

Abstract
TT1426, from Thermus thermophilus HB8, is a conserved hypothetical protein with a predicted phosphoribosyltransferase (PRTase) domain, as revealed by a Pfam database search. The 2.01 Å crystal structure of TT1426 has been determined by the multiwavelength anomalous dispersion (MAD) method. TT1426 comprises a core domain consisting of a central five-stranded β sheet surrounded by four α-helices, and a subdomain in the C terminus. The core domain structure resembles those of the type I PRTase family proteins, although a significant structural difference exists in an inserted 43-residue region. The C-terminal subdomain corresponds to the “hood,” which contains a substrate-binding site in the type I PRTases. The hood structure of TT1426 differs from those of the other type I PRTases, suggesting the possibility that TT1426 binds an unknown substrate. The structure-based sequence alignment provides clues about the amino acid residues involved in catalysis and substrate binding.
Keywords: structural genomics, Thermus thermophilus, hypothetical protein, phosphoribosyltransferase
TT1426 from Thermus thermophilus HB8 is a conserved hypothetical protein, which consists of 208 amino acid residues (22.4 kDa). It is annotated as a predicted phosphoribosyltransferase (PRTase) in the Pfam database (PF00156) (Bateman et al. 2002), with a sequence conserved among bacteria and archaea (Marchler-Bauer et al. 2003). TT1426 and its homologs share a highly conserved 13-residue sequence, which is known as the 5-phosphoribosyl-1-pyrophosphate (PRPP)–binding motif in the type I PRTases, such as adenine phosphoribosyltransferase (APRTase) (Phillips et al. 1999), uracil phosphoribosyltransferase (UPRTase) (Schumacher et al. 1998), xanthine phosphoribosyltransferase (XPRTase) (Vos et al. 1997), orotate phosphoribosyltransferase (OPRTase) (Scapin et al.1995), hypoxanthine-guanine phosphoribosyltransferase (HGPRTase) (Eads et al. 1994), hypoxanthine-guanine-xanthine phosphoribosyltransferase (HGXPRTase) (Schumacher et al. 1996), and glutamine PRPP amidotransferase (GAT) (Muchmore et al. 1998). The type I PRTases catalyze the transfer of ribose 5-phosphate from PRPP to the N1 nitrogen of various substrates (uracil in the case of UPRTase). They share a common structural architecture of a core domain, comprising five parallel-β strands and at least three α-helices, and a subdomain, known as the “hood.” The hood contains the substrate-binding site and displays different structures, depending on the substrate. The second type of PRTase lacks the conserved PRPP motif, and its three-dimensional structure is quite different from those of the type I PRTases (Eads et al. 1997).
We now report the crystal structure of a predicted PRTase, TT1426 from T. thermophilus HB8, at 2.01 Å resolution. The structure was determined by the multiwave-length anomalous dispersion (MAD) method. The TT1426 structure shares the common type I PRTase fold, although its large insertion within the core domain is unique. In the C-terminal region, we found a subdomain corresponding to the hood, but its structure is not homologous to those of the other type I PRTases.
Results and Discussion
The crystals of TT1426 belong to the primitive tetragonal space group P41212, with unit cell constants of a = b = 103.97 Å, c = 51.29 Å, and contain one protein molecule per asymmetric unit. The structure was refined to 2.01 Å by the MAD method. The crystallographic data are summarized in Table 1. The final model includes 208 amino acid residues of the TT1426 monomer, 12 MES molecules, and 224 water molecules in the asymmetric unit. Two TT1426 monomers are related by the crystallographic axis. As revealed by the DALI homology search, the TT1426 monomer resembles phosphoribosylpyrophosphate (PRPP) synthetase from Leishmania donovani (Protein Data Bank [PDB] 1DKU, RMSD 2.5 Å over 120 Cα atoms; Eriksen et al. 2000), PyrR from Bacillus subtilis (PDB 1A3C, RMSD 2.6 Å over 131 Cα atoms; Tomchick et al. 1998), UPRTase from Toxoplasma gondii (PDB 1BD3, RMSD 3.0 Å over 144 Cα atoms; Schumacher et al. 1998), APRTase from Saccharomyces cerevisiae (PDB 1G2Q, RMSD 2.9 Å over 120 Cα atoms; Shi et al. 2001), and other type I PRTases, with sequence identities ranging from 13%–22%.
Table 1.
X-ray data collection, phasing, and refinement statistics
| Peak | Edge | Remote | |
| Data collection | |||
| Wavelength (Å) | 0.9791 | 0.9794 | 0.9740 |
| Resolution (Å) | 50–2.01 | 50–2.01 | 50–2.01 |
| Unique reflections | 18,107 | 18,095 | 18,482 |
| Redundancy | 7.4 | 7.8 | 7.7 |
| Completeness (%) | 95.3 (67.4) | 95.2 (67.2) | 95.6 (68.5) |
| I/σ (I) | 15.4 (2.8) | 17.8 (2.9) | 17.5 (2.9) |
| Rsym (%)a | 10.7 (30.3) | 10.1 (29.5) | 10.2 (29.6) |
| MAD analysis | |||
| Resolution (Å) | 20–2.1 | ||
| No. of sites | 3 | ||
| FOMMIRb | 0.36 | ||
| FOMRESOLVEc | 0.65 | ||
| Refinement | |||
| Resolution (Å) | 2.01 | ||
| No. of reflections | 31,382 | ||
| No. of protein atoms | 1578 | ||
| No. of MPD molecules | 12 | ||
| No. of water molecules | 224 | ||
| Rwork (%) | 19.5 | ||
| Rfree (%)d | 22.7 | ||
| RMSD bond length (Å) | 0.009 | ||
| RMSD bond angles (°) | 1.2 | ||
All numbers in parentheses represent last outer shell statistics.
aRsym = ∑Iavg − Ii|/∑Ii, where Ii is the observed intensity and Iavg is the average intensity.
b Figure of merit after SOLVE phasing.
c Figure of merit after RESOLVE.
dRfree is calculated for 10% of randomly selected reflections excluded from
refinement.
The core region of TT1426 consists of a parallel β sheet (β2, β1, β6, β7, and β8) surrounded by α-helices (α1, α2, α5, and α6), which is the common fold in the type I PRTases (Fig. 1A ▶). The core domain contains the conserved PRPP-binding motif (residue 125–135), which is located in the β6–α5 loop (Fig. 1B ▶). The β3–β4 loop corresponds to the “flexible loop,” which closes the active site during catalysis in the type I PRTases. Notably, the flexible loop is followed by a 43-residue insertion (β5, α3, and α4), which is not present in the structures of the type I PRTases determined to date. In the inserted region, the β5 strand forms an antiparallel β sheet (β3, β4, and β5), and the α3-helix interacts with the α6-helix of the symmetry-related molecule.
Figure 1.
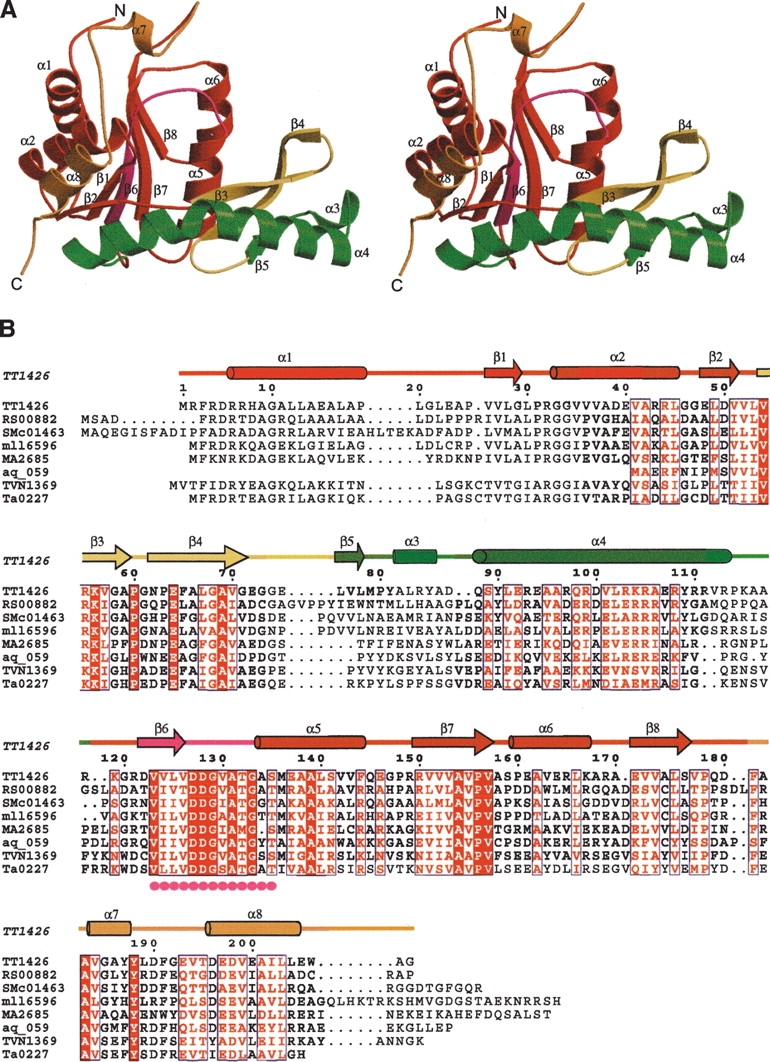
(A) Ribbon representation of TT1426 (stereo view). In the core domain, the PRPP-binding motif is shown in magenta, the flexible loop is yellow, and the inserted region is green. The rest of the core domain is shown in red, and the hood is orange. (B) Sequence alignment of TT1426 and its homologs. The secondary structure for TT1426, shown above the sequences, is colored as in A. Filled magenta circles indicate the conserved PRPP-binding motif. RS00882 indicates Ralstonia solanacearum GMI1000; SMc01463, Sinorhizobium meliloti 1021; mll6596, Mesorhizobium loti MAFF303099; MA2685, Methanosarcina acetivorans C2A; aq_059, Aquifex aeolicus VF5; TVN1369, Thermoplasma volcanium GSS1; and Ta0227, Thermoplasma acidophilum DSM 1728.
In the C-terminal region of TT1426, there is a subdomain (the α7- and α8-helices) that corresponds to the hood in the type I PRTases. The structure-based sequence alignment shows that the hood of TT1426 aligned well with that of T. gondii UPRTase (Fig. 2A ▶). However, the hood structures of TT1426 and the T. gondii UPRTase are different, except that both contain an α-helical region in the C terminus. The hood of TT1426 has no significant sequence similarity to those of the other type I PRTases, and these hood structures are completely different. Thus it is possible that the hood of TT1426 may bind an unknown substrate.
Figure 2.
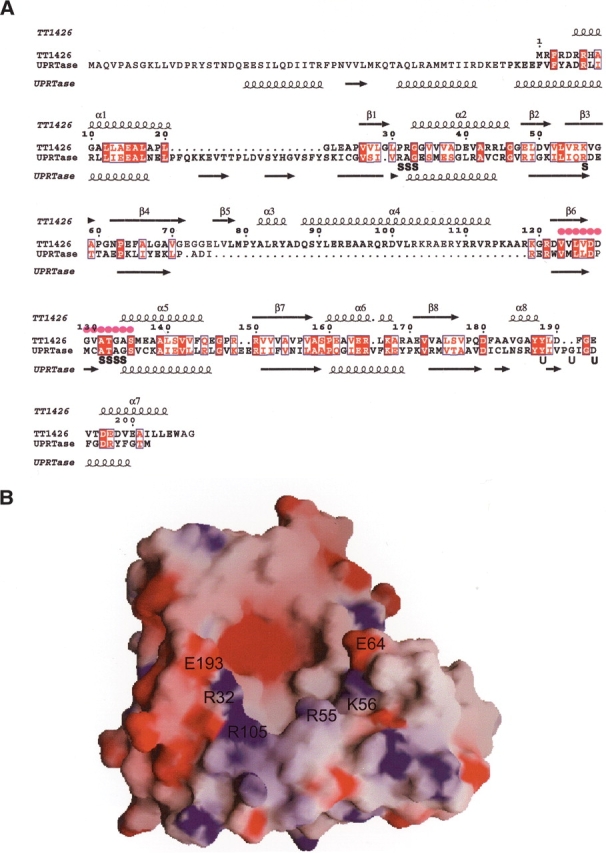
(A) Structure-based sequence alignment of TT1426 and the T. gondii UPRTase. The secondary structures for TT1426 and the T. gondii UPRTase (PDB 1BD3) are shown above and below the sequences, respectively. Filled magenta circles indicate the PRPP-binding motif. Residues involved in sulfate binding (S) and uracil binding (U) in the T. gondii UPRTase structure are labeled below the sequences. (B) Electrostatic surface representation of TT1426. Blue and red surfaces represent positive and negative potentials, respectively. The locations of the conserved hydrophilic residues around the putative active site are labeled.
The electrostatic potential distribution on the solvent-accessible surface of TT1426 revealed the presence of a negatively charged cavity, suggesting the location of a putative active site (Fig. 2B ▶). As found in other type I PRTases, the PRPP-binding motif lies in the bottom of the cavity and confers a negative potential to this area. The negatively charged cavity is surrounded by hydrophilic residues (Arg 32, Arg 55, Lys 56, Glu 64, Arg 105, and Glu 193) (Fig. 2B ▶). Since these residues are highly conserved between TT1426 and its homologs (Fig. 1B ▶), they may serve as catalytic residues. The structure-based sequence alignment suggested that Arg 32 and Lys 56 correspond to the residues involved in the binding of the pyrophosphate moiety of PRPP in the T. gondii UPRTase (Fig. 2A ▶). Glu 193 corresponds to one of the uracil-binding residues in the T. gondii UPRTase and thus may contribute to the binding of an unknown substrate in TT1426.
Materials and methods
Protein expression and purification
The gene encoding TT1426 from T. thermophilus HB8 was cloned into pET-11a (Novagen). The selenomethionine (SeMet)-substituted TT1426 protein was expressed in Escherichia coli B834 (DE3). The E. coli lysate was heated for 30 min at 70°C, and the proteins were purified by a series of HiTrap Q, HiTrap Butyl, Mono Q, and Superdex 75 column chromatography steps (Amersham Biosciences). The yield of purified TT1426 was 0.11 mg per 1 g wet cells.
Crystallization and data collection
The crystals of TT1426 (3.0 mg/mL) were grown at 20°C by the hanging drop vapor diffusion method, against a reservoir solution consisting of 15% PEG20000 and 0.1 M MES (pH 6.3). Crystals with a rod-like morphology (200 × 10 × 10 μm3) were obtained within a week. Data collection was carried out at 100 K with 20% glycerol as a cryoprotectant. The MAD data were collected at three different wavelengths at BL26B1, SPring-8 (Harima), and were recorded on a MAR imaging plate. All diffraction data were processed with the HKL2000 program (Otwinowski and Minor 1997).
Structure determination and refinement
The program SOLVE (Terwilliger and Berendzen 1999) was used to locate the selenium sites and to calculate the phases, and RESOLVE was used for the density modification (Terwilliger 2001). Automatic tracing using Arp/wARP (Perrakis et al. 2001) was used to partially build the model, and the rest of the model was built and refined with the programs O (Jones et al. 1991) and CNS (Brünger et al. 1998). Refinement statistics are presented in Table 1. The quality of the model was inspected by the program PROCHECK (Laskowski et al. 1993). The self-rotation function was calculated with the program MOLREP (Collaborative Computational Project, Number 4, 1994). Structural similarities were calculated with DALI (Holm and Sander 1993). Graphic figures were created using the programs Molscript (Kraulis 1991) and Raster3D (Merritt and Bacon 1997). The molecular surface was created with the program GRASP (Nicholls et al. 1991). The atomic coordinates have been deposited in the PDB, with the accession code 1WD5.
Acknowledgments
We thank Dr. Masaki Yamamoto for data collection at RIKEN beamline BL26B1 at SPring-8. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses, the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041229405.
References
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S.R., Griffiths-Jones, S., Howe, K.L., Marshall, M., and Sonnhammer, E.L. 2002. The Pfam protein families database. Nucleic Acids Res. 30 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macro-molecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Eads, J.C., Scapin, G., Xu, Y., Grubmeyer, C., and Sacchettini, J.C. 1994. The crystal structure of human hypoxanthine-guanine phosphoribosyltransferase with bound GMP. Cell 78 325–334. [DOI] [PubMed] [Google Scholar]
- Eads, J.C., Ozturk, D., Wexler, T.B., Grubmeyer, C., and Sacchettini, J.C. 1997. A new function for a common fold: The crystal structure of quinolinic acid phosphoribosyltransferase. Structure 5 47–58. [DOI] [PubMed] [Google Scholar]
- Eriksen, T.A., Kadziola, A., Bentsen, A.-K., Harlow, K.W., and Larsen, S. 2000. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat. Struct. Biol. 7 303–308. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–118. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Marchler-Bauer, A., Anderson, J.B., DeWeese-Scott, C., Fedorova, N.D., Geer, L.Y., He, S., Hurwitz, D.I., Jackson, J.D., Jacobs, A.R., Lanczycki, C.J., et al. 2003. CDD: A curated Entrez database of conserved domain alignments. Nucleic Acids Res. 31 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Muchmore, C.R., Krahn, J.M., Kim, J.H., Zalkin, H., and Smith, J.L. 1998. Crystal structure of glutamine phosphoribosylphosphate amidotransferase from Escherichia coli. Protein Sci. 7 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis, A., Harkiolaki, M., Wilson, K.S., and Lamzin, V.S. 2001. ARP/wARP and molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 57 1445–1450. [DOI] [PubMed] [Google Scholar]
- Phillips, C.L., Ullman, B., Brennan, R.G., and Hill, C.P. 1999. Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani. EMBO J. 18 3533–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapin, G., Ozturk, D.H., Grubmeyer, C., and Sacchettini, J.C. 1995. The crystal structure of the orotate phosphoribosyltransferase complexed with orotate and α-d-5-phosphoribosyl-1-pyrophosphate. Biochemistry 43 10744–10754. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Carter, D., Roos, D.S., Ullman, B., and Brennan, R.G. 1996. Crystal structures of Toxoplasma gondii HGXPRTase reveal the catalytic role of a long flexible loop. Nat. Struct. Biol. 3 881–887. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Carter, D., Scott, D.M., Roos, D.S., Ullman, B., and Brennan, R.G. 1998. Crystal structures of Toxoplasma gondii uracil phosphoribosyltransferase reveal the atomic basis of pyrimidine discrimination and prodrug binding. EMBO J. 17 3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, W., Tanaka, K.S.E., Crother, T.R., Taylor, M.W., Almo, S.C., and Schramm, V.L. 2001. Structural analysis of adenine phosphoribosyltransferase from Saccharomyces cerevisiae. Biochemistry 40 10800–10809. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2001. Map-likelihood phasing. Acta Crystallogr. D Biol. Crystallogr. 57 1763–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomchick, D.R., Turner, R.J., Switzer, R.L., and Smith, J.L. 1998. Adaptation of an enzyme to regulatory function: structure of Bacillus subtilis PyrR, a pyr RNA-binding attenuation protein and uracil phosphoribosyltransferase. Structure 15 337–350. [DOI] [PubMed] [Google Scholar]
- Vos, S., De Jersey, J., and Martin, J.L. 1997. Crystal structure of Escherichia coli xanthine phosphoribosyltransferase. Biochemistry 36 4125–4134. [DOI] [PubMed] [Google Scholar]