

Abstract
The interaction of bovine microsomal ferricytochrome b5 with yeast iso-1-ferri and ferrocytochrome c has been investigated using heteronuclear NMR techniques. Chemical-shift perturbations for 1H and 15N nuclei of both cytochromes, arising from the interactions with the unlabeled partner proteins, were used for mapping the interacting surfaces on both proteins. The similarity of the binding shifts observed for oxidized and reduced cytochrome c indicates that the complex formation is not influenced by the oxidation state of the cytochrome c. Protein–protein docking simulations have been performed for the binary cytochrome b5–cytochrome c and ternary (cytochrome b5)–(cytochrome c)2 complexes using a novel HADDOCK approach. The docking procedure, which makes use of the experimental data to drive the docking, identified a range of orientations assumed by the proteins in the complex. It is demonstrated that cytochrome c uses a confined surface patch for interaction with a much more extensive surface area of cytochrome b5. Taken together, the experimental data suggest the presence of a dynamic ensemble of conformations assumed by the proteins in the complex.
Keywords: cytochrome c, cytochrome b5, docking, HADDOCK, NMR, electron transfer
Interprotein electron transfer (ET) reactions are at the heart of many biological processes such as oxidative phosphorylation and photosynthesis. These reactions can be described as a sequence of events that include association of the molecules into an encounter complex, the formation of an active ET complex, ET, and the subsequent dissociation of the complex (Crowley and Ubbink 2003). Protein–protein ET complexes can be classified as transient, in which a compromise between specific binding and fast dissociation must be reached in order to achieve high turnover rates.
The cytochrome c (cyt c)–cytochrome b5 (cyt b5) complex was the first ET complex for which a detailed structural model was proposed (Salemme 1976). Docking of the available X-ray structures of the individual proteins (Mathews et al. 1972; Takano et al. 1973) was performed by optimization of the electrostatic contacts via visual inspection and least-square minimization of the heme-to-heme distance. The resulting model suggested the existence of a single, well-defined complex of 1:1 stoichiometry exhibiting four intermolecular complementary charge interactions [E48-K13, E44-K27, D60-K72, and the (6-Pr)-K79; cyt b5 residues listed first], nearly coplanar heme groups separated by 8 Å, and water exclusion from the binding interface (Salemme 1976). This model stimulated the development of experimental approaches to test the structural properties and ET mechanism of the complex.
The first direct evidence for formation of the nonphysiological complex between cyt b5 and cyt c came from the gel permeation and ultracentrifugation studies (Stoneheurner et al. 1979) and was later supported by electronic spectroscopy (Mauk et al. 1982) and NMR (Eley and Moore 1983). Early attempts to identify the residues involved in complex formation were concerned with chemical modification of specific lysyl residues on the surface of cyt c (Ng et al. 1977; Smith et al. 1980), esterification of cyt b5 heme propionates (Reid et al. 1984; Mauk et al. 1986), and mutagenesis of the cyt b5 carboxylate groups (Rodgers et al. 1988; Rodgers and Sligar 1991) combined with steady-state kinetics, spectrophotometry, and hyperbaric spectroscopy, respectively. Although these studies supported the general validity of the Salemme model both in terms of stoichiometry and the residues involved in the interaction, it was suggested that several other interprotein electrostatic contacts might contribute to complex formation (Ng et al. 1977; Stoneheurner et al. 1979; Mauk et al. 1986). The latter suggestion went in line with the earlier findings that five to seven intermolecular charge interactions would account for the observed ionic strength dependence of the cyt c reduction by cyt b5 (Stoneheurner et al. 1979).
In the course of further investigation of the complex, it became clear that the accumulating experimental data, from 1D NMR spectroscopy (Miura et al. 1980; Eley and Moore 1983; Burch et al. 1990; Whitford et al. 1990), spectrophotometry (Mauk et al. 1986), and kinetics of the ET from cyt c to cyt b5 (Willie et al. 1992) could not be accurately interpreted in the frame of the static Salemme model. The existence of at least two structurally similar complexes or of a dynamic ensemble of nearly isoenergetic adducts of 1:1 stoichiometry was suggested (Eley and Moore 1983; Mauk et al. 1986; Burch et al. 1990; Willie et al. 1992), and the structure of an alternative complex proposed (Mauk et al. 1986). Brownian dynamics simulation of the ET complex led to the conclusion that there is “a single binding domain contributing to electron-transfer dynamics, but not a single conformation as would be observed in a crystal” (Northrup et al. 1993). Indeed, two predominant classes of complexes were observed as follows: the more dominant one, involving four interactions E48-R13, E56-K87, D60-K86, and Pr-K72 (cyt b5 residues listed first), and the other being of Salemme type.
From the analysis of NMR titration curves and the line-broadening change at higher cyt c concentrations, it has been suggested that in addition to a binary complex, a ternary (cyt b5)–(cyt c)2 adduct is also formed (Miura et al. 1980; Whitford et al. 1990). Despite the strong criticism of the proposed 1:2 stoichiometry (Mauk et al. 1995), a recent NMR study of the complex by Banci et al. (2003) supported the concept of the ternary complex. In particular, analysis of the molecular tumbling correlation rates of the complex in the millimolar concentration range has indicated the presence of multiple equilibria with stoichiometric ratios of 1:1 and 1:2.
Recently, the 2D NMR mapping of the interactions in ferrous/ferrous (Hom et al. 2000; Banci et al. 2003; Shao et al. 2003) and ferric/ferric (Hom et al. 2000) complexes were reported. In the study of Banci et al. (2003), chemical-shift perturbation maps for both cyt b5 and cyt c were determined. These studies indicate involvement of somewhat different residues in the complex formation, but they all show that the cyt b5 surface region that is involved in the interaction is more extensive than that predicted by any of the static models (e.g., Salemme 1976; Mauk et al. 1986; Rodgers et al. 1988).
In the present work, the complexes of ferric bovine cytochrome b5 with ferric and ferrous yeast iso-1-cytochrome c are investigated, and the interaction surfaces are mapped for both proteins. Analysis of the binding maps allows us to define the surface areas involved in the complex formation, and to perform protein–protein docking simulations using HADDOCK (Dominguez et al. 2003). This novel approach, unlike traditional docking algorithms, allows direct incorporation of the experimental restraints to drive the docking. Both binary (cyt b5–cyt c) and ternary [cyt b5–(cyt c)2 ] complexes are described. The resulting orientations, adopted by the proteins in the various complexes, represent an ensemble of structures most likely assumed by cyt b5 and cyt c in solution.
Results
In order to generate docked structures of the complex between cyt b5 and cyt c using HADDOCK, chemical-shift mapping of the binding interfaces of both interacting proteins is a prerequisite. For this purpose, four titration experiments, differing in the 15N-labeled protein and oxidation state of cyt c, were performed. In each case, complex formation is evidenced by an increase in line-width (7–9 Hz) for all resonances in the NMR spectrum, as well as chemical-shift perturbations of certain resonances. As seen for all titrations, the magnitude of the line broadening is the same for both shifted and unaffected peaks in the bound form, with a single set of amide peaks in the 2D [15N, 1H] HSQC spectrum being observed. These findings indicate that the cyt b5–cyt c complex is in fast exchange on the NMR time scale. The estimate for the lower limit of the dissociation rate (koff >> √2πΔδ/2, where Δδ is the maximal observed Δδbinding in Hz) of 200 sec−1 for both the ferricyt b5–ferricyt c and the ferricyt b5–ferrocyt c complexes is comparable to the recently reported lower limit value of 180 sec−1 for the ferrous complex of cyt b5 with cyt c (Shao et al. 2003).
Interaction of 15N ferricyt b5 with ferricyt c
In the downfield region of the 1D 1H NMR spectrum of ferricyt b5 (Fig. 1 ▶), a number of well-resolved hyperfine-shifted heme resonances and several less-intense signals are observed. The latter are due to the minor form of cyt b5, in which the heme is rotated by 180° about the α-γ-meso axis in respect to the major form (La Mar et al. 1978, 1981; Keller et al. 1980). The peaks of the major form assigned to the 1-CH3 (~ 11.7 ppm), 6-α-Pr CH2 (~ 14.2 ppm), 5-CH3 (~21.5 ppm), and 2-CαH (~27.5 ppm; data not shown) shift during the titration with ferric cyt c (Fig. 1 ▶), while the position of 7-α-Pr CH2 signal (18.8 ppm) does not change. The chemical-shift perturbations of the resonances corresponding to the 1-CH3 and 5-CH3 in the major form fit well to a 1:1 binding model with the shared equilibrium association constant (Ka) of (5 ± 3) × 104 M−1 (Fig. 2A ▶), in good agreement to a value of (6 ± 3) × 104 M−1 reported recently for the ferrous–ferrous complex (Shao et al. 2003). Chemical-shift changes of the signal attributed to the 6-α-Pr CH2 do not follow the 1:1 binding model (Fig. 2B ▶).
Figure 1.

Downfield hyperfine shifted signals of ferricyt b5 in the absence (0) and the presence of increasing amounts of ferricyt c. The cyt c/cyt b5 ratio is given next to each spectrum. The assignments of several peaks of the major form of cyt b5 are indicated. The resonances marked with an asterisk arise from the protons of ferricyt c. The resonances marked with a hash are due to the minor form of cyt b5.
Figure 2.

Chemical-shift perturbations of ferricyt b5 plotted as a function of the ferricyt c/ferricyt b5 ratio and fitted to a 1:1 binding model. (A) Titration profiles for several cyt b5 nuclei that fit well to the 1:1 binding model with a shared Ka of (5 ± 3) × 104 M−1. (B) Titration profiles for several cyt b5 nuclei that do not fit well to the 1:1 binding model. The curves represent the best shared fit to a 1:1 binding model.
Chemical-shift perturbations were also monitored for the amide resonances in a series of 2D [15N, 1H] HSQC spectra. Small Δδbinding for 41% of ferricyt b5 amides were observed, with the largest averaged chemical-shift perturbation (Δδavg) of 0.054 ppm for Y27. The Δδavg values for cyt b5 amides in the presence of 2.5 molar equivalents of ferricyt c were plotted as a function of cyt b5 residue numbers (Fig. 3A ▶). Several representative titration curves were plotted and fitted globally to a 1:1 binding model. A number of residues fit well with the resulting shared binding constant of (5 ± 3) × 104 M−1 (Fig. 2A ▶), indicating that, at the protein concentrations used, ~80% of cyt b5 is bound to ferricyt c at the molar ratio of 1.0. For most residues, however, the titration curves do not fit well to the 1:1 binding model (Fig. 2B ▶).
Figure 3.

Averaged amide chemical-shift perturbations (Δδavg) plotted as a function of the residue number. (A) 15N ferricyt b5 in the presence of 2.5 molar equivalents of ferricyt c. (B) 15N ferricyt c in the presence of 2.5 molar equivalents of ferricyt b5. The lines denote the significance levels of the mean Δδavg value, the mean plus one half of the standard deviation, and the mean plus one standard deviation.
The chemical-shift changes for ferricyt b5 were mapped onto surface representations of the protein and colored according to increasing Δδavg values (Fig. 4A ▶). Two adjacent surface areas on cyt b5 that are influenced by binding of ferricyt c are found on the front and left sides of the protein. The patch on the front side of cyt b5 contains the affected residues E43–E44, R47–E48, N57, D60–V61, S64, and D66, which surround the heme, and L9 and D53, which are located somewhat farther from the heme group (Fig. 4A ▶). On the left side, the affected surface residues H26–Y27 and E59–D60 form a single patch, while the amino acids V4 and D82 are spatially distant from other interacting residues (Fig. 4A ▶). In addition to the solvent-exposed cyt b5 amino acids described above, several buried residues show significant Δδbinding upon interaction with cyt c. These are the heme ligands (H39 and H63), the residues that form part of the hydrophobic heme-binding pocket (L25, L46, A54, and F58), and two amino acids (I12 and I24) that are located toward the bottom of the protein (Fig. 4B ▶). Amide chemical-shift perturbations of the buried cyt b5 residues cannot be explained in terms of direct binding of cyt c and likely arise due to secondary effects.
Figure 4.

Chemical-shift mapping of 15N cyt b5 in the presence of ferricyt c, and 15N ferricyt c in the presence of cyt b5. Surface representations of cyt b5 (A) (PDB entry 1cyo; Durley and Mathews 1996), with residues colored according to the magnitude of their Δδavg experienced upon binding of ferricyt c (Δδavg ≥ 0.029 ppm in red, Δδavg ≥ 0.023 ppm in orange, Δδavg ≥ 0.017 ppm in yellow) and of ferricyt c (C) (PDB entry 2ycc; Berghuis and Brayer 1992), with residues colored according to the magnitude of their Δδavg experienced upon binding to ferricyt Δδavg ≥ 0.030 ppm, red, Δδavg ≥ 0.022 ppm, orange, Δδavg ≥ 0.014 ppm in yellow). Unassigned and proline residues are shown in dark gray; the heme group is in green. Ribbon representations of cyt b5 (B) and ferricyt c (D) show the buried residues affected upon the interaction (labeled ball-and-stick). Coloring scheme is the same as in A and B. The positions of the N and C termini are indicated. Surface representations in this figure were generated with GRASP 1.3 (Nicholls et al. 1991); ribbon representations in this figure and Figures 5 ▶–7 ▶ were created using MOLSCRIPT (Kraulis 1991) and rendered in Raster3D (Merritt and Bacon 1997).
In order to ascertain whether intermolecular pseudocontact shifts arising from the paramagnetic ferricyt c contribute to the observed chemical-shift perturbations of cyt b5 resonances, ferricyt c was reduced by adding a stoichiometric amount of sodium ascorbate at the end of the titration. Comparison of the HSQC spectrum of the ferric–ferric with that of the ferric–ferrous complex did not show any significant differences, which would be attributed to pseudocontact shifts. Therefore, it was concluded that the observed shifts arise solely from complex formation.
Interaction of 15N ferricyt b5 with ferrocyt c
To investigate the influence of the oxidation state of cyt c on complex formation, 15N ferricyt b5 was titrated with ferrocyt c. Reduced cyt c is diamagnetic and does not give rise to signals in the downfield region of the 1D NMR spectrum. Therefore, several cyt b5 peaks that overlapped with cyt c resonances in the ferric–ferric experiment could be observed. In addition to the 1-CH3 (~11.7 ppm), 6-α-Pr CH2 (~14.2 ppm), 5-CH3 (~21.5 ppm), and 2-CαH (~27 ppm; data not shown), chemical-shift perturbations of 7-α-Pr CH2 (15 ppm) of the minor form and an unassigned signal at 16.5 ppm could be monitored. Again, these spectral changes were attributed to complex formation and fitted to a 1:1 binding model. The signals arising from 1-CH3, 5-CH3, and the 7-α-Pr CH2 of the minor form fit well to this model (data not shown). Analogously to the ferric–ferric titration, 2D [15N, 1H] HSQC spectra were recorded and small Δδbinding for various 15N and 1H nuclei observed. Values of Δδavg plotted as a function of residue number give results that are very similar to those for the ferric–ferric titration, with the main difference being the amino acids I24, D53, and N57, which are not affected during the titration with ferrocyt c (see Fig. 1A ▶ in supplemental material). The values of Δδbinding for several amide resonances together with those for three 1D peaks were fitted globally to a 1:1 binding model with a resulting Ka of (4 ± 1) × 104 M−1. The value of the association constant implies that, for the protein concentrations used, ~80% of cyt b5 is bound to ferrocyt c at the molar ratio of 1.0. Although a number of resonances do not fit accurately to the 1:1 binding model, in general, the deviation from 1:1 stoichiometry is less pronounced than in the ferric–ferric titration.
Interaction of 15N ferricyt c with ferricyt b5
Complex formation between cyt b5 and cyt c was also investigated from the cyt c side. For this purpose, the same set of experiments as for the 15N cyt b5–cyt c system was performed, but with 15N cyt c as the observed protein. In the 1D NMR spectra of ferricyt c, two signals at 35.0 and 31.5 ppm, assigned to the heme 8-CH3 and 3-CH3 groups, respectively (Gao et al. 1990), exhibit shifts during titration with ferricyt b5. The Δδbinding for the two peaks can be fitted to a 1:1 binding model with a Ka of (3 ± 1) × 104 M−1, in agreement with the value obtained for the corresponding 15N cyt b5–cyt c titration. Upon addition of cyt b5, chemical-shift perturbations of ferricyt c amides were monitored in the [15N, 1H] HSQC spectra. Small Δδbinding for 32% of the ferricyt c amides were observed, with the largest Δδavg of 0.123 ppm for K86. The Δδavg values for ferricyt c in the presence of 2.5 equivalents of cyt b5 are plotted as a function of ferricyt c residue number in Figure 3B ▶. The chemical-shift perturbations in both 15N and 1H dimensions fit well to the 1:1 binding model with a Ka from the global fit of 10 different binding curves (6 ± 3) × 104 M−1, again consistent with the corresponding cyt b5 titration. The Ka value suggests that, at the protein concentrations used in the experiment, 75% of ferricyt c is bound to cyt b5 at the molar ratio of 1.0.
Using the same procedure as for 15N cyt b5, the residues of cyt c were color-coded and mapped onto the surface representation of the protein (Fig. 4C ▶). Residues exhibiting the largest Δδavg are T8-L9, T12, Q16, K72, G77, K79, A81-F82, and K86, all of which are located in a patch surrounding the heme edge. A few weakly affected amides are found on the opposite side of the protein (Fig. 4C ▶). Several buried residues also exhibit significant chemical-shift perturbations during the titration with cyt b5 (Fig. 4D ▶). These are G6, F10, C14, Y48, V57, and L85, all of which sit in the core of the protein.
Interaction of 15N ferrocyt c with ferricyt b5
Analysis of the [15N, 1H] HSQC spectra for the 15N ferrocyt c–ferricyt b5 titration shows that the oxidation state of cyt c has little effect on the binding properties of the two cytochromes. Plotting Δδavg for ferrocyt c in the presence of 2.5 molar equivalents of cyt b5 against the cyt c residue numbers gives results very similar to those for the ferricyt c (see Fig. 1B ▶ in supplemental material). The main difference is found for the amides of K79 and K86, with the former being more affected when the cyt c is reduced and the latter when cyt c is oxidized. Several residues of ferrocyt c, which exhibit significant Δδbinding, fit well to the 1:1 binding model with a shared Ka of (3 ± 1) × 104 M−1 (data not shown). The value of the association constant indicates that, at the protein concentrations utilized in the experiment, 70% of the ferrocyt c is bound to cyt b5 at the molar ratio of 1.0.
Finally, to check for intermolecular pseudocontact shifts arising from ferricyt b5 and affecting the amides of ferrocyt c, sodium dithionite was added at the end of the titration in order to generate the completely reduced complex. Similarly to what was observed for the 15N ferricyt b5–cyt c titration, no evidence for the presence of intermolecular paramagnetic effects was obtained.
Effects of ionic strength on complex formation
Complex formation between cyt b5 and cyt c, with net charges in the ferric form of −7 and +10, respectively (Margoliash et al. 1961; Tsugita et al. 1970), is electrostatically driven. In order to assess the influence of the ionic strength on the complex formation, two salt titrations, differing in the 15N-labeled protein, were performed. In each experiment, the sample containing the 15N-labeled protein and 2.1 molar equivalents of the unlabeled partner was titrated by adding small aliquots of NaCl solution to a total salt concentration of 20–220 mM (see Materials and Methods for details). To correct for the ionic strength effect on the free protein, the Δδbinding values for each titration point were determined by using the isotonic sample containing only 15N-labeled protein as a reference. For both ionic strength titrations, increasing salt concentration leads to the uniform decrease of the Δδbinding for all affected residues of both proteins (data not shown).
Protein–protein docking
NMR experiments performed in this study have allowed for the chemical-shift mapping of the interacting surfaces on both proteins upon complex formation. However, as a fast-exchange NMR signal is averaged over all molecular orientations, the observed maps represent an average of the multiple orientations of the proteins in the complex. In order to gain a structural insight into the possible orientations that are adopted by the protein molecules in the complex, in silico docking simulations were carried out, using the binding maps as input. Two docking runs for the binary complexes of cyt b5–ferricyt c and cyt b5–ferrocyt c and two runs for the ternary complexes of cyt b5–(ferricyt c)2 and cyt b5–(ferrocyt c)2 were performed. The results of the docking simulations for the complexes with reduced and oxidized cyt c are very similar; therefore, only the latter results are discussed below. The data concerning the runs with the reduced cyt c can be found in the supplemental material.
For both binary and ternary complexes of cyt b5 with ferricyt c, the ambiguous intermolecular restraints (AIRs) were generated using the active and passive residues specified for both proteins. According to the procedure described in Materials and Methods, 13 cyt b5 residues exhibiting significant Δδavg and high solvent accessibility were defined as active, as well as two solvent-accessible heme groups, 5-CH3 and 6-α-Pr CH2, which shift during the titration with cyt c. Twenty-one surface-exposed amino acids that occur close to the active residues were defined as passive. For ferricyt c, 11 active and 22 passive residues were specified in an analogous way (for the complete list of active and passive residues and the flexible segments for both proteins, see Table 1 in supplemental material). The docking process, which is governed by the combination of electrostatic, van der Waals, and AIR restraints, generates solutions that are clustered using a backbone RMSD cutoff criterion (for details, see Materials and Methods and Dominguez et al. 2003).
For the binary cyt b5–ferricyt c complex, 10 clusters of solutions were obtained with a 2.0 Å RMSD cutoff. As can be appreciated from the values of the nonbonded energy terms, which are plotted as a function of the backbone RMSD from the lowest-energy structure calculated over five best structures of each cluster (Fig. 5A ▶), the docking solutions consist of several groups of structures that have nearly equal energies (for the structural statistics of all clusters, see Table 2A of the supplemental material). The spatial distribution of the docking solutions is shown in Figure 5 ▶, B and C, in which the centers of mass of cyt c molecules from the lowest-energy structure of each cluster are superimposed on the cyt b5 structure, and vice versa.
Figure 5.

HADDOCK docking solutions for the binary complex of cyt b5 with ferricyt c. (A) Intermolecular nonbonded energy as a function of the backbone RMSD from the lowest energy structure for the docked structures. The clusters averages are indicated by filled circles with error bars, which represent the standard deviation from the mean for the five lowest-energy structures of each cluster. The clusters are numbered according to the increasing energy. Stereo views of the centers of mass of (B) cyt c from each cluster (gray spheres) superimposed onto the ribbon representation of cyt b5, and (C) cyt b5 from each cluster (gray spheres) superimposed onto the ribbon representation of cyt c. The ribbon representations of the proteins in B and C are shadowed from light to dark gray according to the increasing Δδ avg, with the heme group shown in sticks. The centers of mass were calculated for the lowest energy structure of each cluster. The numbering of the clusters corresponds to that in A.
It is evident that the interacting surface of cyt c is confined to a single patch on the front side of the protein, while cyt b5 provides a much more extensive area for the interaction with cyt c. Overall, the docking solutions consist of two subpopulations of clusters: one in which cyt c molecule docks to the front face of cyt b5 (“head-on” orientation) and the other with cyt c binding to the left side of cyt b5 (“side-on” orientation). The ensemble of the five lowest-energy structures for two representative clusters with the “side-on” and “head-on” orientations are depicted in Figure 6, A and B ▶, respectively.
Figure 6.

Docking models of the binary cyt b5–cyt c complexes with “side-on” (A) and “head-on” (B) orientations. Ensembles of the five lowest-energy structures for the cluster 3 (A) and cluster 8 (B) are shadowed from light to dark gray according to the increasing Δδavg, with the heme group shown in sticks. The positions of the protein termini are indicated.
As mentioned above, the cyt b5–cyt c titration profiles for many of the cyt b5 residues cannot be explained in terms of a 1:1 binding model (Fig. 2B ▶). One of the explanations for the observed binding curves could be the formation of higher-order protein complexes with increasing cyt c concentration. In order to verify the possibility of a ternary complex formation, the protein–protein docking of the cyt c b5–(cyt)2 heterotrimer was performed with a modified version of HADDOCK, allowing direct docking of ternary complexes (see Materials and Methods). Again, only the results for the docking run with the oxidized cyt c are discussed, and those with the reduced cyt c can be found in the supplemental material. The set of AIRs used for the docking of the trimer was the same as that for the binary complex (see Table 1 in supplemental material), with the modification such that each active residue of ferricyt b5 had a restraint to all active and passive residues of both ferricyt c or ferrocyt c molecules. The results for the simultaneous docking of two ferricyt c molecules to one cyt b5 are presented in Figure 7 ▶. The increased number of degrees of freedom in a ternary system makes clustering of the docking solutions more difficult, as can be appreciated from Figure 7A ▶, which shows a broader structure distribution compared with that of the binary complex (Fig. 5A ▶). Clustering of the docked structures with a backbone RMSD cutoff of 3.75 Å resulted in 10 partially overlapping clusters (for the structural statistics, see Table 2C in Supplementary material). The distribution of cyt c molecules around cyt b5 is shown in Figure 7B ▶. Compared with the binary complex, the same surface areas on cyt b5 sustain the interactions with cyt c molecules. An ensemble of structures for the ternary complex (Fig. 7C ▶) looks very much like a combination of binary complexes with the “side-on” and “head-on” orientations (Fig. 6 ▶), implying that similar interactions between the partner molecules take place in the binary and ternary complexes.
Figure 7.

HADDOCK docking solutions for the ternary complex of cyt b5 with two ferricyt c molecules. (A) Intermolecular nonbonded energy as a function of the backbone RMSD from the lowest energy structure for the docked structures. The clusters averages are indicated by filled circles with error bars, which represent the standard deviation from the mean for the five lowest-energy structures of each cluster. (B) Centers of mass of ferricyt c from each cluster (gray spheres) superimposed onto the ribbon representation of cyt b5. The centers of mass were calculated for the lowest energy structure of each cluster. The numbering of the clusters corresponds to that in A. (C) Ensemble of the three lowest-energy structures of the lowest-energy cluster (1 in A and B). The positions of the protein termini are indicated. The ribbon representations are shadowed as in Figures 5 ▶ and 6 ▶.
Discussion
NMR study of the cyt b5–cyt c complex
Previous attempts to study by heteronuclear NMR the complex between cyt b5 and cyt c, in which one or both proteins are in the oxidized state, have been severely hampered by the auto-reduction of cyt c (Banci et al. 2003). Under the experimental conditions used in this work (Sigma Chelex pretreated 20 mM sodium phosphate at pH 6.0), both proteins are stable, and no self-reduction of ferricyt c occurs during the experiments. The binding constants, as estimated from fitting the titration curves to a 1:1 binding model, are (5 ± 3) × 104 M−1 and (3 ± 1) × 104 M−1 for ferricyt b5–ferricyt c and ferricyt b5–ferrocyt c complexes, respectively. These are in good agreement with previously reported values, which range between 4–8 × 104 M−1 at low ionic strength and pH 7.0 at 298–303 K (Mauk et al. 1982; Rodríguez-Marañón et al. 1996; Shao et al. 2003). As pointed out by Shao et al. (2003), the variations in these values can be attributed to different protein sources and experimental conditions, especially the ionic strength, which is notoriously difficult to control at high protein concentrations.
The NMR titrations show that the chemical-shift perturbations of some cyt b5 resonances follow a 1:1 binding model, while others clearly deviate (Fig. 2 ▶). At the same time, the binding curves for most cyt c resonances exhibit monophasic behavior (data not shown). A plausible explanation for this phenomenon could be the formation of higher-order protein complexes with increasing cyt c concentrations. Assessing the stoichiometry of the cyt b5–cyt c complex from the NMR titration curves should be done with care, because the complex is sensitive to variations in the solution ionic strength, which arise from addition of the partner protein in the course of the titration (Mauk et al. 1995). In this study, the approximate ionic strength of solution, as estimated using the approach described by Eley and Moore (1983), increased from 45 to 55 mM with the addition of protein aliquots up to two molar equivalents. A further ionic strength increase of ~1 mM at the end of the titration occurred due to the pH correction after each titration step. On the basis of the ionic strength titration curves, it could be calculated that a total ionic strength increase of 11 mM causes negligible effects on the binding curves. Thus, changes of the ionic strength during the titration experiments cannot account for the observed binding curves that deviate from 1:1 binding behavior. The presence of a ternary complex, in which one cyt b5 molecule interacts with two molecules of cyt c, has been suggested before (Miura et al. 1980; Whitford et al. 1990), and later criticized (Mauk et al. 1995). Although the experimental data presented in this work are not sufficient for reliable fitting to a 1:2 binding model, combined with the protein–protein docking, they support the formation of a (cyt b5)–(cyt c)2 complex, in agreement with the results of another recent NMR study (Banci et al. 2003).
Comparison of the amide binding shifts (Δδavg) for the cyt b5 residues upon interaction with either ferric or ferrous cyt c reveals that the only significant differences are found for three amino acids (I24, D53, and N57), which are perturbed by the binding of oxidized, but not reduced cyt c. The same comparison for the residues of ferric and ferrous cyt c upon interaction with cyt b5 shows that the chemical-shift perturbations for all but two amino acids are similar. These two residues, K79 and K86, have the most salt-sensitive resonances in the protein (data not shown; see also Crowley et al. 2002). Therefore, the variations in Δδavg between the ferric and ferrous forms might be due to slight discrepancies in ionic strengths during the two titrations, rather than to the redox-dependent differences in the binding of cyt c to cyt b5. Taken together, it is concluded that the complex formation between ferric cyt b5 and cyt c is not markedly influenced by the oxidation state of the latter.
The data presented in this work suggest that the interaction interface of cyt b5 in the complex with either reduced or oxidized cyt c is more extensive than that reported recently (Shao et al. 2003) or predicted by the static models of Salemme (1976) and Rodgers et al. (1988). The perturbed surface residues of cyt b5 are located at two adjacent regions (Fig. 4A ▶), which are similar to those described by Banci et al. (2003). In contrast to the results of Hom et al. (2000), no binding at the suggested “cleft” site is observed in our experiments. Many affected amino acids are found on the front face of cyt b5, in a patch surrounding the exposed heme edge (Fig. 4A ▶). Among those are the charged groups implied by the Salemme (1976) and Northrup et al. (1993) models, E44, E48, D60, and the most exposed Pr (6-α-Pr CH2). Involvement of these residues in the complex formation, as well as E56, which is not affected in our work, was shown in an earlier study, by a combination of site-directed mutagenesis and NMR techniques (Qian et al. 2001). In a recent report, it was confirmed that these residues play a pivotal role in stabilization of the complex, but are not crucial for the intermolecular ET (Ren et al. 2004).
Another affected surface area is located on the left side of cyt b5, where most of the perturbed residues cluster in a single patch (Fig. 4A ▶). Although the majority of the affected cyt b5 residues are charged, implying that the intermolecular electrostatic interactions play a predominant part in the formation of this complex, several surface-exposed polar and hydrophobic amino acids are also perturbed by the addition of cyt c. Interestingly, the binding surface map of cyt b5 in the complex with cyt c looks very similar to that for the highly dynamic interaction with metmyoglobin (Worrall et al. 2002), with the residues E43, E44, D60, and V61 strongly affected in both, which implies the similarity of the binding mode used by cyt b5 in both interactions.
The cyt c residues experiencing significant chemical-shift changes upon interaction with cyt b5 are confined to a single surface patch on the front side of the molecule (Fig. 4C ▶). This region is similar to that observed by Banci et al. (2003) upon interaction with rabbit cyt b5, with the main difference being the α4 helix (residues 90–101), being unaffected in our study. Among the cyt c residues most affected by the binding to cyt b5 are K72, K79, and K86, which are involved in the models suggested by Salemme (1976) and Northrup et al. (1993). Comparison of the binding map for cyt c bound to cyt b5 with that of cyt c in the complex with the nonphysiological partner adrenodoxin (Worrall et al. 2003) reveals a considerable similarity of the surface areas that are influenced by complex formation. Chemical-shift mapping studies of the interacting surface of cyt c upon binding to yeast cytochrome c peroxidase (Worrall et al. 2001), cyanobacterial cytochrome f (Crowley et al. 2002), and pea plastocyanin (Ubbink and Bendall 1997) indicate that among the most perturbed residues are T12 (Q12 in horse heart cyt c used by Ubbink and Bendall 1997), Q16 and K79. This is also observed for the cyt c in the complex with cyt b5 (present work). This finding suggests that cyt c uses a conserved set of surface-exposed amino acids for the interactions with a variety of partner proteins in solution.
For both cyt b5 and cyt c, there are several solvent-inaccessible amino acids, for which the observed chemical-shift perturbations cannot be caused directly by the binding event. Such phenomenon has been explained in terms of secondary effects, which arise from transmittance of the primary effect of the complex formation from the surface of the protein to its core via chemical bonds and hydrogen bonds (Ubbink and Bendall 1997). In the case of cyt b5 and cyt c, most of the affected buried residues are located in close proximity to the heme group (Fig. 4B,D ▶); therefore, the observed effects could be attributed to small changes in heme environment induced by complex formation.
When compared with each other, it is evident that the interaction surface of cyt b5 is more extensive than that of cyt c. Although the NMR experiments used in this study cannot discriminate between the chemical-shift changes induced by either the direct-binding events or the associated secondary effects, it is believed that no significant structural rearrangement of the partner proteins occurs upon cytb5–cyt c complex formation (Mauk et al. 1995). In this case, the binding of cyt c to cyt b5 in a single, well-defined orientation with a 1:1 stoichiometry does not account for the observed chemical-shift perturbation maps. As there are two simultaneously affected areas on the surface of cytb5, the interaction implies the coexistence of at least two different complexes with cyt c, or the presence of an ensemble of structures with similar energies as has been suggested before (Mauk et al. 1986, 1995; Burch et al. 1990; Banci et al. 2003). It seems likely that, during the complex formation, cyt c uses a specific and confined region to explore a much wider area on cyt b5. The reason for this behavior is probably the much more dispersed charge distribution on the latter.
The presence of a dynamic ensemble of structures rather than a single, well-defined cyt b5–cyt c complex is further confirmed by the fact that no intermolecular pseudocontact shifts are observed. Contrary to previously published data (Guiles et al. 1996), comparison of the HSQC spectra of the ferricyt b5–ferricyt c complex with those of the complexes in which either only cyt c or both proteins were reduced, shows no significant differences. This implies that no paramagnetic effect arising from the ferric heme group of cyt c is experienced by the amide protons of cyt b5 and vice versa. As previously proposed (Ubbink and Bendall 1997), the absence of the intermolecular pseudocontact shifts can be explained by assuming that the proteins adopt a multitude of different orientations in the complex, all of which are in fast exchange. In this way, the paramagnetic, orientation-dependent effects would average to zero (Ubbink and Bendall 1997).
The magnitude of the chemical-shift perturbations for both cyt b5 and cyt c is also consistent with the existence of multiple protein orientations within the complex. These orientations, which have nearly equal energies and are in fast exchange, result in averaging of the chemical-shift perturbations over all orientations. In combination with the absence of close contacts and extensive desolvation, this would explain the observed small Δδbinding in the complex (Worrall et al. 2003). Comparison of the absolute values of Δδavg for the cyt b5–cyt c complex with those for other dynamic systems places it between that of cyt c–cytochrome c peroxidase (Worrall et al. 2001), in which the proteins occupy a single orientation for a significant fraction of the complex lifetime, and the highly dynamic complex of cyt b5–metmyoglobin (Worrall et al. 2001). The values of the Δδavg for the cyt b5–cyt c complex are of the same magnitude as those for the dynamic complexes of cyt c with adrenodoxin (Worrall et al. 2003) and horse heart cyt c with pea plastocyanin (Ubbink and Bendall 1997), suggesting that cyt b5 and cyt c adopt different orientations within the complex, rather than form a single, well-defined structure. Addition of salt to the protein complex leads to the uniform decrease of the Δδbinding for the affected residues, reaching the values of the chemical shifts of the free protein at the end of the titration. Such behavior is typical of a complex dominated by electrostatic interactions, and implies that, at higher ionic strength, both dynamics of the complex formation and the range of orientations sampled by the proteins in the complex remain similar to those at low ionic strength.
Protein–protein docking
HADDOCK is a new in silico docking approach, which allows direct incorporation of biochemical and/or biophysical information to drive the docking process (Dominguez et al. 2003). Unlike traditional docking algorithms, which use ab initio docking simulations, followed by a posteriori scoring based on the experimental information, HADDOCK a priori includes the available experimental data that drive the subsequent docking simulations.
The distribution of the centers of mass of docked proteins around either cyt b5 or cyt c (Fig. 5B,C ▶) shows that cyt c uses a single surface patch for interaction with the two areas on the surface of cyt b5, one with cyt c binding to the front side of cyt b5 (“head-on” orientation, Fig. 6B ▶), and the other with cyt c docking to the left side of the protein (“side-on” orientation, Fig. 6A ▶). Only a combination of both docking solutions satisfies all restraints in accordance with a dynamic nature of the complex. Docking of the ternary (cyt b5)–(cyt c)2 complex resulted in a broader distribution of clusters (Fig. 7A ▶) than that of the binary complex (Fig. 5A ▶), which is not unexpected for a system with more degrees of freedom. A clustering based on RMSDs calculated over only two of the three components would lead to much better defined solutions. The resulting structures of the ternary complex combine the features of the “head-on” and “side-on” orientations of the binary complex. For each docking cluster, cyt b5 interacts with two cyt c molecules, one of which is located close to the front part of cyt b5, while the other is binding to the side of the protein (Fig. 7B,C ▶). These results indicate that one cyt b5 can sustain the interaction with two cyt c molecules, and the geometry of the ternary complex would account for the observed chemical-shift perturbation maps of the interacting proteins. As has been mentioned above, the formation of the ternary complexes may indeed take place at higher cyt c concentrations. Interestingly, cluster distributions for both binary (Fig. 5A ▶) and ternary (Fig. 7A ▶) complexes of cyt b5 with cyt c are much broader than those for the static complexes, recently analyzed with HADDOCK (Arnesano et al. 2004; van Drogen-Petit et al. 2004). This finding is consistent with the presence of a highly dynamic complex, in which the partner proteins adopt multiple orientations.
Binary and ternary HADDOCK solutions are optimized for interaction energy, yielding four to five salt bridges and 10 to 12 H-bonds between the proteins. However, it is unrealistic to assume that such extensive intermolecular contacts are formed. The apparent binding constant of 5 × 104 M−1 is equivalent to a binding energy of a mere 6 kcal/mol, which suggests that only a few weak interactions are formed on average. Furthermore, the small averaged chemical-shift perturbations could be interpreted as evidence that desolvation of surface residues is limited (Crowley and Ubbink 2003). Extensive H-bonding and desolvation would result in much larger chemical-shift perturbations, as observed in many cases for specific complexes of biomolecules. Thus, the HADDOCK structures should not be considered as detailed snapshots of the complex. Rather, the docking clusters provide a picture of the orientations that both proteins sample within this highly dynamic complex.
Our conclusions concerning the mobility of the complex are consistent with the results of a very recent study, which supported a dynamic docking paradigm for cyt b5–cyt c complex (Ren et al. 2004). This paradigm, initially suggested for cyt b5–myoglobin complex (Liang et al. 2002), indicates that a multitude of weakly bound conformations of the docked complex contribute to the overall binding of cyt c to cyt b5. However, only a few of these conformations are ET active, and they need not be the most favorable for binding. If one assumes that only the conformations with the two proteins in close heme-to-heme contact are ET favorable (Ren et al. 2004), the docked structures with the “head-on” orientation (Fig. 6B ▶) might represent ET-productive complexes.
Materials and methods
Preparation of protein and NMR samples
Both unlabeled and isotopically enriched 15N bovine microsomal cyt b5 and 15N T-5A/C102T variant of yeast iso-1-cyt c were produced in Escherichia coli and purified as reported previously (Funk et al. 1990; Pollock et al. 1998; Morar et al. 1999). Cyt b5 with a UV-vis peak ratio A412.5/A280 ≥4.0 was used for the NMR experiments. Concentrations of the ferricyt b5 were determined according to the absorbance peak at 412.5 nm (ɛ = 117 mM−1cm−1). Ferricyt c with a UV-vis peak ratio A410/A280 ≥ 4.0 was used throughout. It was prepared by addition of an excess (two to threefold) of K3[Fe(CN)6], followed by ultrafiltration under nitrogen against 20 mM sodium phosphate (pH 6.0) (Amicon; YM3 membrane). Ferrocyt c was prepared in an analogous way, except that sodium ascorbate was used as a reductant. Protein concentrations were determined according to the absorbance peak at 410 nm (ɛ = 106.1 mM−1cm−1) for ferricyt c and 550 nm (ɛ = 27.5 mM−1cm−1) for ferrocyt c.
All NMR samples contained 0.49–0.53 mM of the 15N-labeled protein and varying amounts of the unlabeled partner protein in 20 mM sodium phosphate (pH 6.0), 6% D2O for lock, and 100 μM CH3CO15 NH2 as an internal reference. The buffer used for preparing samples and protein stock solutions was pretreated with Chelex 100 (Sigma) chelating agent in order to remove traces of metals that could catalyze self-reduction of cyt c. The pH of the samples was checked before and after each titration step and adjusted, if necessary, with small aliquots of 0.1 M NaOH or 0.1 M HCl solutions.
NMR titrations
All NMR titrations were carried out by adding microlitre aliquots of the stock solution of the unlabeled protein (1.76 and 1.86 mM of ferricyt b5 in titrations with 15N ferric and 15N ferrocyt c, respectively, and 2.15 mM of ferricyt c and 2.2 mM of ferrocyt c in titrations with 15N ferricyt b5) to the sample containing 500 μL of the 15N-labeled protein, with an initial concentration of 0.5 mM. Each titration consisted of 10 experimental points with 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.5, 1.9, and 2.5 molar equivalents of the unlabeled partner protein present. For the ionic strength titrations, 500 μL of a sample, containing 0.5 mM of the 15N-labeled protein and 2.1 molar equivalents of the unlabeled partner protein in 20 mM sodium phosphate (pH 6.0), was titrated by adding aliquots of 2 M NaCl to the total salt concentrations of 20, 70, 120, 170, and 220 mM. For each salt titration, a sample containing only 15N-labeled protein was titrated in the same manner and used as a reference.
NMR experiments
All NMR experiments were performed at 301 K on a Bruker DMX600 spectrometer equipped with a TXI-Z-GRAD (1H, 13C, and 15N) probe. For each titration, 1D 1H spectra were acquired with a spectral width of 48.08 kHz. The spectral widths (in kiloHertz) for 2D [15N, 1H] HSQC spectra were 1.9 (15N) and 9.62 (1H) for 15N cyt b5 titration; 2.68 (15N) and 8.39 (1H) for 15N cyt c titration. Data processing of the 1D 1H and 2D [15N, 1H] HSQC spectra was performed in XWINNMR and AZARA (available from ftp://ftp.bio.cam.ac.uk/pub/azara), respectively. Assignments of the 15N and 1H nuclei of the free ferricyt b5 and ferric and ferrocyt c were taken from previous work (Worrall et al. 2001, 2002). The amides, which were not observed in the present work, were A3, S18, S20, F35, G41, L70, and I76 for ferricyt b5; F-4, A3, H33, H39, N56, E66, M80, G83-G84, K87, and I95 for fer-ricyt c and A3, K11, K22, R38, A43, N52, N56, K73, M80, G83, G84, and K99 for ferrocyt c. Chemical-shift perturbations of 15N and 1H nuclei were analyzed by overlaying the spectra of bound 15N-labeled protein with that of the free protein in the assignment program ANSIG (Kraulis 1989; Helgstrand et al. 2000). The averaged amide chemical-shift perturbations (Δδavg) were derived from:
 |
where ΔδNbinding is the chemical-shift perturbation of the amide nitrogen and ΔδHbinding is that of the amide proton. Chemical-shift titration curves were analyzed with a two-parameter nonlinear least squares fit of the data using a one-site binding model, which corrects for the dilution effect (Kannt et al. 1996):
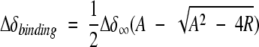 |
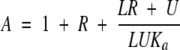 |
where R is the ratio between the unlabeled and the 15N-labeled proteins, Δδbinding is the chemical-shift perturbation at a given protein ratio, Δδ∞ is the chemical-shift perturbation at R→∞, L is the initial concentration of the 15N-labeled protein, U is the concentration of the unlabeled protein stock solution used in the titration, and Ka is the association constant for a 1:1 complex.
Protein–protein docking simulations
The coordinates for ferricyt b5, ferricyt c, and ferrocyt c were taken from the RCSB Protein Data Bank, entries 1cyo (Durley and Mathews 1996), 2ycc (Berghuis and Brayer 1992), and 1ycc (Louie and Brayer 1990), respectively. The docking was performed with HADDOCK 1.2 (Dominguez et al. 2003). The average relative solvent-accessible surface area for each residue was calculated using NACCESS (http://wolf.bms.umist.ac.uk/naccess/). In HADDOCK, the docking is driven by ambiguous intermolecular restraints (AIRs), which are defined as follows. For both proteins, the residues with Δδavg above the mean plus half of the standard deviation and with a relative solvent accessibility above 50% are selected and called ‘active’. Then, all neighbor residues of active residues exhibiting a relative surface accessibility above 50% are defined as ‘passive’. The AIRs are defined as ambiguous distance restraints between the active residues of one protein and both the active and the passive residues of the other (see Dominguez et al. 2003 for details). The flexible segments for docking are defined from the active and passive residues used in the definition of AIRs ± two sequential residues.
The docking of ferricyt b5 with ferricyt c or ferrocyt c was performed following the standard HADDOCK 1.2 protocols (Dominguez et al. 2003) with the modification that, due to the expected high dynamical nature of these complexes, the docking was performed during the semiflexible simulated annealing part of the protocol rather than during the rigid-body energy minimization. For this, the separation distance between the respective molecules was decreased to 50 Å, and only rotational minimization was allowed in the initial rigid body energy minimization (EM) to allow the molecules to face each other properly prior to docking. The 200 solutions from the rigid-body EM were then subjected to the standard semiflexible simulated annealing (SA) in torsion angle space in HADDOCK, but with an increased number of integration steps to allow for the docking during the SA. This part consisted of the following:
A rigid-body Molecular Dynamics search (12,000 steps at 2000 K);
a first rigid-body simulated annealing (8000 steps from 2000 to 500 K);
a second semiflexible simulated annealing, during which side chains at the interface are free to move (2000 steps from 2000 to 50 K); and
a third semiflexible simulated annealing, during which both side chains and backbone at the interface are free to move (1000 steps from 500 to 50 K).
The resulting structures were finally subjected to a final refinement in explicit water, clustered using a 2.0 Å backbone RMSD cutoff criterion and sorted according to the intermolecular energy (sum of the van der Waals, electrostatic, and AIRs energy). The five lowest-energy structures of each cluster were selected for further analysis.
The docking of the tertiary complex, ferricyt b5 with two ferricyt c or two ferrocyt c, was performed with a special version of HADDOCK, allowing direct and simultaneous docking of tertiary systems. The protocol followed is the standard HADDOCK 1.2 protocol (Dominguez et al. 2003) extended to deal with three molecules. A total of 2000 docking solutions were generated during the rigid-body energy minimization step, and the best 200 were further subjected to the semiflexible simulated annealing and final refinement in explicit solvent. For docking of the tertiary complex, the AIR restraints were modified such that each active residue of ferricyt b5 will have a restraint to all active and passive residues of both ferricyt c or ferrocyt c molecules.
Electronic supplemental material
Supplementary material contains chemical-shift perturbation (Δδavg) plots and HADDOCK docking solutions for the ferricyt b5–ferrocyt c complex, and a list of AIR restraints and structural statistics for all docking simulations.
Acknowledgments
The Netherlands Organization for Scientific Research is acknowledged for financial support (grant nos. 700.50.512 [A.M.J.J.B.], 98S1010 [J.A.R.W.], and 700.50.514 and 700.52.425 [M.U.]). D.F. received a fellowship from the Marie Curie Training Site MAGRES-BIOMAC (grant no. HPMT-CT-2000-00120).
Abbreviations
AIR, ambiguity-driven intermolecular restraints
cyt b5, cytochrome b5
cyt c, cytochrome c
EM, energy minimization
ET, electron transfer
HSQC, heteronuclear single quantum coherence
NMR, nuclear magnetic resonance
Pr, heme propionate
RMSD, root-mean-square deviation
SA, simulated annealing
1D, one-dimensional
2D, two-dimensional
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041150205.
Supplemental material: see www.proteinscience.org
References
- Arnesano, F., Banci, L., Bertini, I., and Bonvin, A.M.J.J. 2004. A docking approach to the study of copper trafficking proteins: Interaction between metallochaperones and soluble domains of copper ATPases. Structure 12 669–676. [DOI] [PubMed] [Google Scholar]
- Banci, L., Bertini, I., Felli, I.C., Krippahl, L., Kubicek, K., Moura, J.J.G., and Rosato, A. 2003. A further investigation of the cytochrome b5–cytochrome c complex. J. Biol. Inorg. Chem. 8 777–786. [DOI] [PubMed] [Google Scholar]
- Berghuis, A.M. and Brayer, G.D. 1992. Oxidation state-dependent conformational changes in cytochrome c. J. Mol. Biol. 233 959–976. [DOI] [PubMed] [Google Scholar]
- Burch, A.M., Rigby, S.E.J., Funk, W.D., Macgillivray, R.T.A., Mauk, M.R., Mauk, A.G., and Moore, G.R. 1990. NMR characterization of surface interactions in the cytochrome b5–cytochrome c complex. Science 247 831–833. [DOI] [PubMed] [Google Scholar]
- Crowley, P. B., and Ubbink, M. 2003. Close encounters of the transient kind: Protein interactions in the photosynthetic redox chain investigated by NMR spectroscopy. Acc. Chem. Res. 36 723–730. [DOI] [PubMed] [Google Scholar]
- Crowley, P B., Rabe, K.S., Worrall, J.A.R., Canters, G.W., and Ubbink, M. 2002. The ternary complex of cytochrome f and cytochrome c: Identification of a second binding site and competition for plastocyanin binding. Chem-biochem 3 526–533. [DOI] [PubMed] [Google Scholar]
- Dominguez, C., Boelens, R., and Bonvin, A.M.J.J. 2003. HADDOCK: A protein-protein docking approach based on biochemical and/or biophysical information. J. Am. Chem. Soc. 125 1731–1737. [DOI] [PubMed] [Google Scholar]
- Durley, R.C. and Mathews, F.S. 1996. Refinement and structural analysis of bovine cytochrome b5 at 1.5 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 52 65–76. [DOI] [PubMed] [Google Scholar]
- Eley, C.G.S. and Moore, G.R. 1983. 1H-n.m.r. investigation of the interaction between cytochrome c and cytochrome b5. Biochem. J. 215 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk, W.D., Lo, T.P., Mauk, M.R., Brayer, G.D., Macgillivray, R.T.A., and Mauk, A.G. 1990. Mutagenic, electrochemical, and crystallographic investigation of the cytochrome b5 oxidation-reduction equilibrium: Involvement of asparagine-57, serine-64, and heme propionate-7. Biochemistry 29 5500–5508. [DOI] [PubMed] [Google Scholar]
- Gao, Y., Boyd, J., Williams, R.J.P., and Pielak, G.J. 1990. Assignment of proton resonances, identification of secondary structural elements, and analysis of backbone chemical shifts for the C102T variant of yeast iso-1-cytochrome c and horse cytochrome c. Biochemistry 29 6994–7003. [DOI] [PubMed] [Google Scholar]
- Guiles, R.D., Sarma, S., DiGate, R.J., Banville, D., Basus, V.J., Kuntz, I.D., and Waskell, L. 1996. Pseudocontact shifts used in the restraint of the solution structures of electron transfer complexes. Nat. Struct. Biol. 3 333–339. [DOI] [PubMed] [Google Scholar]
- Helgstrand, M., Kraulis, P.J., Allard, P., and Hard, T. 2000. Ansig for Windows: An interactive computer program for semiautomatic assignment of protein NMR spectra. J. Biol. NMR 18 329–336. [DOI] [PubMed] [Google Scholar]
- Hom, K., Ma, Q.F., Wolfe, G., Zhang, H., Storch, E.M., Daggett, V., Basus, V.J., and Waskell, L. 2000. NMR studies of the association of cytochrome b5 with cytochrome c. Biochemistry 39 14025–14039. [DOI] [PubMed] [Google Scholar]
- Kannt, A., Young, S., and Bendall, D.S. 1996. The role of acidic residues of plastocyanin in its interaction with cytochrome f. Biochim. Biophys. Acta 1277 115–126. [PubMed] [Google Scholar]
- Keller, R.M. and Wüthrich, K. 1980. Structural study of the heme crevice in cytochrome b5 based on individual assignments of the H1 NMR lines of the heme group and selected amino acid residues. Biochim. Biophys. Acta 621 204–217. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1989. Ansig—A program for the assignment of protein H1 and 2D NMR spectra by interactive computer graphics. J. Magn. Reson. 84 627–633. [Google Scholar]
- ———. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- La Mar, G.N., Budd, D.L., Viscio, D.B., Smith, K.M., and Langry, K.C. 1978. Proton nuclear magnetic resonance characterization of heme disorder in hemoproteins. Proc. Natl. Acad. Sci. 75 5755–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Mar, G.N., Burns, P.D., Jackson, J.T., Smith, K.M., Langry, K.C., and Strittmatter, P. 1981. Proton magnetic resonance determination of the relative heme orientations in disordered native and reconstituted ferricyto-chrome b5. Assignment of heme resonances by deuterium labeling. J. Biol. Chem. 256 6075–6079. [PubMed] [Google Scholar]
- Liang, Z.X., Jiang, M., Ning, Q., and Hoffman, B.M. 2002. Dynamic docking and electron transfer between myoglobin and cytochrome b5. J. Biol. Inorg. Chem. 7 580–588. [DOI] [PubMed] [Google Scholar]
- Louie, G.V. and Brayer, G.D. 1990. High-resolution refinement of yeast iso-1-cytochrome c and comparisons with other eukaryotic cytochromes c. J. Mol. Biol. 214 527–555. [DOI] [PubMed] [Google Scholar]
- Margoliash, E., Tuppy, H., Smith, E.L., and Kreil, G. 1961. Complete aminoacid sequence. Nature 192 1125–1127. [DOI] [PubMed] [Google Scholar]
- Mathews, F.S., Argos, P., and Levine, M. 1972. Three-dimensional Fourier synthesis of calf liver cytochrome b5 at 2.8 Å resolution. Cold Spring Harbor Symp. Quant. Biol. 36 387–395.4508153 [Google Scholar]
- Mauk, M.R., Reid, L.S., and Mauk, A.G. 1982. Spectrophotometric analysis of the interaction between cytochrome b5 and cytochrome c. Biochemistry 21 1843–1846. [DOI] [PubMed] [Google Scholar]
- Mauk, M.R., Mauk, A.G., Weber, P.C., and Matthew, J.B. 1986. Electrostatic analysis of the interaction of cytochrome c with native and dimethyl ester heme substituted cytochrome b5. Biochemistry 25 7085–7091. [DOI] [PubMed] [Google Scholar]
- Mauk, A.G., Mauk, M.R., Moore, G.R., and Northrup, S.H. 1995. Experimental and theoretical analysis of the interaction between cytochrome c and cytochrome b5. J. Bioenerget. Biomembr. 27 311–330. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Miura, R., Sugiyama, T., Akasako, K., and Yamano, T. 1980. An NMR study on the interaction between cytochrome b5 and cytochrome c. Biochem. Int. 1532–538. [Google Scholar]
- Morar, A.S., Kakouras, D., Young, G.B., Boyd, J., and Pielak, G.J. 1999. Expression of 15N-labeled eukaryotic cytochrome c in Escherichia coli. J. Biol. Inorg. Chem. 4 220–222. [DOI] [PubMed] [Google Scholar]
- Ng, S., Smith, M.B., Smith, H.T., and Millett, F. 1977. Effect of modification of individual cytochrome c lysines of the reaction with cytochrome b5. Biochemistry 16 4975–4978. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Northrup, S.H., Thomasson, K.A., Miller, C.M., Barker, P.D., Eltis, L.D., Guillemette, J.G., Inglis, S.C., and Mauk, A.G. 1993. Effects of charged amino acid mutations on the bimolecular kinetics of reduction of yeast iso-1-ferricytochrome c by bovine ferrocytochrome b5. Biochemistry 32 6613–6623. [DOI] [PubMed] [Google Scholar]
- Pollock, W.B.R., Rosell, F.I., Twitchett, M.B., Dumont, M.E., and Mauk, A.G. 1998. Bacterial expression of a mitochondrial cytochrome c. Trimethylation of Lys 72 in yeast iso-1-cytochrome c and the alkaline conformational transition. Biochemistry 37 6124–6131. [DOI] [PubMed] [Google Scholar]
- Qian, C.M., Yao, Y., Ye, K.Q., Wang, J.F., Tang, W.X., Wang, Y.H., Wang, W.H., Lu, J.X., Xie, Y., and Huang, Z.X. 2001. Effects of charged aminoacid mutation on the solution structure of cytochrome b5 and binding between cytochrome b5 and cytochrome c. Protein Sci. 10 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, L.S., Mauk, M.R., and Mauk, A.G. 1984. Role of heme propionate groups in cytochrome b5 electron transfer. J. Am. Chem. Soc. 106 2182–2185. [Google Scholar]
- Ren, Y., Wang, W.H., Wang, Y.H., Case, M., Qian, W., Mclendon, G.L., and Huang, Z.X. 2004. Mapping the electron transfer interface between cytochrome b5 and cytochrome c. Biochemistry 43 3527–3536. [DOI] [PubMed] [Google Scholar]
- Rodgers, K.K. and Sligar, S.G. 1991. Mapping electrostatic interactions in macromolecular associations. J. Mol. Biol. 221 1453–1460. [DOI] [PubMed] [Google Scholar]
- Rodgers, K.K., Pochapsky, T.C., and Sligar, S.G. 1988. Probing the mechanism of macromolecular recognition: The cytochrome b5 - cytochrome c complex. Science 240 1657–1659. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Marañón, M.J., Qiu, F., Stark, R.E., White, S.P., Zhang, X., Foundling, S.I., Rodriguez, V., Schilling, C.L., Bunce, R.A., and Rivera, M. 1996. 13C NMR spectroscopic and X-ray crystallographic study of the role played by mitochondrial cytochrome b5 heme propionates in the electrostatic binding to cytochrome c. Biochemistry 35 16378–16390. [DOI] [PubMed] [Google Scholar]
- Salemme, F.R. 1976. An hypothetical structure for an intermolecular electron transfer complex of cytochromes c and b5. J. Mol. Biol. 102 563–568. [DOI] [PubMed] [Google Scholar]
- Shao, W., Sang-Choul, I., Zuiderweg, E.R.P., and Waskell, L. 2003. Mapping the binding interface of the cytochrome b5–cytochrome c complex by nuclear magnetic resonance. Biochemistry 42 14774–14784. [DOI] [PubMed] [Google Scholar]
- Smith, M.B., Stoneheurner, J., Ahmed, A.Q.J., Staudenmeyer, N., and Millett, F. 1980. Use of specific trifluoroacetylation of lysine residues in cytochrome c to study the reaction with cytochrome b5, cytochrome c1, and cytochrome oxidase. Biochim. Biophys. Acta 592 303–313. [DOI] [PubMed] [Google Scholar]
- Stoneheurner, J., Williams, J.B., and Millett, F. 1979. Interaction between cytochrome c and cytochrome b5. Biochemistry 18 5422–5427. [DOI] [PubMed] [Google Scholar]
- Takano, T., Kallai, O.B., Swanson, R., and Dickerson, R.E. 1973. The structure of ferrocytochrome c at 2.45 Å resolution. J. Biol. Chem. 248 5234–5246. [PubMed] [Google Scholar]
- Tsugita, A., Kabayashi, M., Tani, S., Kyo, S., Rashid, M.A., Yoshida, Y., Kajihara, T., and Hagihara, B. 1970. Comparative study of primary structures of cytochrome b5 from 4 species. Proc. Natl. Acad. Sci. 67 442–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubbink, M. and Bendall, D.S. 1997. Complex of plastocyanin and cytochrome c characterized by NMR chemical shift analysis. Biochemistry 36 6326–6335. [DOI] [PubMed] [Google Scholar]
- van Drogen-Petit, A., Zwahlen, C., Matthias, P., and Bonvin, A.M.J.J. 2004. Insight into molecular interactions between two PB1 domains. J. Mol. Biol. 336 1195–1210. [DOI] [PubMed] [Google Scholar]
- Worrall, J.A.R., Kolczak, U., Canters, G.W., and Ubbink, M. 2001. Interaction of yeast iso-1-cytochrome c with cytochrome c peroxidase investigated by [15N, 1H] heteronuclear NMR spectroscopy. Biochemistry 40 7069–7076. [DOI] [PubMed] [Google Scholar]
- Worrall, J.A.R., Liu, Y.J., Crowley, P.B., Nocek, J.M., Hoffman, B.M., and Ubbink, M. 2002. Myoglobin and cytochrome b5: A nuclear magnetic resonance study of a highly dynamic protein complex. Biochemistry 41 11721–11730. [DOI] [PubMed] [Google Scholar]
- Worrall, J.A.R., Reinle, W., Bernhardt, R., and Ubbink, M. 2003. Transient protein interactions studied by NMR spectroscopy: The case of cytochrome c and adrenodoxin. Biochemistry 42 7068–7076. [DOI] [PubMed] [Google Scholar]
- Willie, A., Stayton, P.S., Sligar, S.G., Durham, B., and Millett, F. 1992. Genetic engineering of redox donor sites—measurement of intracomplex electron transfer between ruthenium-65 Cytochrome b5 and cytochrome c. Biochemistry 31 7237–7242. [DOI] [PubMed] [Google Scholar]
- Whitford, D., Concar, D.W., Veitch, N.C., and Williams, R.J.P. 1990. The formation of protein complexes between ferricytochtome b5 and ferricytochrome c studied using high-resolution 1H-NMR spectroscopy. Eur. J. Biochem. 192 715–721. [DOI] [PubMed] [Google Scholar]