

Abstract
NMR spin relaxation experiments are used to characterize the dynamics of the backbone of ubiquitin. Chemical exchange processes affecting residues Ile 23, Asn 25, Thr 55, and Val 70 are characterized using on- and off-resonance rotating-frame 15N R1ρ relaxation experiments to have a kinetic exchange rate constant of 25,000 sec−1 at 280 K. The exchange process affecting residues 23, 25, and 55 appears to result from disruption of N-cap hydrogen bonds of the α-helix and possibly from repacking of the side chain of Ile 23. Chemical exchange processes affecting other residues on the surface of ubiquitin are identified using 1H-15N multiple quantum relaxation experiments. These residues are located near or at the regions known to interact with various enzymes of the ubiquitin-dependent protein degradation pathway.
Keywords: protein dynamics, chemical exchange, 15N spin relaxation, multiple-quantum relaxation
Ubiquitin, one of the most highly conserved proteins in eukaryotes, is the signature component of the ubiquitin-dependent protein degradation pathway (Hochstrasser 1996; Hershko and Ciechanover 1998). Briefly, ubiquitin is activated by adenylation of its C terminus and linked covalently to an ubiquitin-activating enzyme, E1. Next, ubiquitin is transferred from E1 to an ubiquitin-conjugating enzyme, E2. Finally, ubiquitin is attached covalently to the ɛ-amino group of a lysine residue of the target protein. This last step often involves an ubiquitin–protein ligase, E3. Polyubiquitinated proteins subsequently are targeted to the 26S proteasome for degradation. Deubiquitinating enzymes serve a regulatory role by removing ubiquitin from target proteins.
Investigations by site-directed mutagenesis and NMR spectroscopy have identified amino acid residues in ubiquitin that have crucial roles in interactions with E1, E2, and deubiquitinating enzymes. These residues include the four arginines at positions 42, 54, 72, and 74. In particular, Arg 54 and Arg 72 are involved in the initial binding of free ubiquitin to E1 (Burch and Haas 1994). In addition, Leu 8, Ile 44 and Val 70 form a hydrophobic patch on the surface of ubiquitin that is essential for binding to E1 (Beal et al. 1996; Haas and Siepmann 1997). Many of the residues important for interactions with E1 are also involved in binding to E2 enzymes (Miura et al. 1999; Hamilton et al. 2000, 2001) and to deubiquitinating enzymes (Wilkinson et al. 1999). Residues 7–9, 40–49, and 70–76 are most strongly implicated in the interaction with E2 enzymes and deubiquitinating enzymes.
Conformational flexibility within the interaction surfaces of proteins may facilitate protein–protein interactions through induced fit mechanisms and may allow recognition of more than one binding partner (James et al. 2003). Protein conformational dynamics on microsecond to millisecond timescales are manifest as chemical exchange line-broadening in NMR spectroscopy, and a number of techniques have been developed to characterize enhanced transverse relaxation of nuclear spin resonances in proteins (Palmer 2004).
Previous NMR investigations have identified chemical exchange processes that affect a variety of sites within ubiquitin. At room temperature, 15N chemical exchange broadening is most evident for residues Ile 23 and Asn 25, while the resonance for Glu 24 is extremely weak in HSQC spectra (Schneider et al. 1992; Tjandra et al. 1995; Fushman and Cowburn 1998; de Alba et al. 1999; Carlomagno et al. 2000; Meiler et al. 2001; Tolman et al. 2001). Recent 15N R1ρ relaxation measurements in supercooled water at T = 260 K have established the presence of a chemical exchange process affecting Val 70 with an exchange rate constant (vide infra) of kex = 7500 ± 1600 sec−1 (Mills and Szyperski 2002). Relaxation of zero-quantum (ZQ) and double-quantum (DQ) coherences was used by Bodenhausen and coworkers to identify exchange processes affecting residues Ile 23 and Asn 25 (Dittmer and Bodenhausen 2004; Wist et al. 2004). CPMG measurements reported exchange rate constants of kex = 4200 sec−1 for Ile 23 at 296 K using 1H-15N ZQ/DQ relaxation (Dittmer and Bodenhausen 2004) and kex = 1064 sec−1 for Asn 25 at 300 K using 15N-13C′ ZQ/DQ relaxation (Wist et al. 2004). Novel TROSY-based ZQ and DQ experiments were used by Majumdar and Ghose (2004) to identify Glu 24, Asn 25, Glu 51, and Asp 52 as a continuous surface-exposed patch likely to be subject to correlated motions.
This article reports the results of on- and off-resonance 15N R1ρ (Massi et al. 2004) and 1H-15N ZQ/DQ (Kloibert and Konrat 2000) relaxation experiments. Newly developed decoupling schemes were applied to allow the use of weak radiofrequency fields in the R1ρ experiments (Massi et al. 2004). R1ρ relaxation dispersion at 280 K is used to quantify chemical exchange processes affecting residues Ile 23, and Asn 25, located in the N terminus of the α-helix; Thr 55, in the loop between β-strand 4 and the short second 3–10 helix; and Val 70, in β-strand 5. Exchange processes affecting additional residues located on the surface of ubiquitin are identified using ZQ/DQ relaxation.
Results and Discussion
R1ρ relaxation dispersion curves
Figure 1 ▶ shows the conformational exchange contribution 0) to the transverse relaxation rate constant for 15N of ubiquitin at temperatures of 280 K and 298 K, estimated using a Hahn spin-echo experiment (Wang et al. 2001b; Wang and Palmer 2003) (see Materials and Methods). At T = 298 K, only residue Asn 25, located in the α-helix, has a value of Rex (ωe → 0) that is clearly significantly greater than zero. When ηxy values, measured at 298 K, are plotted against R2 values for ubiquitin, both Ile 23 and Asn 25 are identified as subject to chemical exchange linebroadening (data not shown). However, as shown in Figure 2 ▶, Rex(ωe) dispersion curves for these residues are independent of ωe, indicating that the exchange kinetics, kex are too fast, relative to ΔωN, to be refocused even at the highest values of ωe used in the off-resonance R1ρ experiment. Rex(ωe) was calculated for each residue from measured values of R1ρ, R1, and ηxy relaxation rate constants (see Materials and Methods). When the temperature is decreased to 280 K, the exchange contribution for Asn 25 is increased, and significant exchange contributions are observed for three additional residues: Ile 23 at the N terminus of the α-helix, Thr 55 in the turn region between β-strand 4 and second short 3–10 helix, and Val 70 in β-strand 5 of the C-terminal region. The positions of the residues that are observed to undergo chemical exchange at 280 K are mapped onto the three-dimensional structure of ubiquitin in Figure 3A ▶.
Figure 1.

Conformational exchange contribution to transverse relaxation, Rex, for ubiquitin at T = 298 K (open circles) and T = 280 K (filled circles) at B0 = 11.7 T.
Figure 2.

Conformational exchange contribution to transverse relaxation, Rex as a function of the effective field, ωe, at a static magnetic field of 11.7 T and T = 298 K for Ile 23 (circles) and Asn 25 (squares) in ubiquitin.
Figure 3.

Cartoon representation of ubiquitin that highlights the positions of the exchanging residues Ile 23, Asn 25, Thr 55, Val 70 (A). Regions characterized by different secondary structure are represented with different colors: helices ,red; β-strands, yellow; coil and turns, blue. The boxed region is shown as an expansion in B. Each residue is labeled at the backbone N atom. The dashed lines indicate hydrogen bonds.
In order to further characterize the mechanism of exchange broadening for residues Ile 23, Asn 25, Thr 55, and Val 70, the effective field dependence of Rex was measured using on- and off-resonance R1ρ experiments at static magnetic field strengths of 11.7 and 14.1 T. In contrast to the dispersion data recorded at 298 K, relaxation dispersion is observed for all four residues at 280 K, as shown in Figure 4 ▶. The dispersion data is well described by a two-site exchange model that is fast on the chemical shift timescale (equation 2); the results of the dispersion analysis are presented in Table 1. The dispersion curves of Ile 23, Asn 25, Thr 55, and Val 70 are each independently fitted with a value of kex ≈ 25,000 sec−1, suggesting that all four residues are detecting the same conformational transition. The term Φex listed in Table 1 is the product of the site populations and chemical shift changes (equation 3), which in the limit of fast exchange cannot be deconvoluted to yield independent estimates of pApB and ΔωN. However, assuming that all four residues are responding to the same conformational change(s) with similar populations, then the rank order of Φex corresponds to the relative magnitude of |ΔωN |: Asn 25 and Thr 55 experience the largest and smallest absolute chemical shift changes, respectively, in response to the chemical exchange process.
Figure 4.

Conformational exchange contribution to transverse relaxation, Rex, as a function of the effective field, ωe at T = 280 K. Circles and squares represent data collected at a static magnetic field of 11.7 T and 14.1 T, respectively. Lines are the results of simultaneously fitting the data at 11.7 T (circles) and 14.1 T (dashed), using equation 2.
Table 1.
Chemical exchange parameters for Ile 23, Asn 25, Thr 55, and Val 70 of ubiquitin
| Residue | Φex (104 sec−2) | kex (sec−1) | R20 (sec−1) (11.7 T) | R20 (sec−1) (14.1 T) |
| Ile 23 | 8.1 ± 1.0 | 23200 ± 2700 | 10.2 ± 0.1 | 11.1 ± 0.1 |
| Asn 25 | 15.9 ± 2.2 | 23200 ± 2200 | 10.5 ± 0.7 | 11.5 ± 0.7 |
| Thr 55 | 4.1 ± 1.2 | 25000 ± 6000 | 9.0 ± 0.2 | 9.8 ± 0.2 |
| Val 70 | 5.9 ± 0.9 | 26000 ± 4000 | 10.3 ± 0.1 | 11.2 ± 0.1 |
Exchange rate constants for slow conformational processes involving residues Ile 23 and Asn 25 have been characterized by new ZQ and DQ relaxation experiments (Dittmer and Bodenhausen 2004; Wist et al. 2004). Kinetic rate constants, kex, were evaluated to be 4200 sec−1 for Ile 23 at 296 K (Dittmer and Bodenhausen 2004) and 1064 sec−1 for Asn 25 at 300 K (Wist et al. 2004). The rate constant obtained by fitting the 15N single quantum dispersion curves at 280 K is kex ≈ 25,000 sec−1, which is an order of magnitude higher. Even at the lowest field used in the on-resonance 15N R1ρ experiments, a chemical exchange process with a rate constant in the range 1000–4000 sec−1 involving either Ile 23 or Asn 25 is not evident. However, 15N R1ρ experiments are sensitive to motional processes that cause a change in the 15N chemical shift, ΔωN, while multiple quantum coherence experiments are sensitive to processes that affect the chemical shifts of both nuclei involved through the products, ΔωNΔωC′ (Wist et al. 2004), and ΔωN ΔωH (Dittmer and Bodenhausen 2004). Therefore, the observed differences in apparent exchange rate constants can be reconciled by considering the existence of a second slower process that cannot be observed by the 15N R1ρ experiments, but that is instead detected by measurements of the differential relaxation of ZQ and DQ coherences.
Previous work in supercooled water at 260 K has already identified Val 70 as an exchanging residue with a rate constant kex equal to 7500 ± 1600 sec−1 (Mills and Szyperski 2002). In the present study this motional process is determined to have a value of kex = 26,000 ± 4000 sec−1 at 280 K. The data at two temperatures allow the activation barrier for the exchange process to be estimated as ≈ 36 kJ/mol.
Multiple quantum experiments
Amide 1H-15N differential relaxation rates of ZQ and DQ coherences, ΔRMQ, of ubiquitin measured at 280 K are depicted in Figure 5 ▶. ΔRMQ is proportional to both ΔωN and ΔωH. Therefore, this experiment can detect exchange processes that are not detected by the R1ρ experiment, provided that ΔωH is sufficiently large. The residues characterized by the highest values of ΔRMQ—Thr 9, Ile 23, Ile 30, Lys 33, Leu 43, Phe 45, Thr 55, and Val 70—are located in the loop regions, on the α-helix at the N and C termini, and on β-strands 3 and 5. Exchange broadening for residues Ile 23, Leu 43, Phe 45, and Thr 55 have been identified previously in ZQ/DQ CPMG experiments (Dittmer and Bodenhausen 2004). Asn 25, which shows the highest ΔωN from the R1ρ experiment, has ΔRMQ ≈ 0, indicating that the 1H chemical shift for Asn 25 does not change as a result of exchange. Using R1ρ and ΔRMQ measurements, the ratio of ΔωH /ΔωN can be evaluated for a given residue (as described in Materials and Methods). Relative values of ΔωH/ΔωN are 1.81 ± 0.03, 2.0 ± 0.2, and 0.78 ± 0.05 for residues Ile 23, Thr 55, and Val 70, respectively. Values of ΔωH/ΔωN in the range of 1–3 suggest that the exchange process results from dihedral angle and/or hydrogen bonding fluctuations, rather than variations in ring current shifts (Ishima et al. 1998; Wishart and Case 2001).
Figure 5.

Amide 1H-15N differential relaxation rate constants for ZQ and DQ coherences, ΔRMQ, for ubiquitin measured at 11.7 T and T = 280 K.
Correlations between structural changes and ΔωN
The conformational changes that result in chemical exchange broadening are manifest in the structurally dependent chemical shift changes, ΔωN. A common means of estimating ΔωN is to consider the structure dependent secondary chemical shift as an upper limit on the chemical shift change expected for the loss of all native interactions in the minor state conformation. For the case of chemical exchange characterized here, Val 70 has a secondary chemical shift (relative to random coil) of 6 ppm; using this value as an upper limit for ΔωN yields a site population for the minor species of pB = 0.02. On the other hand, such an approach is certainly not valid for Ile 23, where the secondary chemical shift of 0.9 ppm clearly cannot reproduce the observed exchange broadening, even in the limit of pA = pB. Therefore, a more comprehensive analysis of the structure dependence of 15N chemical shift is required to infer potential mechanism of chemical exchange.
The values of ΔωH/ΔωN obtained from residues Ile 23, Thr 55, and Val 70 suggest that changes in hydrogen bonding and dihedral angles are the most likely sources of conformational dynamics. Residues Ile 23 and Asn 25 are located at the N terminus of the α-helix in ubiquitin and form capping interactions with Thr 22 and the loop comprising residues Glu 51–Leu 56 (Fig. 3B ▶). Disruption of the helix capping interactions provides one possible mechanism for chemical exchange broadening. Hydrogen bonding provides a significant contribution to the structure dependent 15N chemical shifts in proteins (Xu and Case 2002). Xu and Case and Oldfield and coworkers (deDios et al. 1993; Xu and Case 2002) have shown that the hydrogen bond status of the amide group itself (direct effect) and the carbonyl group of the preceding residue (indirect effect) both contribute to the 15N chemical shielding. Model calculations suggest that removal of the Ile 23 (NH)–Arg 54 (CO) hydrogen bond results in a -1.5 ppm change in the 15N chemical shift of Ile 23 due to the direct hydrogen bonding effect and a −4.2 ppm change in the 15N chemical shift of Thr 55 due to the indirect hydrogen bonding effect. Assuming that the disruption of the Ile 23–Arg 54 hydrogen bond accounts for the relaxation dispersion observed for Thr 55, a chemical shift change of that magnitude would correspond to a minor state population pB ≈ 0.02.
In addition to the hydrogen bond contribution, 15N chemical shifts exhibit a strong dependence on local backbone and side-chain conformation. Although there is no clear correlation between 15N chemical shifts and φ/ψ angles, such as for 13Cα chemical shifts (Wishart and Case 2001; Xu and Case 2002), trends have been observed for variation in 15N chemical shift with side chain χ1 dihedral angles for particular amino acid types. For the case of Ile, an average upfield shift of ≈ 4.5 ppm is expected for a change in χ1 from −60° to +60° (Fig. 6A ▶). Examination of the local structure of Ile 23 in ubiquitin reveals that such a χ1 isomerization for the completely buried side chain could not proceed without additional conformational changes of the backbone in the vicinity and would likely disrupt the Ile 23–Arg 54 hydrogen bond. Assuming a −4.5 ppm change in chemical shift associated with the χ1 transition and a −1.5 ppm change calculated for disruption of the hydrogen bond as a potential mechanism of exchange broadening for Ile 23 would correspond, again, to a minor state population pB ≈ 0.02. The estimate pB ≈ 0.02 indicates that the forward rate constant for transitions from major to minor states, k1 is ≈ 500 sec−1.
Figure 6.

Dependence of the 15N chemical shift on side chain χ1 dihedral angle. 15N secondary chemical shifts are plotted as a function of side chain χ1 torsional angle for all isoleucine (A) and asparagine (B) residues in the RefDB chemical shift database (Zhang et al. 2003).
Support for the proposed mechanism comes from two sources. First, the ratio of the total relative values of ΔωN2 estimated for residues Ile 23 and Thr 55 (6.0/4.2)2 = 2.0 is a close match to the observed ratio of Ψex for the same residues from Table 1 of 2.0 ± 0.4. Second, the amide proton solvent exchange protection factors for residues Ile 23–Asn 25 are reduced relative to residues more central in the α-helix (Zhang 1995; Cordier and Grzesiek 2002), suggesting a degree of structural plasticity in the capping interactions.
Glu 24, which is broadened extensively and essentially unobservable, and Asn 25 also form N-cap interactions. Based on the minor site population pB = 0.02 derived above and the value of Ψex reported in Table 1, a value of ΔωN ~9 ppm is estimated for Asn 25, and an even larger value would be necessary to account for the extreme broadening of Glu 24. The dependence of the 15N chemical shifts on χ1 dihedral angles is clearly residue-type specific. For example, χ1 isomerization for asparagine residues does not perturb the 15N chemical shift average, although considerable variation about zero is possible (Fig. 6B ▶). Disruption of the N-cap hydrogen bonds would contribute only ~2 ppm to the observed chemical shift changes. The secondary chemical shift for Asn 25 is only 3 ppm. Thus, the large values of ΔωN for Glu 24 and Asn 25 must result from multiple sources, including changes in dihedral angles and redistribution of electrostatic and hydrogen bonding interactions. Model calculations, database analysis, and comparison to random coil shifts cannot uniquely identify the multiple mechanisms of exchange broadening for these residues.
Interestingly, a site population for the minor species of pB = 0.02 for Val 70, estimated from the secondary chemical shift, is in good agreement with the site populations estimated for residues Ile 23 and Thr 55.
Biological implications
An increasing number of investigations have identified sites in ubiquitin that are known to be involved in protein–protein interactions and are subject to chemical exchange linebroadening in NMR experiments (Mills and Szyperski 2002; Majumdar and Ghose 2004). In the present work, R1ρ experiments quantify exchange processes with a rate constant of 25,000 sec−1 at 280 K affecting residues Ile 23, Asn 25, Thr 55, and Val 70. Arg 54 has been found to be important in the interaction of free ubiquitin with E1 (Burch and Haas 1994). Both Ile 23 and Thr 55 are connected to Arg 54, either covalently or through hydrogen bonding, suggesting that a single correlated kinetic process affects all three residues. As noted above, the exchange process may involve the disruption of the hydrogen bond between Ile 23 and Arg 54. Val 70 is located in β-strand 5 at the C terminus of ubiquitin, and together with Leu 8 and Ile 44 is part of a hydrophobic patch essential for the recognition of E1. Val 70 also is known to have a role in the interaction with E2. The exchange rate constant for conformational dynamics of Val 70 has been measured previously by Mills and Szyperski at 260 K (Mills and Szyperski 2002); the present results allow an apparent activation energy for the conformational process to be estimated. Residues with large ΔRMQ relaxation rate constants cover an extended part of the exposed surface of the protein and are clustered together in two main regions. As Figure 7 ▶ shows, one of these regions, consisting of residues Thr 9, Ile 23, Leu 43, Phe 45, Thr 55, and Val 70, is contiguous and partly overlapped with the region of ubiq-uitin that is involved in the binding to the ubiquitin activating and conjugating enzymes E1 and E2 (Burch and Haas 1994; Beal et al. 1996; Haas and Siepmann 1997; Miura et al. 1999; Wilkinson et al. 1999; Hamilton et al. 2000, 2001). This is a distinct surface patch from Glu 24, Asn 25, Asp 52, and Glu 51 previously identified by Majumdar and Ghose (2004). The observation and quantification of chemical exchange processes affecting two groups of residues in ubiq-uitin located near or at the region of interactions with E1 and E2 give insight into the conformational dynamics of ubiquitin that may be necessary for recognition of diverse E1 and E2 partners.
Figure 7.

Amide 1H-15N ΔRMQ for ubiquitin measured at 11.7 T and T = 280 K. Data are represented with different colors on the surface of the protein. Residues with ΔRMQ < 3 are represented as gray, residues with 3 ≤ ΔRMQ < 5 are yellow, residues with 5 ≤ ΔRMQ < 8 are orange, residues with 8 ≤ ΔRMQ < 15 are red, and residues with ΔRMQ ≥ 15 are purple. The opaque residues are those known to be directly involved in the interaction with E1 and E2 enzymes. All other residues are depicted as transparent.
Conclusion
The chemical exchange process affecting residues Ile 23, Asn 25, Thr 55, and Val 70 of ubiquitin has been characterized using on- and off-resonance R1ρ experiments to have a kinetic exchange rate of 25,000 sec−1 at 280 K. Other residues located on the surface of the protein have been identified as undergoing conformational transitions using differential ZQ/DQ relaxation. These residues are located near or at the regions of ubiquitin that interact with E1 and E2. Conformational flexibility in binding interfaces may be important in allowing ubiquitin to recognize different binding partners.
Materials and methods
Theory
The relaxation rate constant, R1ρ for decay of the magnetization component parallel to the effective field in the rotating frame, ωe = (ω12 + Ω2)1/2, is a function of the amplitude of the applied rf field, ω1, and of the resonance offset from the spin-lock carrier, Ω The relaxation rate constant in the rotating frame is given by Deverell et al. (1970) and Abragam (1983),
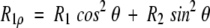 |
(1) |
in which θ = arctan(ω1/Ω) is the tilt angle between the static magnetic field and the effective field in the rotating frame. Thus, 15N R2 rate constants can be calculated using measured values of R1ρ and R1 relaxation rate constants. In the presence of a conformational exchange process, R2 = R20 + Rex, in which R20 is the relaxation rate constant due to relaxation mechanisms other than exchange. The conformational exchange contribution to transverse relaxation, Rex, depends on the effective field, ωe. The kinetic parameters that characterize the exchange process can be obtained from the variation of Rex as a function of ωe. For two-site exchange that is fast on the chemical shift timescale (Deverell et al. 1970),
 |
(2) |
in which kex is the exchange rate constant (given as the sum of the forward and reverse kinetic rate constants for two-site exchange),
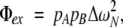 |
(3) |
pA and pB are the fractional populations of sites A and B, and ΔωN is the difference between the 15N chemical shifts of the two sites A and B. Relaxation dispersion is observed for values of ωe that are experimentally accessible over the range ωe < kex to ωe > kex. If kex >>ωe, then Rex is approximately independent of ωe.
The difference between the relaxation rate constants for 1H-15N ZQ and DQ coherences is given by (Kloibert and Konrat 2000),
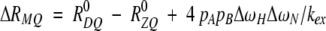 |
(4) |
in which R0ZQ and R0DQ are the relaxation rate constants for ZQ and DQ coherences from processes other than chemical exchange, respectively, and ΔωH is the difference between the 1H chemical shifts of the two sites A and B. Thus, if both R1ρ and RMQ measurements are available for a given residue:
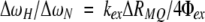 |
(5) |
and R0ZQ - R0DQ is assumed to be small.
NMR spectroscopy and data processing
NMR 15N relaxation measurements were performed on a 1.25 mM [U-15 N] sample of ubiquitin (90% H2O/10% D2O, 10 mM sodium phosphate buffer (pH 5.8), T = 280 K and 298 K). Relaxation data were collected at static magnetic field strengths of 11.7 T and 14.1 T, using Bruker DRX500 and DRX600 spectrometers. Temperatures were calibrated using a sample of 100% methanol. Relaxation rate constants were determined from series of two-dimensional spectra recorded with different relaxation delays. Intensities of cross-peaks were fitted to mono-exponential or hyperbolic tangent decay functions as appropriate to give spin relaxation rate constants; uncertainties were estimated by jackknife simulations (Mosteller and Tukey 1977). Spectra were processed using nmr-Pipe (Delaglio et al. 1995), Sparky (University of California, San Francisco), and Curvefit (http://www.palmer.hs.columbia.edu).
15N R1 and R2 values were measured at 298 K and 280 K as described previously using inversion recovery and CPMG experiments (Farrow et al. 1994). Maximum relaxation delay values of 800 msec and 200 msec were used for R1 and R2, respectively. The interval between 15N 180° pulses in the CPMG sequence of the R2 experiment was equal to 1 msec. Twelve spectra, including duplicates, are recorded for each measurement.
R20 values were determined at 280 K and 298 K from measurements of the rate constant for 15N-1H dipole–dipole/15N chemical shift anisotropy, CSA, cross-correlated transverse relaxation, ηxy, by an established method (Kroenke et al. 1998). Five pairs of spectra, each one corresponding to a different relaxation time delay, were recorded; the maximum value of the relaxation delay used was 106.8 msec. ηxy rates were calculated by fitting the ratio of the signal intensities 〈Ny〉〈2HzNy〉 (t) to the function tanh(ηxyt). The average value of the ratio κ = R20/ηxy was obtained from data for residues in ubiquitin that were unaffected by chemical exchange broadening, assuming constant 15N CSA (Tjandra et al. 1996; Fushman et al. 1998; Wang et al. 2001a). R20 values for the residues undergoing conformational exchange were calculated as R20 = κηxy using values of κ obtained at 11.7 T and 14.1 T.
Rex(ωe→0) was estimated using a Hahn spin-echo experiment (Wang et al. 2001b; Wang and Palmer 2003). Five spectra were acquired with t = 0 msec, and five spectra with t = 115.8 msec. WALTZ 16 1H decoupling was employed during the relaxation period t. The apparent relaxation rate constant, R2HE, was calculated by fitting the ratio of the signal intensities I(t)/I(0) to an exponential decay function. Rex(ωe→0) was calculated for each residue as Rex(ωe→0)=R2HE − R20.
15N1ρ rate constants were measured as previously described (Mulder et al. 1998; Massi et al. 2004). The maximum length of the spin-lock period in all experiments was equal to 160 msec. Twelve spectra, including duplicates, were recorded. On-resonance R1ρ experiments were performed at 280 K using different spin-lock field strengths (370 Hz, 1000 Hz, and 1300 Hz at 11.7 T, and 370 Hz and 1000 Hz at 14.1 T). Off-resonance R1ρ experiments, where the spin-lock rf frequency was set outside the spectral region of interest were performed at 280 K using nine and seven different effective fields, respectively, at 11.7 T and 14.1 T. The different effective fields were obtained using several values of the spin-lock field strength (1270 Hz, 1765 Hz, 1790 Hz, 1830 Hz, and 1870 Hz at 11.7 T; 1000 Hz and 1700 Hz at 14.7 T) and different resonance offsets. The nominal tilt angles used, calculated from the middle of the spectrum, were 69.8°, 52.0°, 51.8°, 47.3°, 46.3°, 40.1°, 35.1°, 30.5°, 26.9°, and 69.5°, 50.5°, 45.5°, 40.3°, 34.3°, 29.5°, and 25.9°, respectively, at 11.7 T and 14.1 T. The tanh/tan adiabatic pulse, used in the off-resonance R1ρ experiments, had a duration of 10 msec and the frequency sweep began at −15,000 Hz from the carrier frequency. On- and off-resonance R1ρ experiments were performed at 300 K using a static magnetic field strength of 11.7 T. On-resonance R1ρ experiments were performed using spin-lock field strengths equal to 250 Hz, 500 Hz, and 1000 Hz. The off-resonance R1ρ experiment used a field strength of 1257 Hz and an offset of 2600 Hz.
The spin-lock field strengths used in the R1ρ experiments were calibrated by off-resonance continuous wave decoupling as previously described (Palmer et al. 2001). Reported uncertainties are obtained from curve fitting. Variation in ω1 due to B1 inhomogeneity is ~10%, as measured by a transient nutation experiment (Guenneugues et al. 1999).
Amide 15N-1H differential relaxation of the DQ and ZQ coherences, ΔRMQ, was measured at 11.7 T as previously described (Kloibert and Konrat 2000; Wang and Palmer 2002). The spin echo delays, T, for the multiple quantum coherence relaxation period were 10, 20, 30, 40, and 50 msec, and five measurements were recorded for each delay. ΔRMQ, rates were calculated by fitting the ratio of the signal intensities 〈2NyHy〉(t)/〈2NxHx〉 (t) the function tanh(ΔRMQ T/2) (Kloibert and Konrat 2000).
Dispersion curve fitting
R2 rate constants were determined from measured values of R1ρ and R1 using equation 1. Use of R2 rather than R1ρ is preferable because R2 does not depend on the tilt angle, which varies with field strength and the resonance offset from the spin-lock carrier for any given resonance. The resulting R2 values, from all experiments at the two static magnetic fields, 11.7 T and 14.1 T, were used together with the R20 values to simultaneously optimize kex, Φex and R2opt using the following equations:
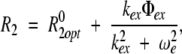 |
(6) |
in which & R02 = R02opt. As a first approximation
 |
(7) |
in which d = (μ0/4π)ħ γHγN〈 r−3NH 〉, C = γNB0Δσ/ √3μ0 is the permeability of vacuum; ħ is Planck’s constant divided by 2π; γH and γ N are the gyromagnetic ratios of 1H and 15N, respectively; f = B/B0 〈r−3NH〉 −1/3 = 1.02 Å, Δσ = −172 ppm is the difference between the principal components of the 15N CSA tensor, σ‖ and σ⊥ (Kroenke et al. 1999); J(ω) is the spectral density function evaluated at the frequency ω, Bo = 11.7 T; and B is either 11.7 T or 14.1 T.
Nonlinear least-squares optimization used the Levenberg-Marquardt algorithm implemented in Mathematica (Wolfram). Uncertainties in fitted parameters were estimated by jackknife simulations.
Structural dependence of 15N chemical shifts
The structural dependence of 15N chemical shifts was analyzed using the RefDB database of uniformly referenced chemical shifts (Zhang et al. 2003) as previously described (Grey et al. 2003). Briefly, for each protein entry in the RefDB with a three-dimensional structure determined at high resolution, a structure-dependent secondary chemical shift was calculated for each residue as δN − δrc, where δN is the referenced 15 N chemical shift and δrc is the sequence-corrected random coil chemical shift (Braun et al. 1994). Structural parameters φ, ψ, χ1, and χ2 were calculated for each residue based on the atomic coordinates of the corresponding PDB file.
Model chemical shifts calculations were performed using the program SHIFTS (Xu and Case 2001, 2002). To examine the contribution of the Ile 23–Arg 54 hydrogen bond to the 15N chemical shifts of Ile 23 and Thr 55, a fragment of ubiquitin corresponding to the residues Thr 22–Val 26 (A) and Asp 52–Leu 56 (B) was used. 15N chemical shifts were calculated for both fragments together (presence of hydrogen bond) and on each fragment separately to mimic the effect of removing the Ile 23–Arg 54 hydrogen bond.
Acknowledgments
This work was supported by NIH grants GM59273 (A.G.P.) and GM08281 (M.J.G.). We thank Clay Bracken (Weill Medical College of Cornell University) for providing the 15N sample of ubiquitin.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041139505.
References
- Abragam, A. 1983. Principles of nuclear magnetism. Oxford University Press, Oxford.
- Beal, R., Deveraux, Q., Xia, G., Rechsteiner, M., and Pickart, C. 1996. Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc. Natl. Acad. Sci. 93 861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, D., Wider, G., and Wüthrich, K. 1994. Sequence corrected 15N random coil chemical-shifts. J. Am. Chem. Soc. 116 8466–8469. [Google Scholar]
- Burch, T.J. and Haas, A.L. 1994. Site-directed mutagenesis of ubiquitin. Differential roles of arginine in the interaction with ubiquitin-activating enzyme. Biochemistry 33 7300–7308. [DOI] [PubMed] [Google Scholar]
- Carlomagno, T., Maurer, M., Hennig, M., and Griesinger, C. 2000. Ubiquitin backbone motion studied via NHN-C′ Cα dipolar-dipolar and C′-C′ C′/ NHN CSA-dipolar cross-correlated relaxation. J. Am. Chem. Soc. 122 5105–5113. [Google Scholar]
- Cordier, F. and Grzesiek, S. 2002. Temperature-dependence of protein hydrogen bond properties as studied by high-resolution NMR. J. Mol. Biol. 317 739–752. [DOI] [PubMed] [Google Scholar]
- de Alba, E., Barber, J.L., and Tjandra, N. 1999. The use of residual dipolar coupling in concert with backbone relaxation rates to identify conformational exchange by NMR. J. Am. Chem. Soc. 121 4282–4283. [Google Scholar]
- de Dios, A.C., Pearson, J.G., and Oldfield, E. 1993. Secondary and tertiary structural effects on protein NMR chemical shifts: An ab initio approach. Science 260 1491–1496. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe—A multidimentional spectral processing system based on unix pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Deverell, C., Morgan, R.E., and Strange, J.H. 1970. Studies of chemical exchange by nuclear magnetic relaxation in rotating frame. Mol. Phys. 18 553–559. [Google Scholar]
- Dittmer, J. and Bodenhausen, G. 2004. Evidence of slow motion in proteins by multiple refocusing of heteronuclear nitrogen/proton multiple quantum coherence in NMR. J. Am. Chem. Soc. 126 1314–1315. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Fushman, D. and Cowburn, D. 1998. Model-independent analysis of 15N chemical shift anisotropy from NMR relaxation data. Ubiquitin as a test example. J. Am. Chem. Soc. 120 7109–7110. [Google Scholar]
- Fushman, D., Tjandra, N., and Cowburn, D. 1998. Direct measurement of 15N chemical shift anisotropy in solution. J. Am. Chem. Soc. 120 10947–10952. [Google Scholar]
- Grey, M.J., Wang, C., and Palmer, A.G. 2003. Disulfide bond isomerization in basic pancreatic trypsin inhibitor: Multi-site chemical exchange quantified by CPMG relaxation dispersion and chemical shift modeling. J. Am. Chem. Soc. 125 14324–14335. [DOI] [PubMed] [Google Scholar]
- Guenneugues, M., Berthault, P., and Desvaux, H. 1999. A method for determining B1 field inhomogeneity. Are the biases assumed in heteronuclear relaxation experiments usually underestimated? J. Magn. Reson. 136 118–126. [DOI] [PubMed] [Google Scholar]
- Haas, A.L. and Siepmann, T.J. 1997. Pathways of ubiquitin conjugation. FASEB J. 11: 1257–1268. [DOI] [PubMed] [Google Scholar]
- Hamilton, K.S., Ellison, M.J., and Shaw, G.S. 2000. Identification of the ubiquitin interfacial residues in a ubiquitin–E2 covalent complex. J. Biomol. NMR 18 319–327. [DOI] [PubMed] [Google Scholar]
- Hamilton, K.S., Ellison, M.J., Barber, K.R., Williams, R.S., Huzil, J.T., McKenna, S., Ptak, C., Glover, M., and Shaw, G.S. 2001. Structure of a conjugating enzyme–ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure 9 897–904. [DOI] [PubMed] [Google Scholar]
- Hershko, A. and Ciechanover, A. 1998. The ubiquitin system. Annu. Rev. Biochem. 67 425–479. [DOI] [PubMed] [Google Scholar]
- Hochstrasser, M. 1996. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30 405–439. [DOI] [PubMed] [Google Scholar]
- Ishima, R., Wingfield, P.T., Stahl, S.J., Kaufman, J.D., and Torchia, D.A. 1998. Using amide 1H and 15N transverse relaxation to detect millisecond time-scale motions in perdeuterated proteins: Application to HIV-1 protease. J. Am. Chem. Soc. 120 10534–10542. [Google Scholar]
- James, L.C., Roversi, P., and Tawdik, D.S. 2003. Antibody multispecificity mediated by conformational diversity. Science 299 1362–1367. [DOI] [PubMed] [Google Scholar]
- Kloibert, K. and Konrat, R. 2000. Differential multiple-quantum relaxation arising from cross-correlated time-modulation of isotropic chemical shifts. J. Biomol. NMR 18 33–42. [DOI] [PubMed] [Google Scholar]
- Kroenke, C.D., Loria, J.P., Lee, L.K., Rance, M., and Palmer, A.G. 1998. Longitudinal and transverse 1H-15N dipolar 15N chemical shift anisotropy relaxation interference: Unambiguous determination of rotational diffusion tensor and chemical exchange effect in biological macromolecules. J. Am. Chem. Soc. 120 7905–7915. [Google Scholar]
- Kroenke, C.D., Rance, M., and Palmer, A.G. 1999. Variability of the 15N chemical shift anisotropy in Escherichia coli ribonuclease H in solution. J. Am. Chem. Soc. 121 10119–10125. [Google Scholar]
- Majumdar, A. and Ghose, R. 2004. Probing slow backbone dynamics in proteins using TROSY-based experiments to detect cross-correlated time-modulation of isotropic chemical shifts. J. Biomol. NMR 28 213–227. [DOI] [PubMed] [Google Scholar]
- Massi, F., Johnson, E., Wang, C., Rance, M., and Palmer, A.G. 2004. NMR R1ρ rotating-frame relaxation with weak radio frequency fields. J. Am. Chem. Soc. 126 2247–2256. [DOI] [PubMed] [Google Scholar]
- Meiler, J., Prompers, J.J., Peti, W., Griesinger, C., and Brüschweiler, R. 2001. Model-independent analysis of 15N chemical shift anisotropy from NMR relaxation data. Ubiquitin as a test example. J. Am. Chem. Soc. 123 6098–6107. [DOI] [PubMed] [Google Scholar]
- Mills, J.L. and Szyperski, T. 2002. Protein dynamics in supercooled water: The search for slow motional modes. J. Biomol. NMR 23 63–67. [DOI] [PubMed] [Google Scholar]
- Miura, T., Klaus, W., Gsell, B., Miyamoto, C., and Senn, H. 1999. Characterization of the binding interface between ubiquitin and class I human ubiquitin-conjugating enzyme 2b by multidimensional heteronuclear NMR spectroscopy in solution. J. Mol. Biol. 290 213–228. [DOI] [PubMed] [Google Scholar]
- Mosteller, F. and Tukey, J.W. 1977. Data analysis and regression. A second course in statistics. Addison-Wesley, Reading, MA.
- Mulder, F.A.A., deGraaf, R.A., Kaptein, R., and Boelens, R. 1998. An off-resonance rotating frame relaxation experiment for the investigation of macromolecular dynamics using adiabatic rotations. J. Magn. Reson. 131 351–357. [DOI] [PubMed] [Google Scholar]
- Palmer, A.G. 2004. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 104 3623–3640. [DOI] [PubMed] [Google Scholar]
- Palmer, A.G., Kroenke, C.D., and Loria, J.P. 2001. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological molecules. Methods Enzymol. 339 204–238. [DOI] [PubMed] [Google Scholar]
- Schneider, D.M., Dellwo, M.J., and Wand, A.J. 1992. Fast internal main-chain dynamics of human ubiquitin. Biochemistry 31 3645–3652. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Feller, S.E., Pastor, R.W., and Bax, A. 1995. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117 12562–12566. [Google Scholar]
- Tjandra, N., Szabo, A., and Bax, A. 1996. Protein backbone dynamics and 15N chemical shift anisotropy from quantitative measurement of relaxation interference effects. J. Am. Chem. Soc. 118 6986–6991. [Google Scholar]
- Tolman, J.R., Al-Hashimi, H.M., Kay, L.E., and Prestegard, J.H. 2001. Structural and dynamic analysis of residual dipolar coupling data for proteins. J. Am. Chem. Soc. 123 1416–1424. [DOI] [PubMed] [Google Scholar]
- Wang, C., and Palmer, A.G. 2002. Differential multiple quantum relaxation caused by chemical exchange outside the fast exchange limit. J. Biomol.NMR 24: 263–268. [DOI] [PubMed]
- ———. 2003. Solution NMR methods for quantitative identification of chemical exchange in 15N-labeled proteins. Magn. Reson. Chem. 41 866–876. [Google Scholar]
- Wang, C., Grey, M.J., and Palmer, A.G. 2001a. CPMG sequences with enhanced sensitivity to chemical exchange. J. Biomol. NMR 21 361–366. [DOI] [PubMed] [Google Scholar]
- Wang, L.C., Pang, Y.X., Holder, T., Brender, J.R., Kurochkin, A.V., and Zuiderweg, E.R.P. 2001b. Functional dynamics in the active site of the ribonuclease binase. Proc. Natl. Acad. Sci. 98 7684–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, K.D., Laleli-Sahin, E., Urbauer, J., Larsen, C.N., Shih, G.H., Haas, A.L., Walsh, S.T.R., and Wand, A.J. 1999. The binding site for UCH-L3 on ubiquitin: Mutagenesis and NMR studies on the complex between ubiquitin and UCH-L3. J. Mol. Biol. 291 1067–1077. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. and Case, D.A. 2001. Use of chemical shifts in macromolecular structure determination. Methods Enzymol. 338 3–34. [DOI] [PubMed] [Google Scholar]
- Wist, J., Frueh, D., Tolman, J.R., and Bodenhausen, G. 2004. Triple quantum decoherence under multiple refocusing: Slow correlated chemical shift modulations of C′ and N nuclei in proteins. J. Biomol. NMR 28 263–272. [DOI] [PubMed] [Google Scholar]
- Xu, X.-P. and Case, A.D. 2001. Automated prediction of 15N, 13Cα, 13Cβ, 13C′ chemical shifts in proteins using a density functional database. J. Biomol. NMR 21: 321–333. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Probing multiple effects on 15N, 13Cα, 13Cβ, 13C′ chemical shifts in peptide using density functional theory. Biopolymers 65 408–423. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.-P. 1995. “Protein and peptide structure and interactions studied by hydrogen exhange and NMR”. Ph.D. thesis, University of Pennsylvania, Philadelphia, PA.
- Zhang, H., Neal, S., and Wishart, D.S. 2003. RefDB: A database of uniformly referenced protein chemical shifts. J. Biomol. NMR 25 173–195. [DOI] [PubMed] [Google Scholar]