

Abstract
L-serine dehydratase (SDH), a member of the β-family of pyridoxal phosphate-dependent (PLP) enzymes, catalyzes the deamination of L-serine and L-threonine to yield pyruvate or 2-oxobutyrate. The crystal structure of L-serine dehydratase from human liver (hSDH) has been solved at 2.5 Å-resolution by molecular replacement. The structure is a homodimer and reveals a fold typical for β-family PLP-dependent enzymes. Each monomer serves as an active unit and is subdivided into two distinct domains: a small domain and a PLP-binding domain that covalently anchors the cofactor. Both domains show the typical open α/β architecture of PLP enzymes. Comparison with the rSDH-(PLP-OMS) holo-enzyme reveals a large structural difference in active sites caused by the artifical O-methylserine. Furthermore, the activity of hSDH-PLP was assayed and it proved to show catalytic activity. That suggests that the structure of hSDH-PLP is the first structure of the active natural holo-SDH.
Keywords: serine dehydratase, pyridoxal phosphate-dependent (PLP) enzyme, human liver, deamination, crystal structure
L-serine dehydratase (SDH) (EC 4.2.1.17) catalyzes the deamination of L-serine to yield ammonia and pyruvate (Fig. 1 ▶). The enzyme from a number of sources also acts on L-threonine to yield 2-oxobutanoate (Ogawa et al. 2000). SDH is a pyridoxal 5′-phosphate (PLP) dependent enzyme and has been assigned to the β family of PLP enzymes (Mehta and Christen 2000). L-threonine dehydratase (TDH) (EC 4.2.1.19) (Nakagawa 1971) also catalyzes the degradation of L-threonine and L-serine by the same mechanism and, therefore, is thought to be similar to SDH.
Figure 1.

The enzymatic reaction of serine dehydratase.
SDH is widely distributed in nature, but its physicochemical properties are considerably different from species to species. For example, rat liver serine dehydratase (rSDH) is a dimer (Xue et al. 1999; Ogawa et al. 2002), whereas yeast and Escherichia coli biosynthetic threonine dehydratase (eTDH), the first enzyme in the isoleucine synthesis pathway, is a tetramer and is feedback-inhibited by isoleucine and heterotropically activated by valine. E. coli biodegradative threonine dehydratase (d-eTDH) induced anaerobically in tryptone-yeast extract medium is a tetramer and is allosterically activated by AMP (Madison and Thompson 1976). However, E. coli D-SDH is a monomer (Marceau et al. 1988b). On the other hand, the sequence alignment of SDHs shows only moderate sequence homology.
In spite of the great variety in primary and quaternary structures of SDHs, two conserved sequence motifs have been identified. One is Ser-Xaa-Lys-Ile-Arg-Gly, which is well conserved among SDHs from human (Ogawa et al. 1989a), rat (Ogawa et al. 1989b), tomato (Samach et al. 1991), E. coli (Datta et al. 1987; Eisenstein et al. 1995), and yeast (John 1995). The lysine in rat liver SDH has been identified as the residue that forms a Schiff base with PLP. The active sites are highly homologous with those of the microbial threonine dehydratases, such as yeast and E. coli TDH. Moreover, these enzymes have a glycine-rich sequence in a region 100–130 amino acid residues downstream of the PLP binding lysyl residue (Datta et al. 1987). The importance of this motif in the interaction with the coenzyme was first assessed by the finding that substitution of the glycine residues with aspartic acid residues impairs PLP binding to E. coli D-SDH (Marceau et al. 1988a) and further demonstrated by the crystal structure of E. coli TDH (Gallagher et al. 1998).
In mammals SDH is found predominantly in the liver. Extensive studies have been carried out on SDH from rat and the enzyme has been found to play an important role in gluconeogenesis since its activity is remarkably induced by the consumption of high-protein diets, starvation, and other conditions (Snell 1984; Ebara et al. 2001). Little is known about the physicochemical and enzymatic properties of human SDH because human livers exhibit low SDH activity (Yoshida and Kikuchi 1969). However, some important functions have been found. It has been reported that a case of nonketotic hyperglycinemia was apparently due to hereditary deficiency of threonine dehydratase (Krieger and Booth 1984). In addition, the serine dehydratase activities were absent in human colon carcinoma and rat sarcoma (Snell et al. 1988). As a result, SDH is thought to be related to nonketotic hyperglycinemia and tumors. This prompted us to examine the properties of human SDH.
Here we report the crystal structure of recombinant human liver SDH holo-enzyme with bound PLP cofactor, or SDH-PLP. The structure is a homodimer and reveals a fold typical for β-class PLP-dependent enzymes. The active-site residues can be readily correlated with the predictions based on the results of biochemical and mutagenesis studies of rat SDH. Recently the structures of the rat liver SDH apo-enzyme and a holo-enzyme with O-methyl serine, SDH-(PLP-OMS) have been determined (Yoshida and Kikuchi 1969). Although human SDH shares a high sequence identity of 82% with rat SDH, the comparison with the rat SDH apo- and holo-enzymes reveals interesting structural differences. In particular, comparison with the rSDH-(PLP-OMS) holo-enzyme reveals a large structural difference in active sites caused by the artifical O-methylserine. Furthermore, the activity of hSDH-PLP was assayed and it proved to show catalytic activity. That suggests that hSDH-PLP is the first structure of the active natural holo-SDH.
Results and Discussion
The hSDH monomer
The current monomer model of hSDH comprises 319 (4–322) residues, 1 PLP molecule, and 131 water molecules. The N-terminal three residues and the C-terminal six residues were not visible in the electron density map and, consequently, were not included in the final model. Most of the 319 residues were clearly defined. During the refinement, the R-factor dropped from 45.8% to 18.9%, while the free R-factor decreased continuously from 52.8% to 24.7%. Stereochemistry is good with 93.7% of residues in the most favored region of the Ramachandran plot (Table 1). A further 6.3% of residues lie in additionally allowed regions, and there are no residues in the generously allowed or disallowed regions. The overall fold of the monomer is very similar to that of other PLP-dependent enzymes of the β-family (Fig. 2 ▶). Briefly, the hSDH monomer consists of two domains: a small domain consisting of residues 42–137, and a large domain consisting of residues 1–41 and 138–322. The large domain is also referred to as the catalytic domain since it contains the essential PLP cofactor. The secondary structure of the hSDH monomer as defined by the program DSSP (Kabsch and Sander 1983) is shown in Figure 4 ▶, below. The small domain folds as a twisted α/β structure consisting of a central four-stranded (β3–6) parallel β-sheet and four surrounding helices (helices 3–6). The PLP-binding domain contains 10 α-helices (helices 1 and 7–15), one -strands (β1, β2, and β7–10). The core 310-helix 2, and six β of this domain is an open twisted α/β structure with a central β-sheet surrounded by seven helices (helices 7–11 and 13–14), similar to the small domain. The central β-sheet is basically parallel, but one N-terminal short strand, strand 1, is antiparallel. These strands are strongly twisted so as to make the sixth strand nearly perpendicular to the first. There is a crevice at the carboxyl ends of the β-strands 7 and 10, which makes a space for bound PLP. Except for strand 1, all β-strands of the two domains are directed toward the molecular center, and all α-helices have a chain direction toward the molecular surface. The 310-helix 2 at the N terminus consists of three residues. The 310-helix of hSDH is at the interface region between the two domains. The two domains are connected by two linkers (residues 32–35 and 138–146). Between the domains, there is a large internal gap that includes the crevice mentioned above. This large internal gap provides a space for the active site.
Table 1.
Crystallographic data and refinement statistics
| Crystal parameters | ||
| Space group | I422 | |
| Unit cell dimensions | a = 157.4 Å, b = 157.4 Å, c = 59.2Å α= β= γ = 90° | |
| Estimated solvent content (%) | 46 | |
| Vm (Å3 da−1)a | 2.7 | |
| Data collection | ||
| Wavelength (Å) | 1.5418 | |
| Resolution limit (Å) | 50–2.5 | |
| Total reflections | 94,265 | |
| Unique reflections | 12,739 | |
| Completeness (%) | 96.3 (98.5)e | |
| Rmerge (%)b | 9.9 (36.6) | |
| Ave I/σ | 20.0 (5.6) | |
| Refinement statistics | ||
| Resolution range (Å) | 50–2.5 | |
| I/σ cutoff | 0.0 | |
| Rworkingc/Rfreed (%) | 18.9/24.7 | |
| RMS deviation | bonds (Å) | 0.011 |
| angles (°) | 1.62 | |
| Baverage (no. of atoms) | protein | 30.3 (2345) |
| water | 24.5 (131) | |
| PLP | 26.7 (1) | |
| Residues in most favored regions of Ramachandran plot (%) | 93.7 | |
a Vm = Matthews’ coefficient (volume of asymmetric unit/molecular weight).
b Rmerge = ∑hkl|<I> I|∑hkl|I|.
c Rworking = ∑(||Fp(obs)| − |Fp(calc)||)/∑|Fp (obs)|.
d Rfree = R factor for a selected subset (10%) of the reflections that was not included in prior refinement calculations.
e Values in parentheses correspond to the highest resolution shell 2.6–2.5 Å.
Figure 2.

(A) Ribbon diagram of the hSDH dimer. Green represents the small domain (residues 58–169); purple (residues 1–57) and cyan (residues 170–341) represent the PLP-binding domain. Both domains fold as an open twisted α/β structure consisting of a central β-sheet and surrounding helices. The Lys41 and PLP molecule are shown as ball-and-stick.
Figure 4.
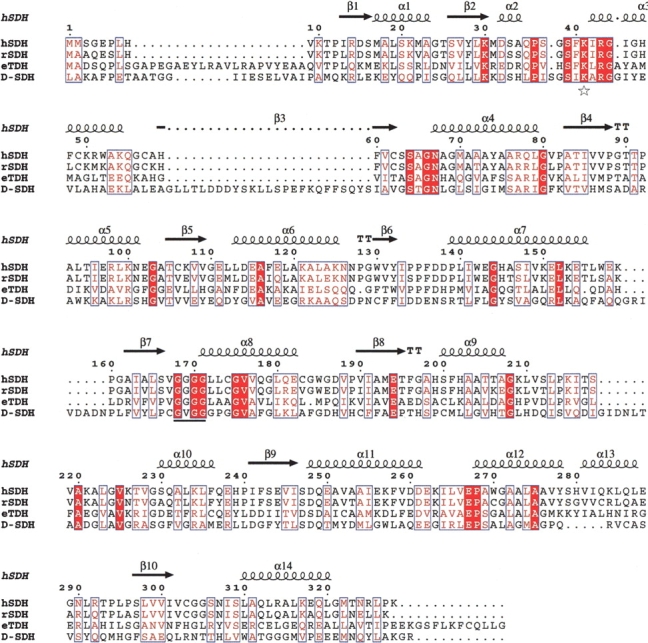
Sequence alignment of hSDH and other serine/threonine dehydratases. The sequences displayed are hSDH (from human liver), rSDH (from rat liver), eTDH (from E. coli), and D-SDH (from E. coli). The sequence of hSDH shares 82% identity with rSDH, 27% identity with eTDH but has low (11%) identity with D-SDH. The amino acids in blue boxes represent conserved changed residues. The completely identical amino acid residues are shaded red. The star represents the Lysine residue to which PLP is bound. The line represents the glycine-rich region by which PLP is fixed. The secondary structure elements indicated correspond to the hSDH structure.
Dimer structure
Two monomers of hSDH interact with each other, forming a dimer with a dyad axis running through the interacting surfaces (Fig. 2 ▶). The interface between the two monomers is formed through hydrogen bonds and hydrophobic interactions. The accessible surface area buried between the two monomers is 5.7% of the total molecular accessible surface. The monomer–monomer contacts involve six pairs of hydrogen bonds formed between 10 residues (Arg98-Asn260, Leu310-Asn260, and Leu265-Lys263 and their dyad symmetry mates). Additional interactions include a number of hydrophobic contacts. The residues Met17, Lys21, Asn101, Glu102, Ser306, Ile308, Ser309, and Ile264 in each monomer form a hydrophobic surface. Most of the residues involved in the contacts are located on helices α2, α5, α11, α14 and a loop of each monomer.
Cofactor binding site
Typical of open twisted α/β structures, the PLP cofactor is positioned at a crevice between the β-strands (β7 and β10) of the PLP-binding domain and lies on the large internal gap between the two domains (Fig. 3 ▶). The PLP cofactor is covalently bound through a Schiff base linkage to NZ of Lys41 with C4′ (the internal aldimine). The cofactor is sandwiched between the side chain of Phe40 and the main chain of Ala222. Each of the polar substituents of PLP is coordinated by an appropriate functional group: the pyridinium nitrogen is hydrogen-bonded to the side chain of Cys303, the C3-hydroxyl group is hydrogen-bonded to the side chain of Asn67, and the phosphate group of PLP is coordinated by main chain amides from the tetraglycine loop (residues 168–171).
Figure 3.
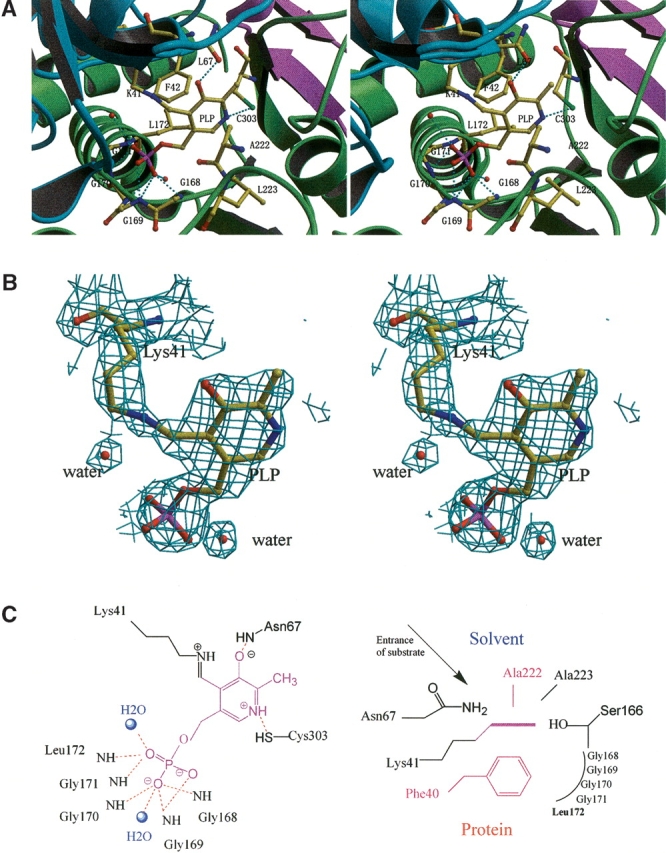
The active site of hSDH. (A) Stereoscopic view of the active site region of hSDH. Hydrogen bonds are shown in cyan. (B) 2Fo − Fc maps showing the electron density peaks of the PLP-aldimine and water molecules in the active site. The contour is drawn at the 1.5σ level. (C) Hydrogen bonding networks around the active site. On the left side, hydrogen bonds (red) between protein, water (blue balls) and cofactor (purple) are indicated, whereas the fixation of the PLP pyridine ring is shown on the right side.
Comparison with other serine/threonine dehydratases
SDH is widely distributed throughout nature, but its physicochemical properties vary considerably from species to species. It is of particular interest that the quaternary structures of all the known SDHs of various organisms can be classified into three types: monomer (D-SDH), dimer (hSDH, rSDH), or tetramer (eTDH, d-eTDH). Furthermore, the sequence similarity among the three types is relatively low and there is wide variation in their molecular masses. Sequence alignment between SDH and TDH shows that two regions are conserved in both types of enzymes: the sequences around the lysyl residue forming a Schiff base with PLP and the glycine-rich sequence which is considered to interact with the phosphate moiety of PLP (Fig. 4 ▶). Since all SDHs have a similar catalytic activity, it is expected that the active sites and catalytic mechanism are similar as well.
The structure of the rat SDH (rSDH) apo-enzyme has recently been determined to 2.8 Å (PDB ID 1PWE), and a holo-enzyme form of rSDH with O-methylserine has also been determined to 2.6 Å (PDB ID 1PWH) (Yamada et al. 2003). Human SDH shares a high sequence identity of 82% with rat SDH (Fig. 4 ▶). While hSDH and rSDH share the same overall fold, a large conformational change in the region 197–228 is observed. Superposition of hSDH with the rSDH apo-enzyme shows an r.m.s. deviation (r.m.s.d.) of 0.97 Å if residues 197–228 are excluded. The large conformation change of residues 197–228 in the rSDH apo-enzyme has been attributed to the lack of PLP and K+ ion in the active site of the apo-enzyme. Consequently, in the rSDH apo-enzyme dimer structure, residues 197–228 are stabilized by residues 105–124 of the partner subunit.
The hSDH holo-enzyme has a different dimeric form to the rSDH apo-enzyme, but has the same dimeric form as the rSDH-(PLP-OMS) holo-enzyme. The hSDH and rSDH holo-enzymes can be superimposed with an r.m.s.d. of 1.50 Å for all Cα atoms. Comparison of the superimposed structures shows that residues 197–228 adopt a similar conformation in both structures (Fig. 5A ▶). However, interesting differences are observed between our hSDH-PLP holo-enzyme structure and the rSDH-(PLP-OMS) holo-enzyme (Fig. 5B ▶). First, while the hydrogen-bonding of the PLP cofactor is preserved in the two holo-enzymes, the PLP cofactor in the hSDH holo-enzyme is tilted by approximately 5° toward Lys41 relative to rSDH. Second, a hydrogen bond is made in the rSDH holo-enzyme between the O-methylserine and Ser64 Oγ. The formation of a hydrogen bond by the O-methylserine effectively pulls strand β3, on which Ser64 is located, toward itself. The β3 strand forms part of a small four stranded β-sheet covering residues 65–130. Consequently, there is a large cooperative movement of residues 65–130 in the small domain of the rSDH holo-enzyme toward the PLP-OMS aldimine, thus reducing access to the active site. At its largest, the deviation between this region of the rSDH holo-enzyme is more than 6 Å relative to the hSDH holo-enzyme. Since the presence of OMS in the rSDH holo-enzyme is entirely artificial, we can conclude that the hSDH structure reported here is the natural SDH holo-enzyme.
Figure 5.

Superposition of hSDH, rSDH. (A) A stereo figure showing superposition of the hSDH-PLP and rSDH-(PLP-OMS) holo-enzymes. hSDH-PLP is shown in blue, and rSDH-(PLP-OMS) is shown in green. (B) A stereo figure showing the large difference in the small domain regions (residues 65–130) of hSDH-PLP and rsDH-(PLP-OMS) holo-enzymes. hSDH is shown in blue and its PLP cofactor, in yellow; rSDH is shown in green and its PLP-OMS cofactor, in magenta.
The crystal structure of E. coli TDH (eTDH) has also been determined (PDB ID 1TDJ) (Gallagher et al. 1998). Sequence identity between hSDH and eTDH is a moderate 25%. Although the activity of human SDH is not regulated by any known effector molecule, the tetrameric E. coli TDH is subject to allosteric regulation by isoleucine and valine. To illustrate the main structural differences of various SDHs and TDHs, a detailed structural comparison was performed between hSDH and eTDH.
Each monomer of eTDH contains one catalytic domain (the PLP-binding domain) and one regulatory domain. The structure of hSDH is similar to the catalytic domain of eTDH (Fig. 6A ▶). In the eTDH structure, PLP is accommodated in a pocket where it is maintained in the correct orientation by a number of covalent and noncovalent interactions. Figure 6B ▶ compares the PLP binding sites of eTDH and hSDH. The pocket consists mainly of residues conserved between the two enzymes. The orientation of PLP within hSDH is very similar to that observed in the eTDH. In both cases, the cofactor phosphate group is hydrogen-bonded to an invariant tetraglycine loop, Gly168–Gly171 in hSDH and Gly188–Gly191 in eTDH. The pyridoxyl ring is packed between two hydrophobic groups, namely Phe40 and Ala222, corresponding to Phe61 and Gly241, respectively. On the other hand, the hydrogen bond that C3-hydroxyl establishes with Asn89 in eTDH corresponds with Asn67 in hSDH. The main differences between the two binding sites regard the interactions of the polar moiety of the PLP ring. The nitrogen forms a hydrogen bond with Ser288 in TD, whereas it is coordinated by the sulfhydryl of Cys303 in the hSDH.
Figure 6.
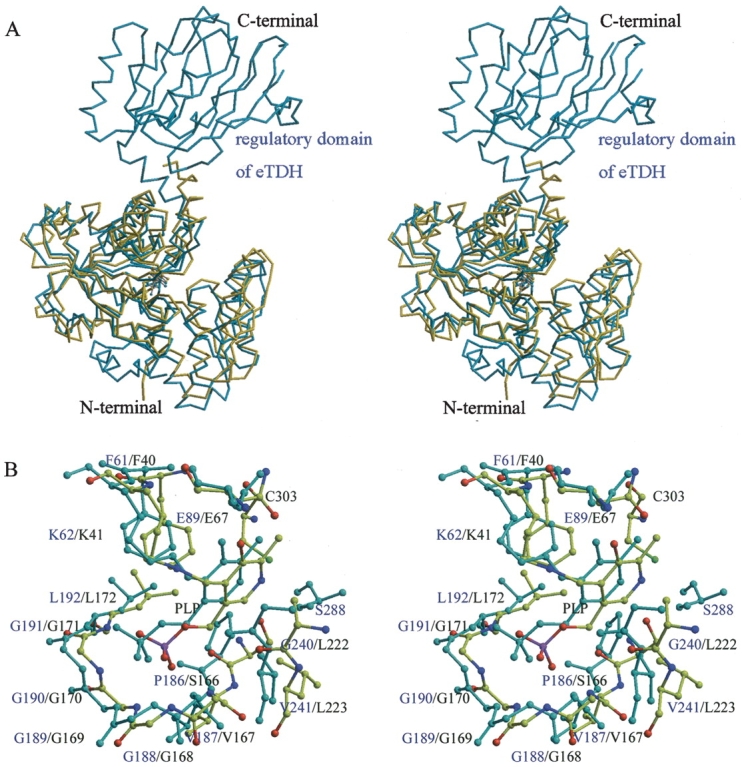
Stereo view of comparison between hSDH and eTDH. (A) Superposition of hSDH (yellow) and eTDH (blue). The structure of hSDH is very similar with the catalytic domain of eTDH. (B) The active sites of hSDH (yellow) and eTDH (blue).
Unlike eTDH, hSDH has no regulatory domain. This is the main difference between the two enzymes. In the absence of this domain, hSDH shows complete catalytic activity but no allosteric regulation. The result is consistent with the former data that showed the main effect of regulatory domain on allosteric regulation and feedback inhibition (Chinchilla et al. 1998). Despite low sequence homology, the catalytic domains of the two enzymes are highly conserved and the same result can be expected among other SDHs.
From the comparison, we can conclude that despite the low sequence homology and the striking different quaternary structures of various SDHs, their catalytic active sites are highly conserved. The result strongly suggests that different SDHs may perform their catalytic activity in the same way.
Enzyme activity
The enzyme activity assay was carried out to test the activity of human SDH. The assay showed an obvious increase in absorbance at 265 nm when L-serine was added. This increase represents the formation of pyruvate and thus confirms that recombinant human SDH shows catalytic activity for L-serine. When potassium phosphate was added to the standard enzyme assay solution, the rate of reaction was increased, suggesting that K+ ions might have an important role in the catalysis of SDH. Previous reports have confirmed that the presence of potassium ions affects the enzyme activity of both rat SDH (Pestana et al. 1971) and D-SDH (Kojiro et al. 1989).
In many PLP-enzymes, monovalent cations such as Na+ and K+ are important effectors of catalytic activity (Woehl and Dunn 1995b). In these systems, the metal ion cofactor is classified as an allosteric effector that does not participate directly in bonding interactions with the reacting substrate. In tryptophan synthase, for instance, Na+ and K+ stabilize different conformational states of the external aldimines. They also affect the equilibrium distribution of enzyme–substrate intermediates in the crystalline state (Woehl and Dunn 1995a). As no potassium was added during the purification and crystallization, no K+ ions are found in the hSDH structure. However, potassium ions have been observed near the phosphate group of PLP cofactor of the β subunit in the structures of the rSDH holo-enzyme (Yamada et al. 2003), tryptophan synthase (Hyde et al. 1988) and tryptophanase (Isupov et al. 1998). The major role of this ion is to stabilize the framework of the active site and it is not believed to be directly involved in catalysis.
Conclusions
In conclusion, the crystal structure of human SDH confirms that the enzyme belongs to the β-class of pyridoxal 5′-phosphate (PLP) dependent enzymes. Human SDH is a dimer of identical polypeptide chains containing 328 amino acid residues. The essential PLP cofactor is located between two loops of the catalytic domain and identifies the active site. Comparison with the rat SDH apo- and holo-enzymes reveals interesting structural differences. In particular, comparison with the rSDH-(PLP-OMS) holo-enzyme reveals a large structural difference caused by the artificial O-methylserine. That suggests that the structure of hSDH-PLP reported here represents the natural holo-enzyme. On the basis of the present structure, amino acids implicated in substrate binding and catalysis can be identified for further site-directed mutagenesis studies. Such research will aid the understanding of the catalytic mechanism of action of this enzyme.
Materials and methods
Crystallization and data collection
The expression of hSDH in E. coli, the purification of the expressed enzyme and the crystallization of the purified enzyme have been reported elsewhere (Sun et al. 2003). Briefly, recombinant hSDH was crystallized by the hanging-drop vapor diffusion method. Crystals were grown at 291 K using (NH4)2SO4 as precipitant. Diffraction data was obtained to a resolution of 2.5 Å from a single frozen crystal using Cu Kα radiation. All data were integrated and scaled using DENZO and SCALEPACK (Otwinowski and Minor 1997). Crystal parameters and data collection-data statistics are summarized in Table 1.
Structure determination
The structure of hSDH was determined by molecular replacement, employing the monomeric form of eTDH (PDB ID 1TDJ) as a search model. eTDH shares 27% sequence identity with hSDH. The search model was composed of residues 32–330 of the eTDH structure based on sequence alignments using CLUSTALW (Thompson et al. 1994). Rotation and translation searches were performed using CNS (Brunger et al. 1998). Following rigid body refinement at 3.0 Å, a σ-weighted map was calculated as a basis for fitting the model. Crystallographic refinement was carried out with molecular dynamics using the program CNS. Ten percent (10%) of the reflection data were set aside for calculation of the free R-factor. Several cycles of simulated annealing at 3000 K, including minimization and B-factor refinement, were performed. After each cycle of refinement, manual rebuilding was carried out with the guidance of σ-weighted 2|Fo| − |Fc| and |Fo| − |Fc| difference maps using the program O (Jones et al. 1991). The PLP molecule from the eTDH structure was manually adjusted and built into the electron density map. Solvent molecules were added during the final cycles of refinement. The crystallographic free R-factor was monitored at each stage and a composite omit map was calculated to eliminate model bias. The quality of the structure was evaluated using a Ramachandran plot calculated by the program PROCHECK (Laskowski et al. 1993). Statistics on the final model are presented in Table 1.
Enzyme assay of human SDH
A direct spectrophotometric assay (Freedland and Avery 1964) was used to determine the SDH activity. Through a wavelength scan, we found that pyruvate had a characteristic absorbance at 265 nm but serine and SDH had not. Therefore, in the enzyme activity assay, we monitored the increase in absorbance at 265 nm, which is associated with the formation of pyruvate. Changes in absorbance were recorded by a spectrophotometer. The standard assay solution contained 990 μL of 0.1 M sodium phosphate, physiological pH 7.5, and 10 μL purified enzyme; 50 μL of 0.5M L-serine was added to start the reaction. Assays were performed at 25°C.
Coordinates
The atomic coordinates and the structure factors (code 1P5J) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Bruns-wick, NJ (http://www.rcsb.org/).
Acknowledgments
We thank Feng Gao from the Rao laboratory for his help during data collection. The research was supported by the Ministry of Science & Technology (MOST) Human Liver Proteomics Project, grant no. 2004CB520801, and the National Natural Science Foundation of China (NSFC), grant no. 30221003.
Abbreviations
SDH, serine dehydratase
TDH, threonine dehydratase
PLP, pyridoxal 5′-phosphate
hSDH human serine dehydratase
rSDH, rat serine dehydratase
eTDH, threonine deaminase from E. coli
d-eTDH, biodegradative threonine dehydratase from E. coli
β-TRPS, tryptophan synthase β-subunit
OASS, O-acetylserine sulfhydrylase
CBS, cystathione β-synthase
ACCD, 1-aminocyclopropane-1-carboxylate deaminase
aTS, threonine synthase from A. thaliana
yTS, yeast threonine synthase
OPHS, O-phosphohomoserine
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041179105.
References
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macro-molecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Chinchilla, D., Schwarz, F.P., and Eisenstein, E. 1998. Amino acid substitutions in the C-terminal regulatory domain disrupt allosteric effector binding to biosynthetic threonine deaminase from Escherichia coli. J. Biol. Chem. 273 23219–23224. [DOI] [PubMed] [Google Scholar]
- Datta, P., Goss, T.J., Omnaas, J.R., and Patil, R.V. 1987. Covalent structure of biodegradative threonine dehydratase of Escherichia coli: Homology with other dehydratases. Proc. Natl. Acad. Sci. 84 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebara, S., Toyoshima, S., Matsumura, T., Adachi, S., Takenaka, S., Yamaji, R., Watanabe, F., Miyatake, K., Inui, H., and Nakano, Y. 2001. Cobalamin deficiency results in severe metabolic disorder of serine and threonine in rats. Biochim. Biophys. Acta 1568 111–117. [DOI] [PubMed] [Google Scholar]
- Eisenstein, E., Yu, H.D., Fisher, K.E., Iacuzio, D.A., Ducote, K.R., and Schwarz, F.P. 1995. An expanded two-state model accounts for homotropic cooperativity in biosynthetic threonine deaminase from Escherichia coli. Biochemistry 34 9403–9412. [DOI] [PubMed] [Google Scholar]
- Freedland, R.A. and Avery, E.H. 1964. Studies on threonine and serine dehydratase. J. Biol. Chem. 239 3357–3360. [PubMed] [Google Scholar]
- Gallagher, D.T., Gilliland, G.L., Xiao, G., Zondlo, J., Fisher, K.E., Chinchilla, D., and Eisenstein, E. 1998. Structure and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure 6 465–475. [DOI] [PubMed] [Google Scholar]
- Hyde, C.C., Ahmed, S.A., Padlan, E.A., Miles, E.W., and Davies, D.R. 1988. Three-dimensional structure of the tryptophan synthase α2 β 2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 263 17857–17871. [PubMed] [Google Scholar]
- Isupov, M.N., Antson, A.A., Dodson, E.J., Dodson, G.G., Dementieva, I.S., Zakomirdina, L.N., Wilson, K.S., Dauter, Z., Lebedev, A.A., and Harutyunyan, E.H. 1998. Crystal structure of tryptophanase. J. Mol. Biol. 276 603–623. [DOI] [PubMed] [Google Scholar]
- John, R.A. 1995. Pyridoxal phosphate-dependent enzymes. Biochim. Biophys. Acta 1248 81–96. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kojiro, C.L., Marceau, M., and Shafer, J.A. 1989. Effect of potassium ion on the phosphorus-31 nuclear magnetic resonance spectrum of the pyridoxal 5′-phosphate cofactor of Escherichia coli D-serine dehydratase. Arch. Biochem. Biophys. 268 67–73. [DOI] [PubMed] [Google Scholar]
- Krieger, I. and Booth, F. 1984. Threonine dehydratase deficiency: A probable cause of non-ketotic hyperglycinaemia. J. Inherit. Metab. Dis. 7 53–56. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Madison, J.T. and Thompson, J.F. 1976. Threonine synthetase from higher plants: Stimulation by S-adenosylmethionine and inhibition by cysteine. Biochem. Biophys. Res. Commun. 71 684–691. [DOI] [PubMed] [Google Scholar]
- Marceau, M., Lewis, S.D., and Shafer, J.A. 1988a. The glycine-rich region of Escherichia coli D-serine dehydratase. Altered interactions with pyridoxal 5′-phosphate produced by substitution of aspartic acid for glycine. J. Biol. Chem. 263 16934–16941. [PubMed] [Google Scholar]
- Marceau, M., McFall, E., Lewis, S.D., and Shafer, J.A. 1988b. D-serine dehydratase from Escherichia coli. DNA sequence and identification of catalytically inactive glycine to aspartic acid variants. J. Biol. Chem. 263 16926–16933. [PubMed] [Google Scholar]
- Mehta, P.K. and Christen, P. 2000. The molecular evolution of pyridoxal-5′-phosphate dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 74 129–184. [DOI] [PubMed] [Google Scholar]
- Nakagawa, H. 1971. Enzymological study of amino acid nutrition. Physiological roles of serine dehydratase and cystathionine synthetase. Seikagaku 43 245–266. [PubMed] [Google Scholar]
- Ogawa, H., Gomi, T., Konishi, K., Date, T., Nakashima, H., Nose, K., Matsuda, Y., Peraino, C., Pitot, H.C., and Fujioka, M. 1989a. Human liver serine dehydratase. cDNA cloning and sequence homology with hydroxyamino acid dehydratases from other sources. J. Biol. Chem. 264 15818–15823. [PubMed] [Google Scholar]
- Ogawa, H., Konishi, K., and Fujioka, M. 1989b. The peptide sequences near the bound pyridoxal phosphate are conserved in serine dehydratase from rat liver, and threonine dehydratases from yeast and Escherichia coli. Biochim. Biophys. Acta 996 139–141. [DOI] [PubMed] [Google Scholar]
- Ogawa, H., Gomi, T., and Fujioka, M. 2000. Serine hydroxymethyltransferase and threonine aldolase: Are they identical? Int. J. Biochem. Cell Biol. 32 289–301. [DOI] [PubMed] [Google Scholar]
- Ogawa, H., Gomi, T., Takusagawa, F., Masuda, T., Goto, T., Kan, T., and Huh, N.H. 2002. Evidence for a dimeric structure of rat liver serine dehydratase. Int. J. Biochem. Cell Biol. 34 533–543. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallogr. A 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pestana, A., Sandoval, I.V., and Sols, A. 1971. Inhibition by homocysteine of serine dehydratase and other pyridoxal 5′-phosphate enzymes of the rat through cofactor blockage. Arch. Biochem. Biophys. 146 373–379. [DOI] [PubMed] [Google Scholar]
- Samach, A., Hareven, D., Gutfinger, T., Ken-Dror, S., and Lifschitz, E. 1991. Biosynthetic threonine deaminase gene of tomato: Isolation, structure, and upregulation in floral organs. Proc. Natl. Acad. Sci. 88 2678–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell, K. 1984. Enzymes of serine metabolism in normal, developing, and neoplastic rat tissues. Adv. Enzyme Regul. 22 325–400. [DOI] [PubMed] [Google Scholar]
- Snell, K., Natsumeda, Y., Eble, J.N., Glover, J.L., and Weber, G. 1988. Enzymic imbalance in serine metabolism in human colon carcinoma and rat sarcoma. Br. J. Cancer 57 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, L., Li, X., Dong, Y., Yang, M., Liu, Y., Han, X., Zhang, X., Pang, H., and Rao, Z. 2003. Crystallization and preliminary crystallographic analysis of human serine dehydratase. Acta Crystallogr. D Biol. Crystallogr. 59 2297– 2299. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. Clustal-W—Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehl, E.U. and Dunn, M.F. 1995a. Monovalent metal ions play an essential role in catalysis and intersubunit communication in the tryptophan synthase bienzyme complex. Biochemistry 34 9466–9476. [DOI] [PubMed] [Google Scholar]
- ———. 1995b. The roles of Na+ and K+ in pyridoxal-phosphate enzyme catalysis. Coordination Chemistry Reviews 144 147–197. [Google Scholar]
- Xue, H.H., Sakaguchi, T., Fujie, M., Ogawa, H., and Ichiyama, A. 1999. Flux of the L-serine metabolism in rabbit, human, and dog livers. Substantial contributions of both mitochondrial and peroxisomal serine:pyruvate/alanine:glyoxylate aminotransferase. J. Biol. Chem. 274 16028–16033. [DOI] [PubMed] [Google Scholar]
- Yamada, T., Komoto, J., Takata, Y., Ogawa, H., Pitot, H.C., and Takusagawa, F. 2003. Crystal structure of serine dehydratase from rat liver. Biochemistry 42 12854–12865. [DOI] [PubMed] [Google Scholar]
- Yoshida, T. and Kikuchi, G. 1969. Physiological significance of glycine cleavage system in human liver as revealed by the study of a case of hyperglycinemia. Biochem. Biophys. Res. Commun. 35 577–583. [DOI] [PubMed] [Google Scholar]