing two IQ-induced liver tumors that were wild-type (wt) and a third with a deletion (middle lane, ΔL); (c) sequencing of ΔL with the reverse primer revealed an in-frame deletion involving codons 32 to 49; (d) representative SSCP patterns for the more common mutations in IQ- and DMH-induced tumors; bands shown with arrows were removed and sequenced. For examples of typical sequencing results, see [16].
In all but one case, primers designed to amplify the region encoding the GSK-3β domain of β-catenin gave a single PCR product of 150 bp. One IQ-induced liver tumor gave two PCR products, one with the expected size of 150 bp and the second approximately 50 bp shorter (Figure 1a). Bands with shifted mobility in the SSCP gel (Figure 1b) were removed and sequenced, revealing an in-frame deletion originating in codon 32 and extending through codon 49 (Figure 1c). This deletion would be expected to remove all four of the critical Ser/Thr residues within the GSK-3β regulatory domain, without affecting other important domains in β-catenin, such as the armadillo repeats that bind TCF, LEF, Apc, and other protein partners [1].
Typical SSCP results are shown in Figure 1d; single base pair substitutions were detected in codons 32, 33, 34, 37, 41, and 45 of Ctnnb1. Sequencing data for the more common mutations in this region of Ctnnb1 have been reported previously [16].
Table 1 summarizes the results for each specific target organ examined, namely colon, small intestine, liver, Zymbal’s gland, and skin. In the colon tumors from groups given DMH or IQ, the overall frequency of β-catenin mutations was 44/119 (37%) and 6/13 (46%), respectively. In the small intestine tumors induced by DMH, 28/81 (35%) harbored mutations in β-catenin, whereas none of the IQ-induced small intestine tumors had such mutations, perhaps due to the small sample set available for analysis (none of five). Screening of more than 100 IQ-induced liver tumors revealed four cases of β-catenin mutation (4/106 = 3.8%; Table 1). The mutation screening also included 14 DMH-induced Zymbal’s gland tumors, 24 IQ-induced Zymbal’s gland tumors, and 29 IQ-induced skin tumors, but mutations in β-catenin were detected in none of these tumors. Thus, taken together, 82 of 391 (21%) of the tumors examined here had mutations in β-catenin, with low or undetectable frequencies in liver, skin, and Zymbal’s gland tumors, but higher frequencies in colon and small intestine tumors.
Inspection of the data in Table 1 reveals that G → A transitions were common within two CTGGA sequences around codons 31–34 (45/82 = 56%). Of the remainder, many involved C → T transitions in codons 33, 37, 41, and 45 (21/37 = 57%). The find-ings are consistent with the known preference of IQ and DMH for forming adducts with guanine bases, leading to a preponderance of G → A or C → T transitions (66/82 = 80%).
Colon tumors with wild-type β-catenin were screened further for their Apc status. None of the IQ-induced colon tumors examined had mutations affecting Apc (none of five), consistent with prior studies that showed few Apc mutations in IQ-induced colon tumors, but a relatively high frequency of such mutations in colon tumors initiated by PhIP [20]. In groups given DMH, the frequency of Apc mutations was as follows: DMH alone, none of five; DMH + CHL, none of four; DMH + I3C, five of 43. In the latter group, four of five of the mutations involved a C → T transition, three of which produced a premature stop codon and one a change from A to V. One tumor had a G → A transition that did not change the corresponding amino acid (Q) (Table 2). Genetic changes were detected only in the central Mutation Cluster Region of Apc, and no mutations were found elsewhere in Apc, such as the N-terminal armadillo repeat region or the C-terminal basic domain [1]. For all groups combined, the overall frequency of mutations in Apc was five of 57 (8.8%).
Table 2.
Summary of Apc Mutations in Colon Tumors*
| Codon affected | Treatment | Nucleotide change detected | Consequence |
|---|---|---|---|
| 1094 | DMH + 0.001% I3C | CAA → TAA | Q → stop |
| 1149 | DMH + 0.1% I3C | CAG → TAG | Q → stop |
| 1234 | DMH + 0.001% I3C | CAA → TAA | Q → stop |
| 1307 | DMH + 0.1% I3C | GCC → GTC | A → V |
| 1722 | DMH + 0.001% I3C | CAG → CAA | Q → Q |
Previously, colon tumors from rats given carcinogen alone were compared with those from animals given carcinogen plus CHL or I3C, and this revealed a shift in the β-catenin mutation spectrum [16]. After completing the mutational analysis of all target organs of IQ- and DMH-induced tumorigenesis in the present study, the shift in the spectrum of β-catenin mutations was equally striking (Figure 2). Thus, in the tumors fromrats given carcinogen alone, most of the amino acids substitutions were immediately adjacent to Ser33 (21 of 24 = 87.5%), and there were only three cases in which a critical Ser/Thr residue was substituted directly, two affecting Ser33 and one substituting Ser45 (three of 24 = 12.5%). However, in groups given IQ or DMH and posttreated with I3C or CHL, 23 of 58 (40%) of the tumors had mutations that substituted critical Ser/Thr residues directly. The increased frequency of mutations affecting critical Ser/Thr residues was highly significant following phytochemical treatment (P < 0.02).
Figure 2.

Amino acid substitutions in β-catenin. The wild-type (wt) sequence for part of the GSK-3β region of β-catenin is shown in the horizontal gray box, including critical Ser/Thr phosphorylation sites (Ser33, Ser37, Thr41, Ser45). Results from groups given carcinogen alone (DMH or IQ) are shown above the horizontal box, whereas those from groups given carcinogen plus CHL or I3C are shown below. Superscripts indicate the target organ: C, colon; SI, small intestine; L, liver. ΔL indicates a liver tumor with an in-frame deletion starting in codon 32 (see Figure 1c). Vertical boxes highlight amino acid substitutions in β-catenin that are rarely seen in colon tumors after carcinogen treatment alone. For more complete information on the mutations and the treatment groups, see Table 1. Modified from a figure (containing the colon tumor data alone) published previously (Figure 2 of [16]).
An important observation from our previous study was that the expression of β-catenin/TCF target genes appears to be higher in colon tumors with certain β-catenin mutations versus others [16]. Specifically, five colon tumors with wild-type β-catenin had low or undetectable levels of c-jun and c-myc, three tumors with mutations in codons 32 or 34 of Ctnnb1 had intermediate expression, and two tumors with mutations in codon 41 of Ctnnb1 (i.e., directly substituting a critical Thr41 residue in β-catenin) had high expression levels of these proteins. In the present investigation, as before [16], RT-PCR analyses confirmed the high expression of cyclin D1, c-myc, and c-jun in colon tumors with β-catenin mutations (data not presented). Consequently, the relative expression of β-catenin and c-jun proteins was examined in greater detail for each specific codon of Ctnnb1 affected; triplicate samples were analyzed in each case in order to generate data that was amenable to statistical analysis. Representative blots are shown for three tumors with mutations in codon 32 and three tumors with mutations in codon 41 of Ctnnb1, as well as three tumors containing wild-type β-catenin (Figure 3, inset). β-Catenin and c-Jun proteins were elevated in all tumors with Ctnnb1 mutations, but they were fourfold to sixfold higher in tumors with mutations in codons 32, 33, 34, or 37 compared with eightfold to tenfold higher in tumors with mutations in codons 41 or 45 (P <0.05).
Figure 3.
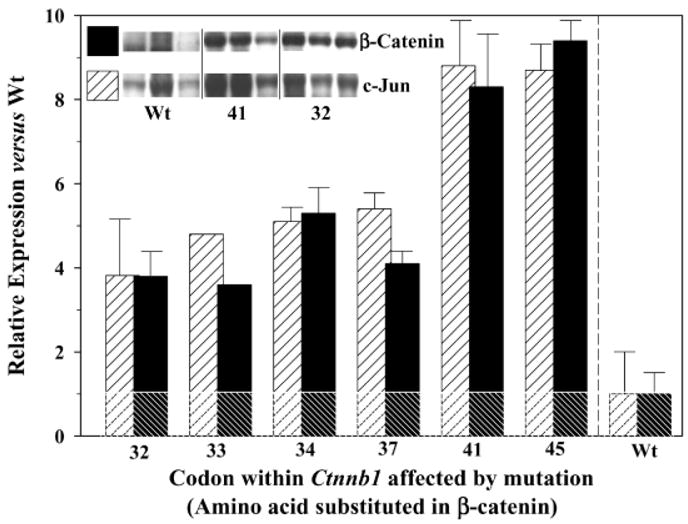
Expression in rat colon tumors of β-catenin (filled bars) and c-jun (hatched bars). Total tissue lysates were prepared from three separate colon tumors for each of the codons mutated in Ctnnb1, and these were subjected to SDS-polyacrylamide gel electrophoresis and probed with antibodies to β-catenin and c-Jun proteins, as reported before [16]. Inset, representative blots from three tumors containing wild-type (wt) β-catenin, three tumors with mutations in codon 41 of Ctnnb1, and three tumors with mutations in codon 32 of Ctnnb1. Normal colonic mucosa and positive controls for the proteins of interest were included in each gel (not shown). Densitometry measurements were obtained with a gel documentation system and associated software; results were first normalized to expression levels of glyceraldehyde-3-phosphate dehydrogenase and then expressed relative to the protein levels in tumors with wild-type β-catenin (assigned an arbitrary expression value of 1.0). Data represent mean ± SD, n = 3 (except for codon 33, which was from a single tumor). The horizontal line is a reference guide indicating the relative expression of 1.0 for c-jun and β-catenin proteins in tumors with wt β-catenin; all of the bars above this line were significantly different from wt controls, and the bars labeled 41 and 45 were significantly different from all others (P < 0.05).
In the case of the small intestine tumors from this study, many were of a size that allowed for histological examination and mutation screening but not Western blotting. However, for samples that were amenable to immunoblot analysis, β-catenin and c-Jun proteins were expressed at highest levels when mutations involved codons 41 and 45 of Ctnnb1 (data not presented). Results for colon and small intestine tumors suggested that mutations in Ctnnb1 may not have been functionally equivalent, and that certain mutations might have activated β-catenin/TCF target genes more effectively than others. Future studies will examine the protein expression by immunohistochemical methods to determine which population of cells (e.g., anaplastic cells versus less anaplastic cells) are associated with these changes in β-catenin and its downstream targets.
DISCUSSION
This report describes the most comprehensive screening to date of β-catenin mutations in the major target organs of DMH- and IQ-induced tumorigenesis in the rat, and provides the first attempt to quantify the relative expression in vivo of β-catenin and one of its target genes, c-jun, according to the specific site of mutation in Ctnnb1. The results confirm and extend previous work in which β-catenin mutants are expressed by transient transfection in cultured cells and are shown to activate gene expression in a reporter assay in vitro [25]. However, data from the present in vivo study revealed that β-catenin and c-jun proteins were more highly elevated in tumors with mutations affecting codons 41 and 45 compared with tumors harboring other types of mutation in Ctnnb1 (Figure 3). The significance of this finding might relate to the recent discovery that GSK-3β does not phosphorylate all four Ser/Thr residues in β-catenin to the same extent, and that a ‘priming’ kinase (possibly casein kinase-1) is required for initial phosphorylation of Ser45 [26]. It is believed that GSK-3β subsequently phosphorylates other Ser/Thr residues in a sequential fashion (Thr41 → Ser37 → Ser33); thus, a mutation in either codon 45 or codon 41 of Ctnnb1wouldlikelyinterfere with phosphorylation of multiple Ser/Thr residues in β-catenin, and thereby effectively interfere with subsequent ubiquitination and proteosomal degradation [12].
These results also might help to explain the response to phytochemicals given postinitiation. Screening of colon and small intestine tumors from rats treated with carcinogen alone showed hotspots for mutation around codon 33, and a high preponderance of the substitutions affected residues on either side of Ser33 (Figure 2). Virtually none of the tumors from rats given DMH or IQ alone had mutations affecting Ser37, Thr41, or Ser45 of β-catenin, yet these mutations were quite common in tumors from rats given carcinogen and treated with I3C or CHL postinitiation.
Our current working hypothesis is that mutations affecting Thr41 and Ser45 so severely upregulate β-catenin expression and β-catenin/TCF target genes that the cells normally respond by undergoing early apoptosis (i.e., these cells would be deleted from the intestinal mucosa rather than progressing to tumors). However, chronic exposure to exogenous agents, including certain phytochemicals, might interfere in the apoptotic mechanisms, thereby enabling a population of cells to progress to tumor formation that might otherwise be deleted by programmed cell death. Inhibition of apoptosis is an important mechanism driving colorectal cancer development in humans [27], as well as in animals treated with chemical carcinogens such as IQ [28], and chemo-preventive agents that are effective against colon cancer frequently enhance the rate of apoptosis in the colonic mucosa [29]. In this context, any phytochemical that overrides the apoptotic mechanism according to the hypothesis stated above would be predicted to act as a tumor promoter rather than a chemopreventive agent, although this could be influenced by the dose administered and the duration of postinitiation exposure. It is noteworthy, therefore, that most of the mutations in Ser37, Thr41 and Ser45 were from I3C and CHL groups in which tumor promotion, not suppression, was previously reported [21]. Whether natural chlorophyll as opposed to synthetic CHL, or I3C as it is present in cruciferous vegetables, acts as a tumor promoter under the same experimental conditions remains to be determined, but a pilot study showed suppression rather than promotion of colonic aberrant crypts by natural chlorophyll [30]. It is also noteworthy that in the original study [21], CHL acted in a concentration-dependent manner to suppress IQ-induced liver tumors. The ability of CHL and I3C to promote in one tissue but suppress in another might be related to the relative importance of the β-catenin signaling pathway in those tissues, but further study is required in order to confirm this hypothesis.
One apparent discrepancy between the working hypothesis and the results presented here is the relatively high frequency of mutations affecting Ser37 (I3C and CHL groups, Figure 2) and the ‘intermediate’ expression of β-catenin and c-jun proteins in tumors bearing those mutations (Figure 3). It is possible that other target genes of β-catenin/TCF signaling may be more strongly overexpressed, such as c-myc, cyclin D1, peroxisome proliferator activator receptor-δ, or matrix metalloproteinase-7 [1], but additional studies are necessary. Nonetheless, the findings from the present investigation clearly indicated that the spectrum of β-catenin mutations in tumors can be influenced by exogenous factors, including certain phytochemicals in the diet. This is consistent with the well-established relationship between diet and cancer, and the overall importance of gene-diet interactions in determining human cancer risk [31–34].
In summary, this report described the mutational analysis of Ctnnb1 in the major target organs of DMH-and IQ-induced tumorigenesis in the rat. Mutations were found at higher frequency in the small intestine tumors and colon tumors, and at low or undetectable frequency in liver, skin, and Zymbal’s gland tumors. More than 50 of the colon tumors with wild-type β-catenin also were screened for genetic changes in Apc; the overall frequency of such mutations was <10%, and they were localized to the Mutation Cluster Region of Apc. β-catenin and c-jun proteins were more strongly overexpressed in tumors with mutations affecting codons 41 and 45 of Ctnnb1 compared with other mutations in β-catenin. Against this background of genetic changes, phytochemicals such as CHL and I3C might select for or against specific β-catenin mutations, and thereby act as suppressing agents or tumor promoters in different tissues [35,36].
Acknowledgments
This work was supported in part by National Institutes of Health grants CA65525 and CA80176. C.A.B. was supported by Toxicology Training grant T32 ES07060 from the National Institute of Environmental Health Sciences. The services of Dr. Pereira were supported in part by the Statistics Core of the Environmental Health Sciences Center at Oregon State University (P30 ES00210).
Abbreviations
- APC
human adenomatous polyposis coli gene
- Apc
murine Apc gene
- CTNNB1
human β-catenin gene
- Ctnnb1
murine β-catenin gene
- TCF
T-cell factor
- LEF
lymphocyte enhancer factor
- GSK-3β
glycogen synthase kinase-3β
- DMH
1,2-dimethyl-hydrazine
- IQ
2-amino-3-methylimidazo[4,5-f]quinoline
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- CHL
chlorophyllin
- I3C
indole-3-carbinol
- PCR
polymerase chain reaction
- SSCP
single-strand conformation polymorphism
References
- 1.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 2.Oving IM, Clevers HC. Molecular causes of colon cancer. Eur J Clin Invest. 2002;32:448–457. doi: 10.1046/j.1365-2362.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- 3.Clements WM, Wang J, Sarnaik A, et al. β-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 2002;62:3503–3506. [PubMed] [Google Scholar]
- 4.Lustig B, Jerchow B, Sachs M, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 6.Morin PJ, Sparks AB, Korinek V, et al. Activation of β-Catenin-Tcf signaling in colon cancer by mutations in β-Catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 7.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β-Catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- 8.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. β-Catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hajra KM, Fearon ER. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer. 2002;34:255–268. doi: 10.1002/gcc.10083. [DOI] [PubMed] [Google Scholar]
- 10.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol. 1998;153:333–339. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-catenin is a target for the ubiquitin-proteosome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He T-C, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 14.Mann B, Gelos M, Siedow A, et al. Target genes of β-catenin-T-cell factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA. 1999;96:1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tetsu O, McCormick F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cell. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 16.Blum CA, Xu M, Orner GA, et al. β-Catenin mutation in rat colon tumors initiated by 1,2-dimethylhydrazine and 2-amino-3-methylimidazo[4,5-f]quinoline, and the effect of post-initiation treatment with chlorophyllin and indole-3-carbinol. Carcinogenesis. 2001;22:315–320. doi: 10.1093/carcin/22.2.315. [DOI] [PubMed] [Google Scholar]
- 17.Dashwood RH, Suzui M, Nakagama H, Sugimura T, Nagao M. High frequency of β-catenin (Ctnnb1) mutations in the colon tumors induced by two heterocyclic amines in the F344 rat. Cancer Res. 1998;58:1127–1129. [PubMed] [Google Scholar]
- 18.Takahashi M, Fukuda K, Sugimura T, Wakabayashi K. β-catenin is frequently mutated and demonstrates altered cellular location in azoxymethane-induced rat colon tumors. Cancer Res. 1998;58:42–46. [PubMed] [Google Scholar]
- 19.Suzui M, Ushijima T, Dashwood RH, et al. Frequent mutations of the rat β-catenin gene in colon cancers induced by methylazoxymethanol acetate plus 1-hydroxyanthraquinone. Mol Carcinog. 1999;24:232–237. doi: 10.1002/(sici)1098-2744(199903)24:3<232::aid-mc10>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 20.Nagao M, Ushijima T, Toyota M, Inoue R, Sugimura T. Genetic changes induced by heterocyclic amines. Mutat Res. 1997;376:161–167. doi: 10.1016/s0027-5107(97)00039-0. [DOI] [PubMed] [Google Scholar]
- 21.Xu M, Orner GA, Bailey GS, Stoner GD, Horio D, Dashwood RH. Post-initiation effects of chlorophyllin and indole-3-carbinol in rats given 1,2-dimethylhydrazine or 2-amino-3-methylimidazo[4.5-f]quinoline. Carcinogenesis. 2001;22:309–314. doi: 10.1093/carcin/22.2.309. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Dixon BM, Al-Fageeh M, Blum CA, Dashwood RH. Sequencing of the rat β-catenin gene (Ctnnb1) and mutational analysis of liver tumors induced by 2-amino-3-methylimidazo[4,5-f]quinoline. Gene. 2002;283:255–262. doi: 10.1016/s0378-1119(01)00839-3. [DOI] [PubMed] [Google Scholar]
- 23.Suzui M, Ushijima T, Yoshimi N, et al. No involvement of APC gene mutations in ulcerative colitis-associated rat colon carcinogenesis induced by 1-hydroxyanthraquinone and methylazoxymethanol acetate. Mol Carcinog. 1997;20(4):389–393. [PubMed] [Google Scholar]
- 24.Orner GA, Dashwood W-M, Blum CA, Díaz D, Li Q, Dashwood RH. Suppression of tumorigenesis in the Apcmin mouse: Down-regulation of β-catenin signaling by a combination of tea plus sulindac. Carcinogenesis. 2003;24:263–267. doi: 10.1093/carcin/24.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porfiri E, Rubinfeld B, Albert I, Hovanes K, Waterman M, Polakis P. Induction of a β-catenin-LEF-1 complex by wnt-1 and transforming mutants of β-catenin. Oncogene. 1997;15:2833–2839. doi: 10.1038/sj.onc.1201462. [DOI] [PubMed] [Google Scholar]
- 26.Hagen T, Vidal-Puig A. Characterisation of the phosphorylation of β-catenin at the GSK-3 priming site Ser45. Biochem Biophys Res Commun. 2002;294:324–328. doi: 10.1016/S0006-291X(02)00485-0. [DOI] [PubMed] [Google Scholar]
- 27.Bedi A, Pasricha PJ, Akhtar AJ, et al. Inhibition of apoptosis during development of colorectal cancer. Cancer Res. 1995;55:1811–1816. [PubMed] [Google Scholar]
- 28.Hayashi R, Luk H, Horio D, Dashwood R. Inhibition of apoptosis in colon tumors induced in the rat by 2-amino-3-methylimidazo[4,5-f]quinoline. Cancer Res. 1996;56:4307–4310. [PubMed] [Google Scholar]
- 29.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–174. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 30.Blum CA, Xu M, Orner GA, et al. Promotion versus suppression of rat colon carcinogenesis by chlorophyllin and chlorophyll: Modulation of apoptosis, cell proliferation, and β-catenin/Tcf signaling. Mutat Res. 2003 doi: 10.1016/s0027-5107(02)00338-x. in press. [DOI] [PubMed] [Google Scholar]
- 31.Go VL, Wong DA, Butrum R. Diet, nutrition and cancer prevention: Where are we going from here? J Nutr. 2001;131:3121S–3126S. doi: 10.1093/jn/131.11.3121S. [DOI] [PubMed] [Google Scholar]
- 32.Rock CL, Lampe JW, Patterson RE. Nutrition, genetics, and risks of cancer. Annul Rev Public Health. 2000;21:47–64. doi: 10.1146/annurev.publhealth.21.1.47. [DOI] [PubMed] [Google Scholar]
- 33.Patterson RE, Eaton DL, Potter JD. The genetic revolution: Change and challenge for the dietetics profession. J Am Diet Assoc. 1999;99:1412–1420. doi: 10.1016/S0002-8223(99)00341-7. [DOI] [PubMed] [Google Scholar]
- 34.Mathers JC, Burn J. Nutrition in cancer prevention. Curr Opin Oncol. 1999;11:402–407. doi: 10.1097/00001622-199909000-00015. [DOI] [PubMed] [Google Scholar]
- 35.Dashwood RH. Indole-3-carbinol: Anticarcinogen or tumor promoter in brassica vegetables? Chem Biol Interact. 1998;110:1–5. doi: 10.1016/s0009-2797(97)00115-4. [DOI] [PubMed] [Google Scholar]
- 36.Dashwood RH. Modulation of heterocyclic amine-induced mutagenicity and carcinogenicity: an ‘A-to-Z’ guide to chemopreventive agents, promoters, and transgenic models. Mutat Res. 2002;511:89–112. doi: 10.1016/s1383-5742(02)00005-4. [DOI] [PubMed] [Google Scholar]