

Abstract
The C-terminal region of focal adhesion kinase (FAK) consists of a right-turn, elongated, four-helix bundle termed the focal adhesion targeting (FAT) domain. The structure of this domain is maintained by hydrophobic interactions, and this domain is also the proposed binding site for the focal adhesion protein paxillin. Paxillin contains five well-conserved LD motifs, which have been implicated in the binding of many focal adhesion proteins. In this study we determined that LD4 binds specifically to only a single site between the H2 and H3 helices of the FAT domain and that the C-terminal end of LD4 is oriented toward the H2-H3 loop. Comparisons of chemical-shift perturbations in NMR spectra of the FAT domain in complex with the binding region of paxillin and the FAT domain bound to both the LD2 and LD4 motifs allowed us to construct a model of FAK–paxillin binding and suggest a possible mechanism of focal adhesion disassembly.
Keywords: NMR, protein structure, focal adhesion kinase, cell migration
Cell migration is promoted by the alternating formation and disassembly of focal adhesions (Sastry et al. 1999); however, the mechanism that governs these dynamics remains unclear (Sastry and Burridge 2000). Two main components of focal adhesions are focal adhesion kinase (FAK) (Ilic et al. 1998; Cary and Guan 1999; Parsons et al. 2000) and paxillin (Turner 2000); they interact directly with one another (Schlaepfer et al. 1999).
FAK is a nonreceptor kinase consisting of a central catalytic domain flanked by large N- and C-terminal noncatalytic domains. The C-terminal region is rich in protein–protein interaction sites. When autonomously expressed, this portion of FAK, called the FAK-related nonkinase (FRNK) (Schaller et al. 1993), acts as a negative regulator of FAK activity (Nolan et al. 1999) and blocks the formation of focal adhesions (Richardson and Parsons 1996). FRNK consists of several proline-rich regions that act as binding sites for many SH3-containing proteins and is adjacent to a C-terminal focal adhesion-targeting (FAT) domain (Hildebrand et al. 1993). The FAT domain targets sites of focal adhesion in the cell (Cary and Guan 1999; Schlaepfer et al. 1999), and microinjections of this domain in cells causes decreased cell motility (Gilmore and Romer 1996). The FAT domain (~15.5 kDa) is composed of four helices that form a right-turn, elongated bundle which is maintained by hydrophobic interactions (Arold et al. 2002; Hayashi et al. 2002; G. Liu et al. 2002). This is also the binding site for paxillin, a focal adhesion protein that binds to FAK early in the formation of focal adhesion complexes (Tachibana et al. 1995; Brown et al. 1996; Shen et al. 1998; Thomas et al. 1999; Hayashi et al. 2002). Therefore, a detailed structural study of the FAK–paxillin complex is an important step in the elucidation of the mechanism responsible for the formation and disassembly of focal adhesions.
Paxillin is a multidomain adaptor protein that localizes in cultured cells primarily to sites of adhesion to the extracellular matrix and is a major target of tyrosine kinases during various cellular events associated with cell adhesion and growth control (Turner 2000). Highly conserved between species, paxillin in chicken is 90% identical to that in humans (Turner and Miranti 1994; Tumbarello et al. 2002). Paxillin contains many protein-binding modules that allow it to bind to various structural and signaling molecules. The C-terminal domain consists of four LIM (double zinc finger) motifs (Brown et al. 1996). The N-terminal domain consists of five protein-binding LD motifs (consensus sequence LDXLLXXL) termed LD1 through LD5 (Brown et al. 1996, 1998; Turner et al. 1999; Sattler et al. 2000). It has been proposed that the binding of paxillin to FAT is achieved by the interaction of LD2 and LD4 with two hydrophobic patches on opposite faces of the four-helix bundle of FAT. (Arold et al. 2002; Hayashi et al. 2002; G. Liu et al. 2002). Although these LD-mediated interactions are apparently important for paxillin function, their structural basis is not fully understood.
By using distance constraints derived from paramagnetic spin-labeling experiments, we previously determined the structure of the FAT domain in a complex with a peptide corresponding to LD2 of paxillin. Recently, a second LD2 peptide-binding site on the FAT domain was detected (Gao et al. 2004), and it has been reported that crystal structures of the FAT domain in complex with either LD2 at 2.8 Å resolution or LD4 at 2.6 Å resolution have been solved (Hoellerer et al. 2003). However, in the crystal structures the quality of the electron density “was not sufficient to determine the directionality of the peptide chain” in the second LD-binding site between the H2 and H3 helices of the FAT domain. Orientation of an LD peptide in the H2–H3 site was therefore inferred from homology with the H1–H4 binding site.
In the present study, we investigated the binding mode of the LD4 motif of paxillin to the FAT domain and the relationship between the FAT-bound LD2 and LD4 motifs. Our results unambiguously show the position and orientation of the LD4 motif when it is bound to FAT and confirm the proposal that the LD4 peptide is in an α-helical conformation upon binding. Furthermore, our chemical-shift perturbation studies demonstrated that LD4 occupies only a single site on the surface of the FAT domain. On the basis of our comparisons of the nuclear magnetic resonance (NMR) spectra of the FAT domain in a complex with a fragment that contains both the LD2 and LD4 motifs of paxillin (residues 128–308) and that of the FAT domain bound to both the LD2 and LD4 motifs, we propose a model of paxillin–FAT domain binding in which a single molecule of paxillin interacts with a single molecule of FAK by simultaneously binding to opposite faces of the four-helix bundle of the FAT domain.
Results
Chemical-shift perturbation studies of the LD2 and LD4 motifs titrated into a solution containing the FAT domain
Chemical-shift perturbation analysis of the LD2 peptide of paxillin (corresponding to residues 139–162) bound to the FAT domain was complicated by the tendency of FAT to dimerize in solution (Arold et al. 2002; G. Liu et al. 2002) and by the fairly weak binding affinity of the LD2 peptide (G. Liu et al. 2002). The heteronuclear single quantum correlation (HSQC) spectrum of the 15N-labeled FAT domain represented the signal average of the monomeric and dimeric forms. Upon addition of 1 eq of LD2 peptide, the HSQC spectrum underwent a drastic change that affected almost every peak in the spectrum (Fig. 1A ▶). This effect was most clearly seen in the “fingerprint” region of 8.5–9.2 ppm in the 1H dimension and 123–130 ppm in the 15N dimension. If the formation of the dimer occurred as the result of a helix swap as previously proposed (Arold et al. 2002), then the binding of LD2 between the H1 and H4 helices would stabilize the monomeric form. Further titration of LD2 showed the motif’s ability to bind to a second site on the FAT domain, but the binding affinity was weaker: 10 eq of LD2 had to be added before no further chemical-shift perturbation was observed. The second LD2-binding site was evidenced by a change in the direction of the chemical shift of most residues affected (Fig. 1B ▶). Only after 10 eq of LD2 were added did the resonances in the HSQC spectrum cease to shift. These experiments confirmed that LD2 bound to two sites on the FAT domain. Because of the nature of fast exchange, it was not possible to observe nuclear Over-hauser enhancements (NOEs) between the FAT domain and LD2. Nevertheless, in our earlier work using paramagnetic spin labeling of the LD2 peptide, we demonstrated that LD2 bound preferentially between the H1 and H4 helices of the FAT domain (G. Liu et al. 2002).
Figure 1.
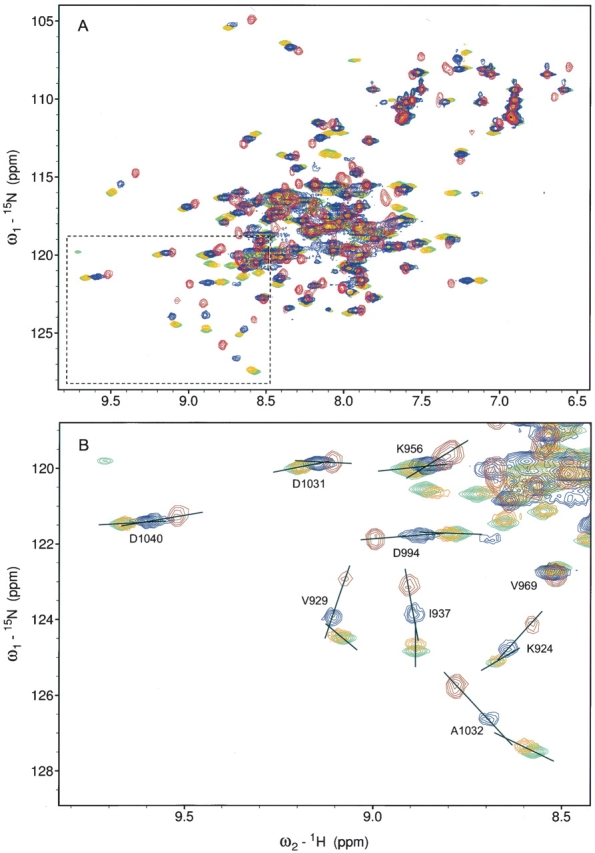
HSQC map of LD2 titration into a solution containing the FAT domain. (A) Overlaid HSQC spectra of the FAT domain (red), FAT with 1 eq of LD2 (blue), FAT with 2 eq of LD2 (orange), and FAT with 10 eq of LD2 (green). (B) Expanded region in A shows changes in the direction of the chemical shifts indicating a secondary binding event.
Using a similar approach, we studied the interaction between the FAT domain and the LD4 motif of paxillin. A 15-amino acid peptide corresponding to residues 262–276 of paxillin (the LD4 motif) was chemically synthesized. When this LD4 peptide was titrated into a solution of 15N-labeled FAT domain, the addition of 1 eq appeared to affect the FAT domain equilibrium in a similar way as 1 eq of LD2 did (Fig. 2A ▶). However, when the spectrum for FAT bound to 1 eq of LD4 was compared with that of FAT bound to 1 eq of LD2, significant differences in the HSQC spectra were observed, suggesting that the two peptides do not bind to FAT in a similar manner (Fig. 2B ▶). Further titration of LD4 (up to 10 eq) showed no additional perturbation of the spectrum; this finding suggests that only a single equivalent of LD4 binds to FAT, presumably at a single site (Fig. 3A ▶). When the spectra for FAT saturated with LD2 (Fig. 3B ▶, green) were compared with that for FAT saturated with LD4 (Fig. 3B ▶, red), the chemical shifts for V929, K924, and A1023 were similar (see Fig. 1B ▶ for assignment). We propose that the binding of a single equivalent of LD peptide (either LD2 or LD4) to the FAT domain serves to stabilize the four-helix bundle and shift the monomer–dimer equilibrium to the monomeric form; therefore, this binding has a similar (and drastic) effect on the chemical shift of residues whose side chains are oriented toward the core of the four-helix bundle (such as V929, K924, and A1023). However, residues that are located in the LD2-binding site and whose side chains are oriented toward the solvent (e.g., I937) show a marked difference in chemical shift when LD2 rather than LD4 is titrated.
Figure 2.

HSQC map of LD4 titration into a solution containing the FAT domain. (A) Overlaid HSQC spectra of the FAT domain (red), FAT with 1 eq of LD4 (orange), and FAT with 10 eq of LD4 (green). (B) Comparison of the HSQC spectra of FAT with 1 eq of LD2 (blue) and with 1 eq of LD4 (red). The difference in the spectra indicates that the two peptides bound to different sites in FAT.
Figure 3.
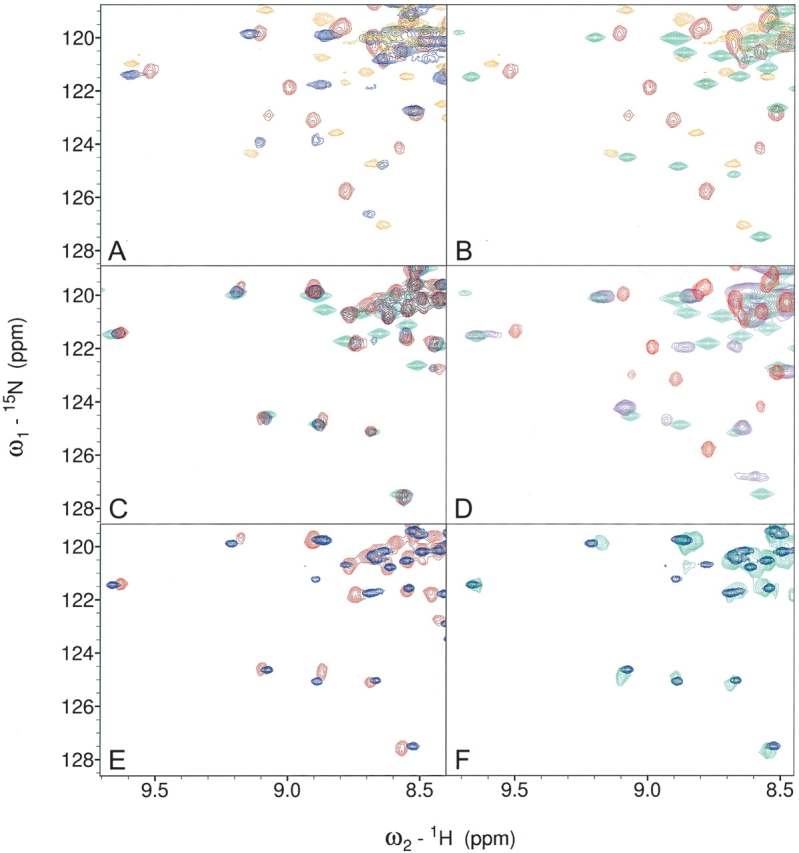
HSQC fingerprints of FAT–LD complexes. The spectra shown are the “fingerprint” regions that highlight the differences between the FAT domain bound to one or two peptides and the monomer–dimer shift. Full spectra are available in the supplemental data section. (A) FAT (red), FAT + LD2 1:1 (blue), and FAT + LD4 1:1 (orange); (B) FAT (red), FAT + LD2 1:10 (green), and FAT + LD4 1:1 (orange); (C) FAT + LD2 + LD4 1:1:1 (red), FAT + LD2 + LD4 1:10:1 (blue), and FAT + LD2 1:10 (green); (D) FAT (red), FAT–LD2 (purple), and FAT + LD2 1:1 (green); (E) FAT + LD2 + LD4 1:1:1 (red) and FAT–LD2 + LD4 1:1 (blue); (F) FAT–LD2 + LD4 1:1 (blue) and 15N FAT in a complex with unlabeled paxillin residues 128–308 (green).
We then examined the effect of the LD2 peptide on the FAT domain–LD2 interaction. Upon addition of a single equivalent of LD2 to the FAT domain–LD4 complex, the spectrum shifts resembled those seen when 10 eq of LD2 were added to FAT, i.e., this spectrum resembled the spectrum of FAT with both LD-binding sites occupied (Fig. 3C ▶). When further equivalents of LD2 (up to 10) were added to FAT/LD4/LD2 (1:1:1), the HSQC spectrum underwent no further chemical-shift perturbations; this result indicates that LD4 has a much stronger affinity for the second LD-binding site than does LD2 (Fig. 3C ▶). These data are consistent with the model of two separate LD-binding sites in which LD2 has a stronger affinity for the first LD-binding site between helices H1 and H4 (the LD2-binding site) of the FAT domain but also has a weaker affinity for the second LD2-binding site. In contrast, LD4 exhibits specific binding to only a single site (the LD4-binding site) that overlaps the second site of LD2 binding and has a much stronger affinity for this site than does LD2.
Chemical-shift perturbation studies of a FAT domain–LD2 construct
To unambiguously study the interaction between LD4 and FAT as it exists in the complex formed by full-length FAK and paxillin, it was necessary to design a construct that simulated the monomer of FAT in which the LD2-binding site is occupied. To achieve this design, we linked the LD2 motif to the C terminus of the FAT domain by a series of Gly-Gly-Ser repeats (termed FAT–LD2) that enabled the LD2 motif to interact with the H1-H4 face of the four-helix bundle but not with the opposite face. The HSQC spectrum of FAT–LD2 was similar to that of the FAT domain to which 1 eq of LD2 was bound (Fig. 3D ▶). When a second LD peptide (either LD2 or LD4) was titrated into a solution of FAT–LD2, the 15N-HSQC spectrum of the complex closely resembled that of the FAT domain bound by free peptides; the addition of 1 eq of LD4 to a solution of FAT–LD2 resulted in an 15N-HSQC spectrum that was similar to that of FAT in solution with 1 eq of both free LD2 and LD4 peptides (Fig. 3E ▶). To further test the validity of FAT–LD2 + LD4 as a model of paxillin binding to the FAT domain, we prepared unlabeled protein corresponding to residues 128–308 of full-length chicken paxillin (~21.0 kDa); this region includes the LD2, LD3, and LD4 motifs. When in complex with 15N-labeled FAT, the resulting HSQC spectrum was very similar to that of the FAT–LD2 + LD4 (1:1) complex (Fig. 3F ▶). We therefore conclude that the LD4 peptide binds to FAT–LD2 in the same way as the LD4 motif of wild-type paxillin.
Circular dichroism spectroscopy of the FAT-bound LD4 peptide
After determining the binding site and the orientation of the FAT-bound LD4 peptide, we sought to construct a model of the FAT domain–LD4 complex, but it was first necessary to obtain information about the conformation of the FAT-bound LD4 peptide. Unlike LD2, which is mostly helical as a free peptide, the circular dichroism (CD) spectra of the free LD4 peptide in an aqueous solution (Fig. 4A ▶) revealed it is mostly random coil with only a small percentage of it in a helical conformation. Nevertheless, titrating trifluoroethanol (TFE) into the solution of LD4 peptide showed that the amount of helicity in the peptide increased in direct proportion to the concentration of TFE in the solution (Fig. 4A ▶, inset). This result is consistent with those of earlier reports that predicted LD4 in wild-type paxillin to be helical (Turner 2000).
Figure 4.

CD spectra of LD4 helix formation. (A) CD spectra showing the molar ellipticity of samples of 20 μM LD4. As the amount of TFE increased, the percent helicity (inset) of the LD4 peptide increased. (B) Change in total helicity of FAT–LD2 samples (50 μM) containing increasing amounts of LD4 (0.2–2 eq). The predictive model that was used is independent of sample concentration.
We then used CD spectroscopy to investigate the conformations of FAT-bound LD4 peptide by titrating LD4 peptide into solutions of the FAT–LD2 construct and calculating the helicities of the system by using the CD spectra at each step during the titration. Because the FAT domain undergoes conformational change upon the binding of LD peptide, the use of the FAT–LD2 construct instead of the wild-type FAT domain allowed us to reasonably assume that the binding of the LD4 peptide to FAT–LD2 does not increase the helical content of FAT–LD2. Therefore, the subtraction of helicities at different titration points was expected to provide the percent helicity of the LD4 peptide in the complex. However, we found that it is prohibitively difficult to perform such a calculation by using traditional methods because the difference between the molar ellipticity of LD4 and that of FAT–LD2 + LD4 was on the same order of magnitude as the errors of the measurements. However, by using a predictive model that is independent of sample concentration and that does not factor chain length into the calculation (Raussens et al. 2003; see Materials and Methods), we measured the change in helicity of the entire complex as the titration progressed. The resulting curve (represented as the change in helicity [%] of the total complex vs. concentration of LD4) showed that the helicity of the complex increased until 1 eq of LD4 had been added (Fig. 4B ▶). Upon addition of excess LD4, the change in total helicity decreased rapidly. As the predictive model is concentration-independent, the observed increase in helicity (%) cannot be accounted for simply by the increasing amount of LD4 peptide added to the solution. Nevertheless, the study allows us to reach the conclusion that the LD4 peptide is folded into a helical conformation upon binding to FAT.
Paramagnetic spin labeling of LD4 to map the LD4-binding site of FAT
Chemical-shift perturbation studies clearly showed that LD4 binds to FAT in a specific way. To obtain detailed structural information about the FAT–LD4 complex, we prepared two spin-labeled mutants of LD4 by chemically synthesizing two LD4 peptides that contained a cysteine mutation at opposite ends of the helix (Ala263Cys and Ser273Cys). The reaction between a cysteine residue and (1-oxy-2,2,5,5-tetramethylpyroline-3-methyl)methane-thiosulfonate (MTSSL) results in an oxidized nitroxide, which serves as a paramagnetic center (G. Liu et al. 2002). Because of the distance-dependent spin lattice relaxation of magnetic nuclei in close proximity to a paramagnetic center, the spin-labeling method provides accurate distance information between residues of the affected resonances and the spin label (Bertini et al. 1997; Gaponenko et al. 2000). By comparison of the 15N HSQC spectra of 15N-labeled FAT–LD2 with 1 eq of unlabeled LD4 with that of FAT–LD2 titrated with spin-labeled LD4 peptide, it was revealed that several resonances of amide protons in the H2 and H3 helices of FAT are in proximity to the spin label. When the analysis included the spin-labeled Ala263Cys LD4 peptide, there was a drastic reduction in the signal intensity of the residues Arg963, Asn992, and Ala996 of the FAT domain, each of which lies midway along the H2 and H3 helices (Fig. 5A ▶). For the spin-labeled Ser273Cys LD4 peptide, the affected residues (Ile999, Asn1000, and Lys1001) lie along the C-terminal end of the H3 helix of FAT (Fig. 5B ▶). Neighboring residues showed slight chemical shifts that are most probably due to paramagnetically induced shift. This study unambiguously located the LD4 peptide-binding site between H2 and H3 helices on the surface of the FAT domain and determined the orientation of the LD4 peptide when it is bound to the FAT domain.
Figure 5.
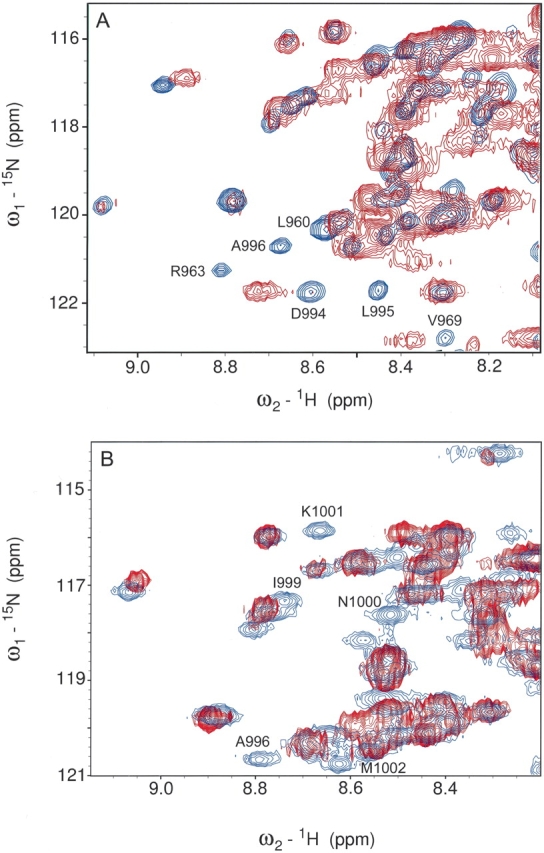
1H-15N correlation maps of 15N FAT–LD2 bound to spin-labeled LD4 peptides. The peaks in blue represent the assigned signals of 15N FAT–LD2. The overlaid image (red) represents the spectrum after titration with spin-labeled Ser273Cys LD4. Peaks for Asn1000 and Lys1001 have been broadened beyond detection via paramagnetic relaxation. Similar spectra for the spin-labeled Ala263Cys LD4 peptide show the disappearance of signals for residues Arg963, Asn992, and Ala996.
By using the distance information derived from spin-labeling experiments to assign the amide protons that were dramatically affected by the spin labeling to be within 10 Å of the paramagnetic center, we constructed the model of the FAT domain–LD4 complex. On the basis of our CD spectroscopy data, LD4 was modeled as an α-helix. When the hydrophobic surface of LD4 was oriented toward the FAT domain and the distance constraints remained, a model structure of the FAT domain–LD4 complex was obtained by using the software SYBYL. This model showed that LD4 is situated between the H2 and H3 helices and is parallel to the LD2-binding site (Fig. 6A ▶). This position of LD4 agrees with the prediction that the LD-binding site is a hydrophobic patch on the H2–H3 face of the FAT domain (Fig. 6B ▶). Also, in the structure of the complex, the Ala263Cys spin label is parallel with the binding leucine residues, whereas the Ser273Cys spin label extends at a right angle to the side of the helix (Fig. 6C ▶).
Figure 6.
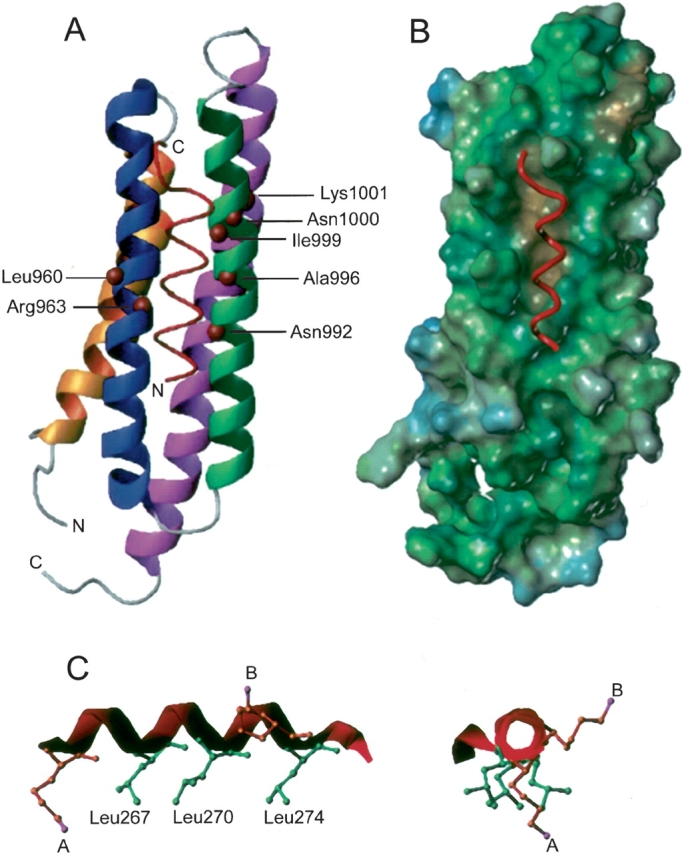
A model of the LD4 motif of paxillin in a complex with the FAT domain of FAK. (A) In the docking of LD4 to the FAT domain, it was assumed that the hydrophobic “leucine face” of the peptide is oriented toward FAT. An upper-limit distance constraint of 10 Å between the amide proton of affected residues and the center of paramagnetism was applied. (B) A surface model of the FAT domain. The hydrophobic patch that is between H2 and H3 and serves as the LD-binding site is in brown, and the LD4 peptide is in red. (C) When LD4 is modeled as an α-helix, the three hydrophobic leucine residues line up on one side of the helix creating a potential binding surface. The two spin-label modifications of Ala263Cys LD4 (A) and Ser273Cys LD4 (B) were estimated by using a lysine residue in which the N atom is the same number of bond lengths away as the unpaired electron on the MTSSL-modified cysteine. Spin-labeled Ser273Cys sticks out to the right of the helix; this position explains the loss of signal only for residues in H3 of FAT.
Discussion
The five LD motifs of paxillin, each of which is in the N-terminal half of the protein, are highly conserved throughout the paxillin superfamily (Tumbarello et al. 2002). On the basis of amino acid sequences, it had been predicted that these LD motifs form amphipathic helices (Sattler et al. 2000). In a previous study, we showed that a chemically synthesized 24-mer peptide whose sequence is identical to that of the LD2 motif of paxillin forms a well-folded α-helical structure in aqueous solution (G. Liu et al. 2002). In the present study, we demonstrated that a synthesized peptide, which corresponds to the LD4 motif, has a strong helical feature in solution and forms a helix when it is in a complex with FAT. We also have data (not shown; G.H. Liu and J. Zheng, unpubl.) that indicates that an LD5 peptide also has a strong helical feature in solution. From these observations, it is reasonable to assume that peptides corresponding to the other two LD motifs, LD1 and LD3, also form helical structure in solution. However, except for the helical elements in the LD motifs, the N-terminal domain of paxillin (residues 51–315) does not appear to be a folded structure in solution (HSQC, CD spectroscopy data not shown). In other words, “beads on a string” is likely the best way to describe the N-terminal domain of paxillin—the five helical LD motifs are the beads connected by unstructured random coils.
The LD motifs of paxillin bind to many different molecules. The flexible nature of the N-terminal domain probably facilitates paxillin’s ability to use different LD motifs to interact with different binding partners. Indeed, the LD2 and LD4 motifs appear to bind simultaneously to the FAT domain at two different sites. Line widths in the HSQC spectrum of the FAT domain–paxillin complex suggest that only a single paxillin molecule interacts with FAT in solution (Fig. 3F ▶). If paxillin were prone to bind to more than one FAT molecule, the resulting complex (≥50 kDa) would result in much broader signals in the HSQC spectrum. The reason for multiple paxillin-binding sites on the FAT domain remains unclear. Undoubtedly, multiple binding sites increase the binding affinity between the two molecules. Perhaps such a feature provides specificity for various binding partners of paxillin utilizing common LD motifs.
The binding affinity of LD2 and LD4 individually to the FAT domain is relatively weak, and the binding of both LD motifs is required to form a stable FAK–paxillin complex. Therefore, the FAK–paxillin complex has potential sites that could be targeted to inhibit complex formation. The FAK–paxillin complex plays an important role in regulation of focal adhesions (Turner 2000). In a migrating cell, the dynamics of focal adhesion formation and disassembly are essential for cell movement. The dissociation of the FAK–paxillin complex could lead to the breakdown of focal adhesions, and such dissociation of the FAK–paxillin complex is most probably achieved by inhibition of the binding of one of the two LD motifs to the FAT domain. Because of the weak interaction between the FAT domain and the individual LD motifs, a disassembly signal, such as that from another type of LD4-binding molecule, would be able to bind to a FAT-bound LD4 motif, thus causing the motif to disassociate from the FAT domain. The consequence of such a binding event would be significant destabilization of the FAK–paxillin interaction and the eventual breakdown of the complex.
Using spin-labeling techniques, we found that the binding site of LD4 is on the surface of the FAT domain of FAK. Furthermore, similar to our previous study of FAT domain–LD2 binding (G. Liu et al. 2002), the present study allowed us to define the orientation of LD4 when it is bound to FAT. Previous reports suggested that both LD2 and LD4 peptides bind to one of the two LD binding sites on the surface of the FAT domain indiscreetly (Hayashi et al. 2002; Gao et al. 2004), but, our evidence clearly demonstrates that LD2 and LD4 peptides preferentially bind to opposite faces of the four-helix bundle of FAT. Furthermore, consistent with earlier reports, we show that the bound LD2 and LD4 peptides lie parallel to the helical bundle of the FAT domain. However, the orientations of the bound peptide had not been known (Hayashi et al. 2002; Gao et al. 2004). In our study, using spin-labeling techniques, we revealed that the two C termini of the bound peptides are both aligned toward the “closed end” of the helix bundle, where the interhelical loops reside. The helices of typical four-helix bundles are staggered at an acute angle of approximately 20° (Fig. 7A ▶). As indicated by the solution structure of the FAT domain (G. Liu et al. 2002), the helices are somewhat distorted: H1 and H4 are almost parallel to each other near the closed end of the bundle. The only other region where the helices are parallel to one another is the region between H2 and H3, which is along the proposed binding site of LD4. This feature may add to the binding affinity of LD motifs that are oriented along the crease formed between these helices and may also explain why no other protein–protein interactions are observed when paxillin binds to the FAT domain.
Figure 7.

A model of paxillin binding to the FAT domain of FAK. The FAT domain is an elongated four-helix bundle with a right-hand twist (A). The LD2 and LD4 motifs of paxillin bind to opposite faces of the four-helix bundle of FAT but are oriented in the same direction (B). The intermediate residues of paxillin remain unstructured in the complex and probably wrap around the H3–H4 side of FAT, although no further binding interactions are observed.
To permit the parallel orientation of the LD motifs, the intermediate paxillin chain would have to wrap around the side of the FAT domain rather than loop over the end. On the basis of the orientation of the helices, we believe that paxillin probably wraps around the H3–H4 side of FAT (Fig. 7B ▶). At this time there is no evidence to support any further binding interactions between paxillin and FAT. However, several potential phosphorylation sites have been identified between the LD2 and LD4 motifs in the paxillin sequence (Bellis et al. 1997; Z.-X. Liu et al. 2002; Huang et al. 2003). It has also been shown that phosphorylation of paxillin alters its binding affinity to FAK (Z.-X. Liu et al. 2002). Undoubtedly, future structural studies of how paxillin phosphorylation affects the formation of the FAK–paxillin complex will yield advantageous insight into the mechanism by which various signaling events regulate focal adhesions.
Materials and methods
Expression and purification of the FAT domain of FAK, paxillin, and the FAT–LD2 construct
The cDNA encoding the FAT domain (residues 916–1053) was subcloned into a pET28a vector. The N-terminal His-tagged FAT domain was subsequently expressed in the Escherichia coli strain BL21(DE3)pLysS. Protein induction, harvest, and purification have been described previously (Wong et al. 2000). To create the FAT–LD2 construct, we inserted oligonucleotide sequence encoding LD2 into a BamHI site downstream of those encoding FAT in the vector pET28a. The oligonucleotide that contained BamHI sites, the LD2 sequence, and 5′ flanking regions encoding the glycine serine repeats (GGS)2GS(GGS)2 (positive and negative strands) were synthesized at the Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital. The cDNA encoding amino acids 51–308 of chicken paxillin was subcloned into a pET28a vector. To generate a pET28a-based construct that encoded amino acids 128–308 of paxillin, we designed oligonucleotide sequences containing 5′ NdeI or 3′ EcoRI sites and amplified the sequences by polymerase chain reaction (PCR). Incorporation of the paxillin-encoding sequence into pET28a was verified by sequencing using a T7 terminator primer. Isotope-labeled protein was prepared by using morpholinepropanesulfonic acid-buffered medium containing 15 NH4Cl (1 g/L) and 13 C6-glucose (2.5 g/L).
LD4 peptide synthesis and spin labeling
The LD4 peptide of chicken paxillin (SATRELDELMASLSD), residues 262–276, and two mutant peptides (Ala263Cys and Ser273Cys) were chemically synthesized by the Hartwell Center. The cysteine-specific spin label MTSSL was purchased from Toronto Research Chemicals. MTSSL was attached to the modified LD4 peptides as described previously (Johansson et al. 1998). In brief, 1 mM of LD4 peptide was stirred for 12 h at room temperature with MTSSL at a ratio of 4:1 in 20 mM sodium phosphate (pH 7.2), 150 mM NaCl, and acetonitrile. Spin-labeled peptide was then purified by reverse-phase high-performance liquid chromatography.
Circular dichroism studies
Circular dichroism spectra were obtained with an Aviv 62DS CD spectrometer (Aviv) and processed by using Igor Pro software (Wavemetrics Inc.). All experiments were performed at room temperature (25°C) by using a quartz cuvette with a 0.1-cm path length. The parameters used for the measurements were as follows: 1-nm step resolution, 10-sec average signaling time, and 1-nm bandwidth. All spectra shown are averages of five scans. Concentration of samples ranged from 20 to 100 μM in 10 mM phosphate buffer (pH 6.5). The total volume of each sample was 400 μL. The CD spectra were expressed as molar ellipticity ([θ]). In the analysis of LD4 folding in response to TFE titration, the helix content was calculated as [θ]222/max[θ]222, where max[θ]222 −-40,000 × [1 - (2.5/n)] and n was the number of amino acid residues (Forood et al. 1993). In the study of LD4 binding to FAT–LD2, the helix content was estimated by using a predictive model developed by Raussens et al. (2003). In the model, ellipticity (E) at each wavelength was normalized to that at 207 nm and the α-helix content = 27.58 − 14.46 × E193 − 5.66 × E2193 + 1.86 × E211− 14.72 × E2211.
NMR spectroscopy
All NMR data were recorded with a Varian Inova 600-MHz and a Bruker Avance AV 800-MHz spectrometer operating at 37°C. Sample concentrations for NMR experiments were typically 1.0–1.5 mM in 10 mM potassium phosphate (pH 6.5), 10% D2O, and 0.1% sodium azide. Spin-labeled LD4 peptides were lyophilized, then dissolved in a solution of 10 mM potassium phosphate. The pH of the spin-labeled peptide solutions was adjusted to 6.5, and these solutions were then titrated into 15N-labeled FAT–LD2 samples in the NMR tube. HSQC spectra were collected at each step of the titration and overlaid to generate a chemical-shift perturbation graph. Backbone assignments of 15N-labeled FAT–LD2 + LD4 were achieved by comparison of the 15N HSQC spectra with previously assigned spectra of 15N-labeled FAT saturated with LD2. Assignments were revised with 3D triple resonance HNCA, HN(CO)CA, CBCANH, and CBCA(CO)NH experiments. The data were processed and displayed by using the programs NMRpipe and NMRDraw (Delaglio et al. 1995) on an SGI Octane workstation. The program SPARKY (T.D. Goddard and D.G. Kneller, University of California, San Francisco) was used for data analysis and assignment.
Structural modeling
The program SYBYL (Sybyl 6.8, Tripos Inc.) was used to build the model of the complex formed by the FAT domain and the LD4 peptide; this model was based upon the previously reported solution structure of FAT (Protein Data Bank accession code 1KTM; G. Liu et al. 2002). LD4 was modeled as an α-helix in the complex, on the basis of the CD data and predicted models of secondary structure. The distance constraints between FAT and LD4 were derived from paramagnetic relaxation effects. An upper-limit distance constraint of 10 Å was assumed between the paramagnetic center of each spin-labeled peptide and amide protons of the FAT domain whose resonances disappeared in the 15N HSQC spectrum upon binding of the spin-labeled peptide. During the fitting, both the backbone conformations of the FAT domain and the peptide were fixed.
Acknowledgments
We thank Dr. J. Thomas Parsons for kindly providing chicken FRNK cDNA and Dr. Christopher Turner for kindly providing chicken paxillin cDNA. We also thank Dr. Weixing Zhang and Dr. William Lewis for technical support, as well as Dr. Julia Cay Jones for editing the manuscript. Dr. Gaohua Liu performed some initial NMR experiments in this study. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), by a grant from the NIH (GM069916), and by grants-in-aid from the American Heart Association (0051073B and 0255413B).
Abbreviations
CD, circular dichroism
FAK, focal adhesion kinase
FAT, focal adhesion-targeting domain
FRNK, FAK-related non-kinase
HSQC, heteronuclear single quantum correlation
MTSSL, (1-oxy-2,2,5,5-tetramethylpyroline-3-methyl)methanethiosulfonate
NMR, nuclear magnetic resonance
NOE, nuclear Overhauser enhancement
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041107205.
Supplemental material: see www.proteinscience.org
References
- Arold, S.T., Hoellerer, M.K., and Noble, M.E. 2002. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 10 319–327. [DOI] [PubMed] [Google Scholar]
- Bellis, S.L., Perrotta, J.A., Curtis, M.S., and Turner, C.E. 1997. Adhesion of fibroblasts to fibronectin stimulates both serine and tyrosine phosphorylation of paxillin. Biochem. J. 325 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini, I., Donaire, A., Luchinat, C., and Rosato, A. 1997. Paramagnetic relaxation as a tool for solution structure determination: Clostridium pasteurianum ferredoxin as an example. Proteins 29 348–358. [PubMed] [Google Scholar]
- Brown, M.C., Perrotta, J.A., and Turner, C.E. 1996. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J. Cell Biol. 135 1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.C., Curtis, M.S., and Turner, C.E. 1998. Paxillin LD motifs may define a new family of protein recognition domains. Nat. Struct. Biol. 5 677–678. [DOI] [PubMed] [Google Scholar]
- Cary, L.A. and Guan, J.L. 1999. Focal adhesion kinase in integrin-mediated signaling. Front. Biosci. 4 D102–D113. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Forood, B., Feliciano, E.J., and Nambiar, K.P. 1993. Stabilization of α-helical structures in short peptides via end capping. Proc. Natl. Acad. Sci. 90 838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, G., Prutzman, K.C., King, M.L., Scheswohl, D.M., DeRose, E.F., London, R.E., Schaller, M.D., and Campbell, S.L. 2004. NMR solution structure of the focal adhesion targeting domain of focal adhesion kinase in complex with a Paxillin LD peptide: Evidence for a two site binding model. J. Biol. Chem. 279 8441–8451. [DOI] [PubMed] [Google Scholar]
- Gaponenko, V., Hwarth, J.W., Columbus, L., Gasmi-Seabrook, G., Yuan, J., Hubbell, W.L., and Rosevear, P.R. 2000. Protein global fold determination using site-directed spin and isotope labeling. Protein Sci. 9 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore, A.P. and Romer, L.H. 1996. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol. Biol. Cell 7 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, I., Vuori, K., and Liddington, R.C. 2002. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol. 9 101–106. [DOI] [PubMed] [Google Scholar]
- Hildebrand, J.D., Schaller, M.D., and Parsons, J.T. 1993. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J. Cell Biol. 123 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoellerer, M.K., Noble, M.E., Labesse, G., Campbell, I.D., Werner, J.M., and Arold, S.T. 2003. Molecular recognition of paxillin LD motifs by the focal adhesion targeting domain. Structure 11 1207–1217. [DOI] [PubMed] [Google Scholar]
- Huang, C., Rajfur, Z., Borchers, C., Schaller, M.D., and Jacobson, K. 2003. JNK phosphorylates paxillin and regulates cell migration. Nature 424 219–223. [DOI] [PubMed] [Google Scholar]
- Ilic, D., Almeida, E.A., Schlaepfer, D.D., Dazin, P., Aizawa, S., and Damsky, C.H. 1998. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 143 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, J.S., Gibney, B.R., Rabanal, F., Reddy, K.S., and Dutton, P.L. 1998. A designed cavity in the hydrophobic core of a four-α-helix bundle improves volatile anesthetic binding affinity. Biochemistry 37 1421–1429. [DOI] [PubMed] [Google Scholar]
- Liu, G., Guibao, C.D., and Zheng, J. 2002. Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol. Cell. Biol. 22 2751–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z.-X., Yu, C.F., Nickel, C., Thomas, S., and Cantley, L.G. 2002. Hepatocyte growth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin-focal adhesion kinase association. J. Biol. Chem. 277 10452–10458. [DOI] [PubMed] [Google Scholar]
- Nolan, K., Lacoste, J., and Parsons, J.T. 1999. Regulated expression of focal adhesion kinase-related nonkinase, the autonomously expressed C-terminal domain of focal adhesion kinase. Mol. Cell. Biol. 19 6120–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons, J.T., Martin, K.H., Slack, J.K., Taylor, J.M., and Weed, S.A. 2000. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 19 5606–5613. [DOI] [PubMed] [Google Scholar]
- Raussens, V., Ruysschaert, J.-M., and Goormaghtigh, E. 2003. Protein concentration is not an absolute prerequisite for the determination of secondary structure from circular dichroism spectra: A new scaling method. Anal. Biochem. 319 114–121. [DOI] [PubMed] [Google Scholar]
- Richardson, A. and Parsons, T. 1996. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature 380 538–540. [DOI] [PubMed] [Google Scholar]
- Sastry, S.K. and Burridge, K. 2000. Focal adhesions: A nexus for intracellular signaling and cytoskeletal dynamics. Exp. Cell Res. 261 25–36. [DOI] [PubMed] [Google Scholar]
- Sastry, S.K., Lakonishok, M., Wu, S., Truong, T.Q., Huttenlocher, A., Turner, C.E., and Horwitz, A.F. 1999. Quantitative changes in integrin and focal adhesion signaling regulate myoblast cell cycle withdrawal. J. Cell Biol. 144 1295–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler, M., Pisick, E., Morrison, P.T., and Salgia, R. 2000. Role of the cyto-skeletal protein paxillin in oncogenesis. Crit. Rev. Oncog. 11 63–76. [PubMed] [Google Scholar]
- Schaller, M.D., Borgman, C.A., and Parsons, J.T. 1993. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol. Cell. Biol. 13 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer, D.D., Hauck, C.R., and Sieg, D.J. 1999. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71 435–478. [DOI] [PubMed] [Google Scholar]
- Shen, Y., Schneider, G., Clouteir, J.F., Veillette, A., and Schaller, M.D. 1998. Direct association of protein-tyrosine phosphatase PTP-PEST with paxillin. J. Biol. Chem. 273 6474–6481. [DOI] [PubMed] [Google Scholar]
- Tachibana, K., Sato, T., D’Avirro, N., and Morimoto, C. 1995. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med. 182 1089–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, J.W., Cooley, M.A., Broome, J.M., Salgia, R., Griffin, J.D., Lombardo, C.R., and Schaller, M.D. 1999. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J. Biol. Chem. 274 36684–36692. [DOI] [PubMed] [Google Scholar]
- Tumbarello, D.A., Brown, M.C., and Turner, C.E. 2002. The paxillin LD motifs. FEBS Lett. 513 114–118. [DOI] [PubMed] [Google Scholar]
- Turner, C.E. 2000. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2 E231–E236. [DOI] [PubMed] [Google Scholar]
- Turner, C.E. and Miranti, C.K. 1994. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motifs and multiple LIM domains: Identification of a vinculin and pp125Fak binding region. J. Cell Sci. 107 1583–1591. [DOI] [PubMed] [Google Scholar]
- Turner, C.E., Brown, M.C., Perrotta, J.A., Riedy, M.C., Nikolopoulos, S.N., McDonald, A.R., Bagrodia, S., Thomas, S., and Leventhal, P.S. 1999. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J. Cell Biol. 145 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, H.C., Mao, J., Nguyen, J.T., Srinivas, S., Zhang, W., Lin, B., Li, L., Wu, D., and Zheng, J. 2000. Structural basis of the recognition of the dishevelled DEP domain in the Wnt signaling pathway. Nat. Struct. Biol. 7 1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]