

Abstract
Post-translational modification by the lipid palmitate is crucial for the correct targeting and function of many proteins. Here we show that huntingtin (htt) is normally palmitoylated at cysteine 214, which is essential for its trafficking and function. The palmitoylation and distribution of htt are regulated by the palmitoyl transferase huntingtin interacting protein 14 (HIP14). Expansion of the polyglutamine tract of htt, which causes Huntington disease, results in reduced interaction between mutant htt and HIP14 and consequently in a marked reduction in palmitoylation. Mutation of the palmitoylation site of htt, making it palmitoylation resistant, accelerates inclusion formation and increases neuronal toxicity. Downregulation of HIP14 in mouse neurons expressing wild-type and mutant htt increases inclusion formation, whereas overexpression of HIP14 substantially reduces inclusions. These results suggest that the expansion of the polyglutamine tract in htt results in decreased palmitoylation, which contributes to the formation of inclusion bodies and enhanced neuronal toxicity.
Post-translational modification of cysteine residues by palmitate has recently emerged as an important and reversible modification that is involved in the trafficking and functional modulation of different membrane proteins and their signaling pathways1–3. In vitro labeling assays have shown that HIP14 is a palmitoyl transferase that palmitoylates numerous neuronal substrates, including htt4. Expansion of the polyglutamine tract of htt is the mutation underlying Huntington disease5.
A characteristic feature of the pathogenesis of Huntington disease5 is neuronal toxicity: an early sign is the formation of intranuclear and cytoplasmic inclusions in the medium spiny neurons of the striatum6, involving the translocation of mutant htt into the nucleus7,8. Htt is normally located on plasma and intracellular membranes and associates with vesicles and different organelles such as the Golgi9,10. Sequences in the N-terminal region of htt are crucial for the binding of htt to membranes11. Disturbed trafficking of mutant htt seems to be an early and consistent feature of Huntington disease. However, the factors influencing the trafficking of htt are largely unknown.
Different post-translational modifications of htt alter its cellular function. Ubiquitination targets htt for degradation12 and SUMOylation promotes its capacity to repress transcription13. Phosphorylation of mutant htt is reduced and is associated with increased toxicity14,15.
Here we show that htt is palmitoylated in vivo and that alterations in the palmitoylation of htt affect its normal distribution and function. We provide evidence that the palmitoylation of htt in vivo is regulated by HIP14 and that this is crucial for its normal trafficking to the Golgi. Furthermore, palmitoylation of mutant htt is markedly reduced in vivo, as a result of its lower interaction with HIP14, leading to increased inclusion formation and enhanced toxicity.
RESULTS
Palmitoylation of htt at cys214 is modulated by CAG size
The observation that palmitoylation is critical for the distribution of proteins to particular membrane locations, combined with the presence of htt in detergent-resistant membranes (Supplementary Fig. 1 online), raised the possibility that the palmitoylation of htt may be crucial for regulating its trafficking and function.
Immunoprecipitation of htt from cortical neurons revealed that full-length htt is indeed palmitoylated (Fig. 1a). Treatment of cells with NMDA, which induces cleavage of htt, revealed that the N-terminal region is modified by palmitate (Fig. 1a). Deletion mapping with N-terminal truncations of htt showed that palmitoylation occurred in the first 224 amino acids (Fig. 1b; N224). Palmitoylation was abolished by treatment with hydroxylamine (NH2OH; Fig. 1c), indicating that the incorporation of palmitate is due to the modification of cysteines through a thioester bond. Further deletion mapping localized the site of palmitoylation to a fragment containing six cysteine residues (Fig. 1d and Supplementary Fig. 1; Htt 79–224). Subsequent mutagenesis localized the palmitoylated residue to three cysteines in the C terminus of this fragment (Fig. 1d and Supplementary Fig. 1; Htt 141–224). Protein sequence alignment5 suggested that C214 was a likely candidate for palmitoylation by virtue of it being the only site in this region that was conserved in all the species we analyzed, including Drosophila (Supplementary Fig. 1). Indeed, C214 was identified as the site at which the N-terminal fragment of htt (Fig. 1d) and full-length htt (Fig. 1e) are palmitoylated, consistent with the existence of a single major site (C214) for palmitoylation within htt.
Figure 1.

Huntingtin is palmitoylated in neurons and COS cells. Palmitoylation is modulated by CAG size. (a) Rat cortical neurons were treated (+) with 100 μM NMDA for 10 min, followed by metabolic labeling. Huntingtin (htt) was immunoprecipitated (IP) and visualized by western blotting and fluorography. A [3H]palmitate band was detected in untreated cells. However, after NMDA treatment, a palmitoylated fragment of htt, detected by the N terminus–specific antibody BKP1, was generated (right). (b) COS cells transfected with fragments of htt (N548, N224) were metabolically labeled and processed as described above. The smallest palmitoylated htt fragment contained six cysteine residues. (c) COS cells were transfected with N548-15 and metabolically labeled. Immunoprecipitates were treated with or without 1 M NH2OH and processed as previously described. Palmitate was removed from htt after NH2OH treatment (right), indicating that it is coupled to htt through a thioester bond. (d,e) COS cells were transfected with plasmids encoding either (i) a GFP-tagged htt fragment containing six cysteines (Htt 79–224), three N-terminal cysteines (Htt 79–149) or three C-terminal cysteines (Htt 141–224), (ii) an N548 htt fragment for the C152S and C204S substitutions, or (iii) a full-length htt construct with and without C214S (Supplementary Fig. 1). Htt from labeled cells was analyzed as described above. Substituting C214 with serine (C214S) abolished the palmitoylation of Htt 79–224 and Htt 141–224 (d), and full-length htt (e). (f) COS cells were transfected with N548, containing 15 or 128Q, and metabolically labeled. Htt from labeled cells was analyzed as described above. (g) Results of four independent experiments were adjusted for protein levels and demonstrated a reduction of ~50% in the palmitoylation of mutant htt (P = 0.001).
Protein palmitoylation is regulated by flanking sequences that can markedly alter the levels of protein palmitoylation2. As the major site of palmitoylation is in the N terminus of htt, close to the polyglutamine tract, this immediately raised the possibility that palmitoylation of htt was modulated by CAG size. Indeed, palmitoylation of the N-terminal fragment of htt in COS cells (N548) was significantly reduced in the presence of a disease-associated expansion of the polyglutamine tract (Fig. 1f,g; P = 0.001). Palmitoylation of other proteins such as SNAP25 was not altered in the presence of the mutation for Huntington disease (Supplementary Fig. 1). The fact that polyglutamine expansion markedly decreases htt palmitoylation raised the question of whether some of the pathogenic effects of mutant htt might be operating through this mechanism.
Palmitoylation regulates distribution and function of htt
We investigated whether the palmitoylation of htt influences its subcellular distribution, frequency of inclusion body formation and toxicity. Palmitoylation-resistant full-length wild-type htt (htt-15(C214S)), mutant htt (htt-128(C214S)) and the N-terminal fragment N548-128(C214S) showed a marked redistribution of the protein in both COS and HEK-293 cells (Fig. 2a,b arrows, Supplementary Fig. 2 online and data not shown). Expression of N548-128(C214S) resulted in a threefold increase in the number of cells containing htt inclusions (Fig. 2b). This tendency for increased inclusion formation appeared specific to the C214S change, as mutation of an adjacent cysteine residue (C204S) did not increase the formation of inclusions (Fig. 2b). Consistent with this, time-lapse imaging in COS cells revealed that inclusions formed three times faster in cells expressing palmitoylation-resistant htt (N548-128(C214S); Fig. 2c,d).
Figure 2.

Increased inclusions of palmitoylation-resistant mutant htt in COS cells. (a–d) COS cells were transfected with N548-128, with or without the C214S substitution, and stained with htt antibody. (a) Inclusion bodies (arrows) were occasionally seen with the expression of N548-128. However, a C214S substitution altered mutant htt distribution and increased the formation of inclusions. Scale bar, 10 μm. (b) The percentage of cells containing inclusions increased in COS cells expressing N548-128 (mean ± s.e.m.: 6.7 ± 0.46%) and was maximal in cells expressing N548-128(C214S) (19.4 ±± 0.58%). This effect was not observed with N548-128(C204S) (8 ± 0.68%). (c) Time-lapse images captured over 2 h revealed that inclusions developed at an accelerated rate in the presence of the C214S substitution in mutant htt (bottom). Scale bar, 10 μm. (d) The rate of inclusion formation in cells transfected with N548-128(C214S) was significantly (P = 0.03) faster than in cells transfected with N548-128.
An important question is whether palmitoylation directly influences the trafficking of htt or whether this effect could be operating via another mechanism. Mutation of the palmitoylated cysteine may have indirectly disrupted other protein-protein interactions or resulted in an altered conformation of htt that promoted inclusion body formation. To exclude this possibility, we assessed whether treatment with drugs that specifically block protein palmitoylation alter htt trafficking. The palmitate modification of htt is transient, with a half life of 2.5 h (Supplementary Fig. 3 online). This rapid turnover allowed us to use the inhibitor 2-bromopalmitate to block palmitoylation and investigate the trafficking of nonpalmitoylated htt in cells. Indeed, 14 h of treatment with 2-bromopalmitate resulted in a significant increase in htt inclusion bodies in cells expressing N548-128 (Supplementary Fig. 3; P =0.006). Taken together, these findings indicate that a defect in palmitoylation specifically alters htt distribution in COS cells. In fact, inclusions in cells expressing palmitoylation-resistant htt colocalized with γ-tubulin, ubiquitin, Lmp2 and Hsp70, indicating that C214S htt is misfolded16,17 in both COS cells and neurons (Fig. 3).
Figure 3.
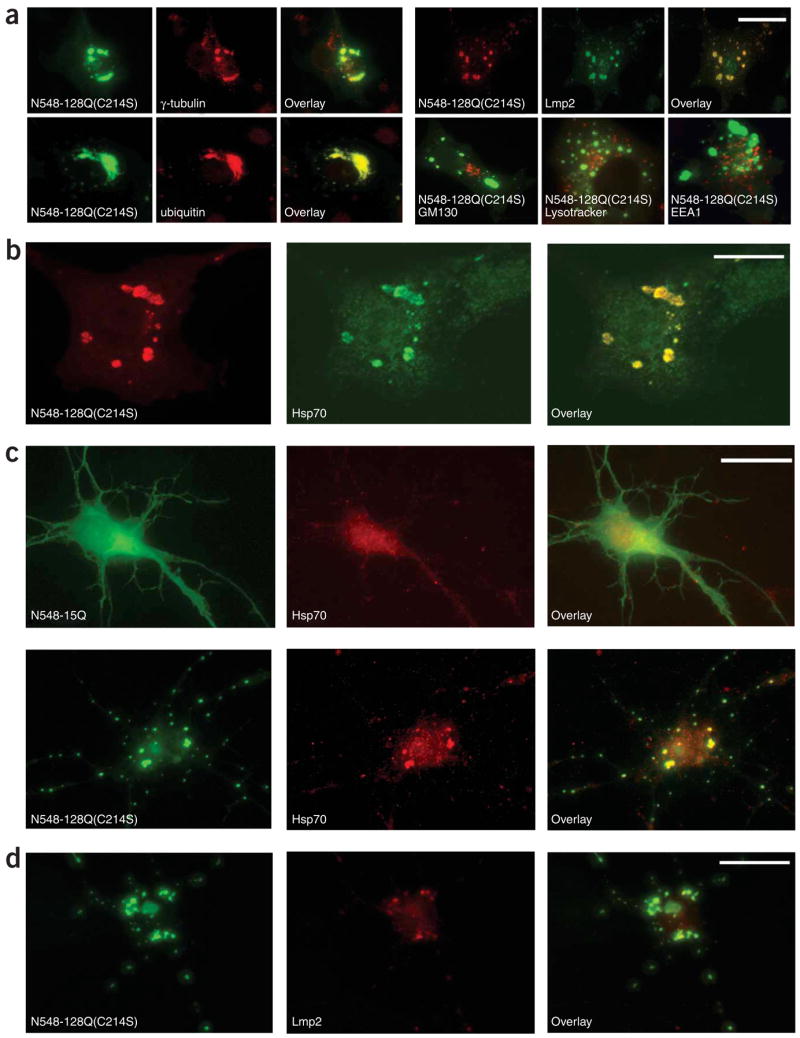
Altered distribution of palmitoylation-resistant mutant htt in COS cells and neurons. (a,b) COS cells transfected with N548-128 (C214S) were immunolabeled with antibodies detecting htt and several markers. Htt inclusions colocalized with the centrosome marker γ-tubulin, the proteasome markers ubiquitin and Lmp2, and misfolded protein marker Hsp70, but not with the Golgi marker GM-130, the lysosomal marker Lysotracker or the early endosome marker EEA1. Scale bar: 10 μm in a, 5 μm in b. (c) Rat cortical neurons transfected with N548-15-GFP or N548-128(C214S)-GFP were immunolabeled with antibodies detecting Hsp70 (red). Mutant htt with the C214S substitution accelerated the formation of inclusions that colabeled with Hsp70. Scale bar, 10 μm. (d) Representative images showing that the inclusions formed in cortical neurons expressing N548-128(C214S) also colocalized with Lmp2. Scale bar, 10 μm.
In an independent assay, we examined what effect the loss of htt palmitoylation has on COS cell survival by analyzing nuclei of transfected cells for fragmentation or pyknosis18. Cell death in the presence of palmitoylation-resistant mutant htt (N548-128(C214S)) was significantly higher than in the presence of htt with polyglutamine expansion alone (Fig. 4a,b; P = 10−5).
Figure 4.

Enhanced toxicity of palmitoylation-resistant mutant htt. (a,b) COS cells transfected with N548-128 and N548-128(C214S) were scored for toxicity by analyzing nuclei for changes associated with cell death. Cell death was significantly (P = 10−5) induced by the expression of N548-128(C214S). Representative images show a normal nucleus and an apoptotic nucleus (arrowhead). (c–e) Rat cortical neurons were transfected with truncated htt (N548) or full-length htt (flhtt) constructs as indicated, and stained with htt antibody. (c) The percentage of neurons containing inclusions increased in neurons expressing N548-15(C214S) (2.5 ± 0.35%), increased still further in neurons expressing N548-128 (15.17 ± 1.3%) and was maximal in neurons expressing N548-128(C214S) (25.08 ± 3.1%). (d) Representative images showing neurons expressing the indicated flhtt constructs. Scale bar, 5 μm. (e) The percentage of neurons containing inclusions increased in neurons expressing flhtt-15Q(C214S) (0.5 ± 0.27%), increased still further in neurons expressing flhtt-128Q (1 ± 0.41%) and was maximal in neurons expressing flhtt-128Q(C214S) (2.1 ± 0.44%). There was a significant (P = 10−5) increase in the percentage of cells with nuclear inclusions in neurons expressing flhtt-128Q(C214S). (f) Rat cortical neurons transfected with the indicated htt constructs were treated with NMDA for 10 min and processed for the TUNEL assay. The percentage of cell death was significantly greater in transfected cells expressing N548-15Q (C214S) (23.16 ± 5.2; P = 0.01) and N548-128Q(C214S) (27.75% ± 4.7; P = 0.03), demonstrating that the loss of palmitoylation significantly reduces cellular viability.
To examine whether these effects are seen in a cell type relevant to Huntington disease, we next investigated the functional effects of a loss of htt palmitoylation on inclusion formation and cell survival in neurons. For both truncated (N548) and full-length htt, inclusion formation was more frequent in the presence of polyglutamine expansion (Fig. 4c–e) and was significantly increased when both wild-type (P = 0.02) and mutant htt (N548: P = 0.01; full-length: P = 10−5) were made palmitoylation resistant (C214S; Fig. 4c–e). We then examined the effects of a loss of htt palmitoylation on the localization of htt in neurons. After transfection of wild-type and mutant full-length htt, htt was predominantly expressed in the cytosol and inclusions were seen only in the presence of mutant htt (Fig. 4d). In contrast, after transfection of palmitoylation-resistant htt, inclusions were occasionally seen even in the presence of a normal polyglutamine tract (Fig. 4d,e). Notably, in the presence of palmitoylation-resistant htt with polyglutamine expansion, the frequency of nuclear inclusions was increased (Fig. 4d,e). Localization of mutant htt in the nucleus is an early reproducible marker of htt toxicity in vivo in animal models for Huntington disease8 and in humans19. This suggests that one mechanism for the localization of mutant htt in the nucleus may be the decreased palmitoylation consequent to the expansion of the polyglutamine tract.
We made use of the fact that neurons expressing mutant htt are more vulnerable to NMDA-induced toxicity20 by transfecting cortical neurons with N-terminal fragments of wild-type and mutant htt, including palmitoylation-resistant htt (C214S), treating the cells with 500 μM NMDA and assessing changes in neuronal viability. Notably, both wild-type and mutant htt became significantly more toxic in the presence of the C214S mutation (Fig. 4f; P = 0.01 and P = 0.03, respectively).
HIP14 regulates the palmitoylation and trafficking of htt
HIP14, a protein that interacts with htt in vitro21, is a conserved mammalian palmitoyl transferase for different neuronal substrates, including htt4. HIP14 is mainly localized in the Golgi21. Sorting of some neuronal proteins, such as Ras, requires palmitoylation for trafficking to Golgi membranes and for delivery to transport vesicles. Palmitoylated Ras shows a pronounced Golgi localization and faster retrograde trafficking from the plasma membrane to the Golgi3. HIP14 alters the distribution of a subset of palmitoylated proteins in a palmitoylation-dependent manner4. We found that HIP14 overexpression resulted in the redistribution of endogenous htt (Fig. 5a), and to a lesser extent of mutant htt (Fig. 5b), to the Golgi. This redistribution was not observed with palmitoylation-resistant htt (Fig. 5c), suggesting that htt trafficking to the Golgi is at least partially regulated by palmitoylation.
Figure 5.
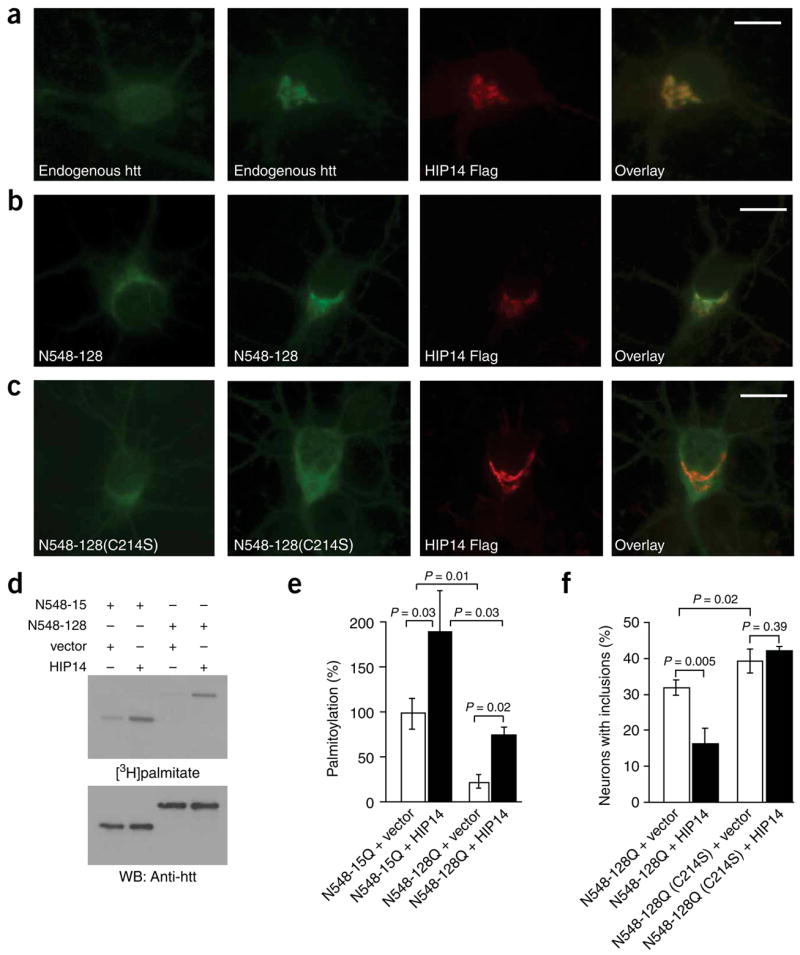
HIP14 influences the distribution and catalyzes the palmitoylation of htt in neurons. (a) Cortical neurons transfected with control vector (left) or with FLAG-tagged HIP14 were labeled with the appropriate antibodies. The overexpression of HIP14 resulted in marked redistribution of endogenous htt to the Golgi. Scale bar, 5 μm. (b,c) Cortical neurons transfected with N548-128Q-GFP or N548-128Q(C214S)-GFP alone (left) or with HIP14-FLAG were labeled with the appropriate antibodies. Scale bar, 5 μm. (b) Partial redistribution of N548-128Q-GFP was observed in the presence of HIP14. (c) No effect in the distribution of palmitoylation-resistant htt (N548-128Q(C214S)-GFP) was observed in the presence of HIP14. (d) COS cells transfected with N548-15Q or N548-128Q, and with a control vector or HIP14, were metabolically labeled. Immunoprecipitated htt was analyzed as described above. (e) Palmitoylation was significantly (P = 0.03) increased in cells coexpressing wild-type htt and HIP14. In addition, HIP14 significantly (P = 0.02) increased palmitoylation of mutant htt. However, in the presence of HIP14, mutant htt was still significantly (P = 0.03) less palmitoylated than wild-type htt. (f) Cortical neurons transfected with GFP-tagged N548-128Q or N548-128Q(C214S), and with a control vector or FLAG-tagged HIP14, were labeled with the appropriate antibodies. The percentage of cells containing htt inclusions was significantly (P = 0.005) lower in the presence of HIP14 whereas no change was detected in the number of C214S inclusions, emphasizing the importance of HIP14 palmitoylation of htt in its distribution.
HIP14’s known interaction with htt, combined with the fact that htt is palmitoylated and its trafficking is regulated at least in part by palmitoylation, raised the possibility that HIP14 is involved in htt palmitoylation in vivo. Overexpression of HIP14 with wild-type and mutant htt significantly (P = 0.03 and P = 0.02, respectively) increased their palmitoylation (Fig. 5d,e), demonstrating that HIP14 catalyzes htt palmitoylation. We obtained further evidence for the role of palmitoylation in the normal distribution of htt by examining the effect of HIP14 on the formation of inclusions in neurons. Over-expression of HIP14 significantly (P = 0.005) reduced the number of inclusions seen in the presence of polyglutamine expansion (Fig. 5f) but had no effect on that seen with palmitoylation-resistant mutant htt (Fig. 5f), highlighting the importance of HIP14 palmitoylation of htt in the intracellular distribution and trafficking of htt.
The reduced palmitoylation of mutant htt (Fig. 1f) suggested that a defect in htt palmitoylation may have resulted from an altered association with HIP14. Our analysis showed that the in vivo interaction of HIP14 with htt was markedly reduced in the presence of mutant htt in brains of YAC128 mice22 (Fig. 6a), indicating that reduced palmitoylation of mutant htt results, in all likelihood, from a decreased association with HIP14. To further test this, we compared palmitoylation23 of full-length wild-type htt and mutant htt obtained from the brains of YAC18 and YAC128 mice, respectively. Similar to our findings with the N-terminal fragment of mutant htt in heterologous cells (Fig. 1f), palmitoylation of full-length mutant htt was significantly (P = 0.01) diminished in YAC128 brain extracts (Fig. 6b,c).
Figure 6.
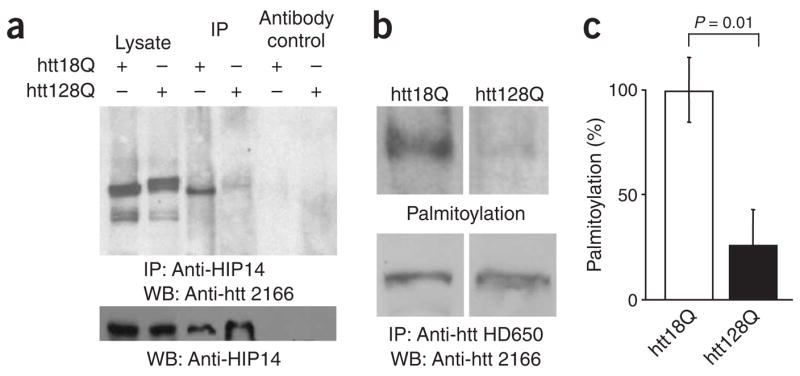
HIP14 associates less with mutant htt, and mutant htt is less palmitoylated in vivo. (a) Coimmunoprecipitation of HIP14 and htt from brains of YAC18 and YAC128 mice demonstrated a weaker interaction between mutant htt and HIP14. (b) Immunoprecipitated htt from YAC18 and YAC128 mouse brain lysate was incubated in the presence or absence of 1 M NH2OH and labeled with Btn-BMCC sulfhydryl-specific reagent. Western blots were probed with streptavidin-conjugated HRP (top) to detect biotin-labeled htt or probed with htt antibody (bottom) to detect total immunoprecipitated htt. (c) Palmitoylation was significantly (P = 0.01) reduced in htt with an expanded polyglutamine tract.
To further investigate the alteration in the palmitoylation of mutant htt and its decreased association with HIP14, and their role in the disturbed trafficking and increased inclusion formation seen in Huntington disease, we used a small interfering RNA (siRNA) that disrupts HIP14 expression as previously described4 (Supplementary Fig. 4 online). Neurons derived from YAC18 and YAC128 brains and transfected with HIP14 siRNA showed significant redistribution of both wild-type and mutant htt (Fig. 7a–f; P = 3 × 10−4 and P = 8 × 10−5, respectively). In YAC128Q neuronal cultures, HIP14 downregulation resulted in the increased formation of htt inclusions (Fig. 7b, arrowhead). Accordingly, we observed a significant increase in inclusions in cortical neurons transfected with N548-128 and HIP14 siRNA (Fig. 7g; P = 0.025). Moreover, reduced levels of HIP14 significantly increased perinuclear distribution of the proteasome marker Lmp2 (Fig. 7c,f; P = 0.02). In contrast, we observed no increase in the perinuclear accumulation of several other proteins examined, including GM130, the NMDAR receptor subunit NR1 and the postsynaptic proteins PSD-95 and SAP-102, upon downregulation of HIP14; this indicated that the altered trafficking of htt is not due to a disruption of Golgi function or a generalized change in protein sorting (Supplementary Fig. 4 and data not shown). We also found that decreasing HIP14 expression in neurons increased their susceptibility to NMDA treatment (Fig. 7h), indicating that reduced palmitoylation is detrimental to neuronal viability. These results establish that HIP14 is critical for the normal targeting and folding of htt in vivo. Our discoveries that HIP14 palmitoylates htt (Fig. 5d) and that polyglutamine expansion markedly decreases the interaction of htt with HIP14 in vivo (Fig. 6a) provide an explanation for the decreased palmitoylation of mutant htt.
Figure 7.

HIP14 regulates the palmitoylation and distribution of huntingtin in vivo. (a–f) Downregulation of HIP14 expression altered the trafficking of wild-type and mutant htt and increased perinuclear accumulation of Lmp2. Cortical neurons (6 DIV) from YAC18 and YAC128 mice were transfected with GFP and either control siRNA or HIP14 siRNA. Arrowheads in panels a,b and c indicate transfected cells. Scale bar, 5 μm. Representative images show an increase in the cytoplasmic accumulation of wild-type htt in YAC18 neurons (a), and mutant htt (b) and Lmp2 (c) in neurons from YAC128 mice expressing HIP14 siRNA. Quantification of htt (d,e) and Lmp2 (f) staining intensity in the cytoplasm of neurons expressing control or HIP14 siRNA demonstrated that htt and Lmp2 intensities were substantially higher in cells expressing HIP14 siRNA. (g) Downregulation of HIP14 in neurons transfected with N548-128 significantly (P = 0.025) increased the percentage of neurons with inclusions. (h) Downregulation of HIP14 in neurons decreased cellular viability following NMDA-induced cell death. Cortical neurons (8 DIV) were infected with lentiviruses expressing either control or HIP14 siRNA for 3 d, followed by NMDA treatment (Methods). The percentage of cell death in neurons infected with HIP14 siRNA virus was significantly (P = 0.002) higher, demonstrating that decreased expression of HIP14 significantly increases cell death.
DISCUSSION
In this study, we demonstrated a critical role for palmitoylation in regulating the trafficking and folding of htt. Palmitoylation may contribute to the sorting of htt to cytosolic transport vesicles or it may serve as a structural signal for proper protein folding and for the association of htt with other molecules required for its proper trafficking. A protein domain in the N terminus of htt, spanning amino acids 172–372, is essential for the membrane association of htt and for targeting wild-type htt to the plasma membrane11. Here we showed that the palmitoylation of htt at cysteine 214 might be contributing to this finding. The importance of palmitoylation for the correct folding and assembly of the α7 nicotinic receptor, for PSD-95–regulated clustering and for the function of AMPA-type glutamate receptors at the synapse has recently been demonstrated24,25. Notably, the reduced palmitoylation of nicotinic receptors results in their aggregation26, similar to that seen in palmitoylation-deficient mutant htt. Also, the reversible nature of palmitoylation makes it subject to regulation by several stimuli that modulate neuronal protein function and synaptic strength. For instance, cycles of palmitoylation and depalmitoylation regulate the localization and function of specific Ras isoforms27 and of R7BP (ref. 28), a membrane anchor for the RGS7 family. Furthermore, the cycling of palmitate on PSD-95 at the synapse is also regulated by neuronal activity, and this modulates the retention of both PSD-95 and specific glutamate receptor subunits at the synapse25. How the disturbance of htt palmitoylation may be contributing to previously described disturbances in synaptic transmission in Huntington disease20 remains to be determined.
The palmitoylation of htt normally occurs as a result of its interaction with HIP14. The presence of polyglutamine expansion disturbed the interaction of htt with HIP14 (Fig. 6a), resulting in both decreased palmitoylation (Figs. 1f and 6b) and altered distribution of htt (Figs. 2–5); consequently, htt accumulated in inclusions and failed to reach its appropriate cellular destinations. Palmitoylation thus is an important regulator of htt trafficking in vivo. When htt is less palmitoylated, as seen in the YAC model for Huntington disease, disturbances in its trafficking are evident; this is associated with enhanced toxicity and increased cell death in neurons.
The presence of insoluble htt inclusions in the brains of individuals with Huntington disease has led to the hypothesis that these inclusions contribute to the neuronal dysfunction and ultimate cell death that are characteristics of the disease. Much research has focused on these inclusions and on the discovery of aggregation inhibitors as possible therapeutic interventions. However, an increasing body of data suggests that these inclusions are not the disease-causing agents. For example, in YAC128 mouse models of Huntington disease, htt inclusions are first observed months after the initial onset of motor and cognitive dysfunction22. In addition, experimental manipulation of mouse models of the disease has revealed the dissociation between insoluble inclusions and neuronal dysfunction and loss29,30. Furthermore, in a transgenic mouse model expressing a short fragment of htt whose CAG size, tissue distribution and level of expression are identical to those in the full-length YAC128 model, inclusions form earlier and are more prevalent, but this model does not manifest the neuronal dysfunction or degeneration present in the YAC mouse31. Results from recent in vitro studies32 indicate that although the insoluble form of htt may not be toxic, it is likely that a soluble, diffuse form of htt is toxic to neurons. In this study, we showed that inclusion formation, resulting from the decreased palmitoylation of htt, serves as a biomarker for the altered trafficking of htt but does not necessarily directly cause cell death or cellular toxicity.
Removal of palmitate from proteins is thought to be mediated by thioesterases, which cleave cysteine linkages33,34. We have shown that the increased expression of HIP14 with increased palmitoylation of mutant htt partially restores the normal trafficking and distribution of htt. Once the enzymes involved in the depalmitoylation of htt are identified, inhibiting their function could result in increased palmitoylation of mutant htt; potentially, this could restore normal trafficking and alleviate the cellular defects induced by polyglutamine expansion in this protein.
METHODS
Antibodies
The generation of the htt-specific antibodies BKP1 and HD650 are described elsewhere12,22. The following antibodies used in this study: BKP1 (1:500 dilution), HD650 (1:85), htt antibody (MAB2166, Chemicon;1:250 for immunoprecipitation, 1:2,000 for western blotting and 1:1,000 for immunofluorescent assays), monoclonal green fluorescent protein (GFP), early endosomal antigen-1 (EEA1) and GM130 (BD Biosciences; 1:5,000, 1:500 and 1:200, respectively), antibody to Hsp70 (Neomarker; 1:200), monoclonal transferrin receptor antibody (Zymed; 1:1,000), polyclonal caveolin antibody (Santa Cruz Biotechnology; 1:500), goat and rabbit anti-mouse horseradish peroxidase (HRP) conjugates (BioRad; 1:5,000), phalloidin, lysotracker, Alexa Fluor 488 and 568 (Molecular Probes; 1:50, 1:7,000 and 1:1,000, respectively), Cy3 donkey anti-mouse antibody (Jackson ImmunoResearch; 1:200), monoclonal antibody to γ-tubulin (Sigma; 1:100), antibody to proteasome Lmp2 (Abcam; 1:400 dilution) and polyclonal antibody to ubiquitin (DakoCytomation; 1:500).
DNA mutagenesis and cloning
Truncated huntingtin constructs N548 and N224 were previously described35. Cysteine substitutions were generated by polymerase chain reaction (PCR)-based site-directed mutagenesis as described previously35. C-terminal GFP-tagged proteins were generated using a similar strategy, with the stop codon of the tagged proteins replaced by the initiation codon of enhanced GFP (EGFP). All mutated DNA constructs were sequence confirmed.
Cell culture and transfections
All reagents for cell cultures were purchased from Invitrogen. COS cells were cultured as previously described4. For live-cell imaging, we used DMEM without phenol red to eliminate autofluorescence. COS cells were transiently transfected with FuGene 6 transfection reagent (Roche) or Lipofectamine 2000 (Invitrogen) as indicated by the manufacturers. Between 24 and 48 h after transfection, cells were processed as described for each experiment. Cultured cortical and striatal neurons were prepared as previously described20 and experiments were performed at 5–12 days in vitro (DIV). Neurons were transfected with a Nucleofector (Amaxa Inc) at day 0.
Statistical analyses
We have used the Student’s t-test throughout the manuscript to compare the means of two samples. Results are plotted as percentage ± s.e.m.
[3H]palmitoylation assay and immunoprecipitation
Transfected COS cells were labeled with 1 mCi ml−1 [3H]palmitic acid (57 Ci mmol−1; Perkin-Elmer) for 3 h and processed as previously described4. For pulse-chase experiments, transfected cells were labeled for 3 h and then chased for 0, 1, 3 and 6 h with cold palmitate.
Htt coimmunoprecipitation with HIP14
Brains from YAC18 and YAC128 transgenic mice were homogenized in phosphate-buffered saline (PBS) containing protease inhibitors and precleared in the presence of 0.2% SDS and 0.8% Triton X-100 for 1.5 h at 4 °C. This was followed by centrifugation at 2,700g for 5 min. Normal mouse IgG or mouse monoclonal antibody to HIP14 was incubated with 3 mg precleared lysate at 4 °C for 1 h. Then 30 μl of equilibrated protein A+G Sepharose 4 fast-flow beads were added and samples were further incubated at 4 °C overnight. Beads were washed with PBS containing 1% Triton X-100, boiled at 95 °C for 5 min and run on NuPAGE Novex 3–8% Tris-acetate gel (Invitrogen). Western blots were probed with the anti-htt monoclonal antibody HD650, which specifically recognizes the transgene.
Immunofluorescence and time-lapse imaging
Transfected cells growing on coverslips were processed as previously decribed4 with the indicated antibodies. For time lapse, images were collected 28 h after transfection, using a 63× oil objective affixed to a Zeiss inverted light microscope and AxioVision software. While images were being acquired, cells were kept in a 37 °C chamber, supplemented with 5% CO2. Images were collected every 15 min for a total time of 2 h.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
Day 0 rat cortical neurons were transfected with different N-terminal truncations of htt. DMEM was replaced with neurobasal medium 1 h after transfection. Neurons (5–11 DIV) were exposed to 500 μM NMDA for 10 min, further incubated with neurobasal medium for 24 h, and fixed for 10 min in 4% paraformaldehyde in PBS (pH 7.4) and then for 5 min in 100% methanol at −20°C. Cells were stained with GFP antibody for 1 h, followed by incubation with a secondary antibody conjugated to Alexa 488 fluorophore for 1 h at 25 °C. Neurons were stained using the ApopTag Red In Situ Apoptosis Detection Kit (Chemicon) according to the manufacturer’s instructions. Nuclei were counterstained with 0.5 μg ml−1 4,6-diamidino-2-phenylindole (DAPI, Molecular Probes). Neurons transfected with various htt constructs were counted under the microscope, and the number of TUNEL-positive (red) neurons was determined as a fraction of DAPI-positive (blue) neuronal nuclei in each transfected cell (green) by an observer blinded to the identity of the samples. Experiments were repeated five times and 100–200 cells were counted in each experiment. The fractions of TUNEL-positive nuclei determined for each experiment were averaged, and the results are presented as means ± s.e.m.
Cell death assay
Cell death and toxicity was monitored by scoring the proportion of transfected COS7 cells with apoptotic nuclear morphology as previously described18.
Imaging and analysis
Images were acquired on a Zeiss Axiovert M200 motorized microscope by using a monochrome 14-bit Zeiss Axiocam HR charge-coupled device camera at 1,300 × 1,030 pixels. Image analysis was performed as previously described4.
Flotation assay
Brains from wild-type and YAC128 mice were homogenized, sonicated and then centrifuged at 9,500g for 10 min in a SW55 Ti rotor (Beckman Coulter) to remove debris. Following further centrifugation at 100,000g for 90 min, the plasma membrane fraction was resuspended in 2 ml solubilization buffer. 5 mg total protein was brought up to 2 ml and further diluted with an equal volume of 80% (wt/vol) sucrose in MES-buffered saline (MBS), and loaded at the bottom of thin-wall ultracentrifuge tubes. 4 ml of 30% sucrose was overlaid on the membrane fraction and 4 ml of 5% sucrose made the top layer. Gradients were centrifuged at 118,000g for 16 h at 4 °C in a SW41 Ti rotor to isolate detergent resistant membranes. Fractions of 1 ml were collected from the top and analyzed using SDS–polyacrylamide gel electrophoresis (SDS-PAGE).
Labeling with biotin-conjugated 1-biotinamido-4-[4-(maleimidomethyl) cyclohexanecarboxamido] butane (Btn-BMCC)
Brains from YAC 18Q and YAC128Q mice were homogenized and processed for immunoprecipitation as described above. The beads were then washed with wash buffer (PBS, containing 1% Triton X-100) supplemented with 10 mM N-ethylmaleimide (NEM), followed by treatment with 1 M hydroxylamine (NH2OH pH 7.4) for 1 h at 25°C. Samples were then processed as previously described23.
Viral infection
Control siRNA and HIP14 siRNA were cloned into human immunodeficiency virus type 1 (HIV-1)-based lentiviral vectors pLL3.7. Production of lentiviral supernatants and infection of dissociated primary cortical neurons were done as previously described36. Briefly, rat cortical neurons (8 DIV) were infected with viruses expressing siRNA for 4 d, followed by exposure to 500 μM NMDA for 10 min; they were then further incubated with neurobasal medium. After 24 h, cells were processed for the TUNEL assay as described above.
Acknowledgments
We thank O. Sadiq and E. Eyu for technical assistance. M.R.H. is supported by grants from the Canadian Institutes for Health Research, the Huntington Disease Society of America, the Jack and Doris Brown Foundation and the Huntington Society of Canada. A.E.H. is supported by grants from the Canadian Institutes for Health Research, the EJLB foundation and Neuroscience Canada. R.C.D. and W.N.G. are supported by funding from the National Institute of Neurological Diseases and Stroke, the National Institute of Drug Abuse and the Alzheimer’s Association. D.C.R. is funded by a Wellcome Trust Senior Fellowship in Clinical Science, an M.R.C. programme grant and E.U. Framework VI (EUROSCA). A.Y. is supported by funding from the Michael Smith Foundation for Health Research. KH is supported by University Graduate Fellowship and HighQ. M.R.H. and A.E.H. are supported by funding from the HighQ foundation and are investigators of the Fundamental Innovation in Neurodegenerative Diseases (FIND) Research Infrastructure Unit, funded by the Michael Smith Foundation for Health Research. M.R.H. holds a Canada Research Chair in Human Genetics and is a University Killam Professor.
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
AUTHOR CONTRIBUTIONS
A.Y. performed all DNA manipulations to generate truncated and full-length mutant htt and HIP14 proteins, and performed most of the [3H]palmitoylation assays and data analyses. K.H. conducted all the Btn-BMCC palmitoylation assays for full-length htt, scored cells for inclusions in htt-transfected COS cells and neurons, designed and characterized HIP14 siRNA, performed all experiments (and corresponding data analysis) in which the alteration of htt trafficking was investigated by knocking down HIP14, conducted the virus production and infection experiments, and scored for TUNEL assay. R.K. performed endogenous htt [3H]palmitoylation assay, scored inclusions in neurons transfected with full-length htt, and performed fragment-htt toxicity assay and the corresponding data analysis. R.R.S. and L.G. performed the htt and HIP14 coimmunoprecipitation experiment. P.C.O. generated HIP14 siRNA lentiviral constructs and supervised the virus production experiment. P.A. conducted the time-lapse experiments. A.M. assisted in the characterization of full-length htt palmitoylation. C.M.C. and L.A.R. were involved in developing the NMDA-induced excitotoxicity TUNEL assay. R.K. and K.H. conducted the TUNEL assays. W.N.G. and R.C.D. developed and supervised the Btn-BMCC palmitoylation assays. B.R. and D.C.R. performed toxicity assays in COS cells. M.R.H., A.E.H., A.Y. and K.H. wrote the manuscript. M.R.H. and A.E.H. supervised the project.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Published online at http://www.nature.com/natureneuroscience
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.El-Husseini AE, et al. Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J Cell Biol. 2000;148:159–172. doi: 10.1083/jcb.148.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Husseini A, Bredt DS. Protein palmitoylation: a regulator of neuronal development and function. Nat Rev Neurosci. 2002;3:791–802. doi: 10.1038/nrn940. [DOI] [PubMed] [Google Scholar]
- 3.Huang K, El-Husseini A. Modulation of neuronal protein trafficking and function by palmitoylation. Curr Opin Neurobiol. 2005;15:527–535. doi: 10.1016/j.conb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Huang K, et al. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron. 2004;44:977–986. doi: 10.1016/j.neuron.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 5.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 6.Hackam AS, et al. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler VC, et al. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet. 2002;11:633–640. doi: 10.1093/hmg/11.6.633. [DOI] [PubMed] [Google Scholar]
- 8.Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- 9.Velier J, et al. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- 10.DiFiglia M, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 11.Kegel KB, et al. Huntingtin associates with acidic phospholipids at the plasma membrane. J Biol Chem. 2005;280:36464–36473. doi: 10.1074/jbc.M503672200. [DOI] [PubMed] [Google Scholar]
- 12.Kalchman MA, et al. Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J Biol Chem. 1996;271:19385–19394. doi: 10.1074/jbc.271.32.19385. [DOI] [PubMed] [Google Scholar]
- 13.Steffan JS, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 14.Humbert S, et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 15.Warby SC, et al. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–1577. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- 16.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 17.Waelter S, et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyttenbach A, et al. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 19.Sapp E, et al. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 20.Zeron MM, et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 21.Singaraja RR, et al. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. 2002;11:2815–2828. doi: 10.1093/hmg/11.23.2815. [DOI] [PubMed] [Google Scholar]
- 22.Slow EJ, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 23.Drisdel RC, Green WN. Labeling and quantifying sites of protein palmitoylation. Biotechniques. 2004;36:276–285. doi: 10.2144/04362RR02. [DOI] [PubMed] [Google Scholar]
- 24.Drisdel RC, Manzana E, Green WN. The role of palmitoylation in functional expression of nicotinic alpha7 receptors. J Neurosci. 2004;24:10502–10510. doi: 10.1523/JNEUROSCI.3315-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Husseini A, et al. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- 26.Rakhilin S, et al. Alpha-bungarotoxin receptors contain alpha7 subunits in two different disulfide-bonded conformations. J Cell Biol. 1999;146:203–218. doi: 10.1083/jcb.146.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rocks O, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 28.Drenan RM, et al. Palmitoylation regulates plasma membrane-nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family. J Cell Biol. 2005;169:623–633. doi: 10.1083/jcb.200502007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrante RJ, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mastroberardino PG, et al. ’Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002;9:873–880. doi: 10.1038/sj.cdd.4401093. [DOI] [PubMed] [Google Scholar]
- 31.Slow EJ, et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci USA. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 33.Soyombo AA, Hofmann SL. Molecular cloning and expression of palmitoyl-protein thioesterase 2 (PPT2), a homolog of lysosomal palmitoyl-protein thioesterase with a distinct substrate specificity. J Biol Chem. 1997;272:27456–27463. doi: 10.1074/jbc.272.43.27456. [DOI] [PubMed] [Google Scholar]
- 34.Verkruyse LA, Hofmann SL. Lysosomal targeting of palmitoyl-protein thioesterase. J Biol Chem. 1996;271:15831–15836. doi: 10.1074/jbc.271.26.15831. [DOI] [PubMed] [Google Scholar]
- 35.Wellington CL, et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 36.Janas J, Skowronski J, Van AL. Lentiviral delivery of RNAi in hippocampal neurons. Methods Enzymol. 2006;406:593–605. doi: 10.1016/S0076-6879(06)06046-0. [DOI] [PubMed] [Google Scholar]