

Abstract
Identification of protein biochemical functions based on their three-dimensional structures is now required in the post–genome-sequencing era. Ligand binding is one of the major biochemical functions of proteins, and thus the identification of ligands and their binding sites is the starting point for the function identification. Previously we reported our first trial on structure-based function prediction, based on the similarity searches of molecular surfaces against the functional site database. Here we describe the extension of our first trial by expanding the search database to whole heteroatom binding sites appearing within the Protein Data Bank (PDB) with the new analysis protocol. In addition, we have determined the similarity threshold line, by using 10 structure pairs with solved free and complex structures. Finally, we extensively applied our method to newly determined hypothetical proteins, including some without annotations, and evaluated the performance of our methods.
Keywords: structure-based function prediction, hypothetical proteins, structural genomics, protein three dimensional structure
The recent progresses in structural genomics projects is now producing many protein structures before their functions are identified (Brenner 2001). In 1998, only four out of the 2150 proteins deposited in the Protein Data Bank (PDB) (Berman et al. 2000, 2003) were hypothetical proteins, but in 2003, 124 out of 4960 entries were annotated as hypothetical ones. One of the aims of structural genomics projects is to obtain some clues about the functions of proteins based on their structural information in the post–genomics-sequencing era. This concept arises from the well-accepted principle that protein three-dimensional (3D) structures are tightly coupled with their functions, especially the molecular functions. However, it is still unknown how the protein 3D structures correlate with the functions, and thus the identification of protein functions using their structural information remains as an essential issue in the field of structural biology.
A similar problem also exists in the sequence–function relationship, where it is still unknown how the protein sequence determines its function. However, fruitful results have been obtained in the sequence analyses field, by putting the ultimate problem aside and using the indirect but strong correlation between sequence similarity and functional similarity, which is possibly a consequence of evolutionary pressure on functional proteins (Durbin et al. 1998). In the same way, in the structural biology of proteins, proteins with similar structures have been analyzed to gain some inferences on their functions from the structural similarity.
Frequently used approaches are based on the global fold similarity (Holm and Sander 1996; Holm and Park 2000; Thornton et al. 2000). However, it is now being gradually accepted that the level of fold similarity does not always correlate with the functional similarity (Todd et al. 2001), as seen in the observation that a limited number of protein folds are used repeatedly and others are not (Orengo et al. 1994; Holm and Sander 1996; Brenner et al. 1997). Thus, several groups have started to focus their attention on the similarity of local structures in proteins. In these approaches, various types of structure representations are used to define the structural similarity, because the type of structural similarity that correlates well with the functional similarity has not been established. For example, the most straightforward representation is the spatial arrangement of atoms (Kinoshita et al. 1999; Kleywegt 1999), where all atomic positions are used explicitly, and thus if some similarity is detected, it can strongly imply the functional similarity. However, since protein structures are flexible, the explicit use of atomic position could be too sensitive to small structural change. Another approach is to use a structural template to handle the small structural changes observed in the functional sites (Moodie et al. 1996; Wallace et al. 1996, 1997; Dawe et al. 2003). From similar viewpoints, abstract side-chain models could be useful to avoid the explicit position of atoms (Artymiuk et al. 1994). These methods seemed to work fine, but quite interestingly, few similarities were found among proteins with different folds, that is, proteins possibly belonging to different evolutionary origins. In other words, the representation of an explicit or semiexplicit atomic position may not be applicable for detecting a similarity beyond the evolutionary relationship, such as in sequence analyses.
Our goal is to develop a method to predict the molecular functions of proteins from their 3D structures. The aim is to introduce the structural information, and the method should detect the similarity among proteins with different folds. We have reported the first trial of the functional annotation with the hypothetical proteins TT1754 (Handa et al. 2003) and MJ0226 (Kinoshita and Nakamura 2003), where we showed that the molecular surface representation (Connolly 1983) of proteins could be a promising method to detect the similarity beyond the fold levels.
There have been several approaches using the molecular surfaces of proteins. In particular, Wolfson and coworkers extensively used molecular surface representation to search for similar functional sites (Lin et al. 1994; Lin and Nussinov 1996; Rosen et al. 1998; Shulman-Peleg et al. 2004), where they tried to reduce the number of vertices on the molecular surface as much as possible, to enhance the search speed. It is important for the search method to work quickly, but reducing the number of vertices could make the search method insensitive to small differences in the molecular surface, because the representative vertexes can change their positions according to small structural changes.
We now describe an expansion of our previous approaches (Kinoshita et al. 2002; Kinoshita and Nakamura 2003) based on similarity searches of the molecular surfaces. In our approach, no reduction of the vertices was carried out, and all of the surface points were used for the similarity search. Although the heavy calculation required the full molecular surface, the number of functional sites to be searched was small and the application range was limited. Here, we extended the database to be searched to almost all of the heterogeneous atom binding sites that appeared within the PDB, and developed a new analysis method for the search results, to solve some problems arising from the database expansion.
The expansion of the database is not a simple task due to the large variety of ligands appeared in PDB. In the small data set, a single index of similarity and a single threshold may work well as in the previous work. However, in the huge data set, as in this study, one single index of similarity measure is not enough; thus, we should search for some other indices to evaluate the similarity as described later. Furthermore, there are some general problems in the study of protein–ligand interaction using PDB: (1) a ligand in crystal structure may not be a cognate ligand, especially in the enzymatic protein, so the interaction can be different from the physiological one; (2) heteroatoms appeared in PDB could be molecules in the buffer of crystallization, and thus such interaction may not be significant in biological context; and (3) crystal contact may create unphysiological binding site. For the first problem, we tried to use as many ligand binding sites as possible, and thus we did not exclude the proteins redundantly appeared from the viewpoint of sequence similarity, where we intend to incorporate as much alternative interactions appeared in PDB as possible. For the second problem, we excluded the binding sites for the molecules commonly used as the crystallization buffer as described later. The third problem remains unsolved in this study. In addition, it is a simple problem but the larger database requires more computation time. This problem is managed by massive parallel computations in the current study.
Results and Discussion
Similarity score normalization
In this study, we used almost all of the heteroatom binding sites within the PDB as the database to be searched (see Materials and Methods). The similarity search method based on the graph theory is the same as that previously described, where the molecular surfaces are represented by a set of triangular meshes, and the electrostatic potential, curvature, and spatial arrangement of the vertices of the meshes are compared (Kinoshita et al. 2002). In the method, the similarity of two molecular surfaces is evaluated by the number of corresponding vertices. Thus, when we made a comparison between a query protein and a set of functional site patches in a data set, a set of similarity scores was obtained. The set of similarity scores was easily converted into Z-scores by using the mean and the standard deviation of the score distribution. The mean and the standard deviation were calculated for each query protein. In this sense, the Z-score normalized the differences between the query proteins when we used the same functional site database.
The normalization with the Z-score worked fine when the variety of functional site patches was not as large as in the previous small data set (Kinoshita and Nakamura 2003), because the number of corresponding vertexes for each patch, as expected from the size of the patch, was not so different, and the normalization for the size differences of the patches was not required. However, if we used a data set with a large variety of ligand binding sites, as in this study, the differences in the sizes of the functional site patches could cause trouble; that is, large patches tend to yield large Z-scores, which are not satisfactory.
To overcome this difficulty, we introduced another index to evaluate the similarity, that is, the coverage. The coverage is the ratio of the number of corresponding vertexes to that of the vertexes in the functional site patch, and so it ranges from 0.0 to 1.0. The aim of the index is to normalize the difference in the expected number of corresponding vertexes. Actually, through the following applications we observed a strong tendency for the binding sites with large Z-scores (large binding sites) to have small coverage value on average, as seen in the left-lower side of the two-dimensional (2D) plot for the coverage and the Z-score (Fig. 1 ▶). Therefore, a binding site with a larger Z-score and larger coverage is considered as a binding site with higher similarity. Hereafter, we use these two indices, the coverage and the Z-score, and the results are shown in the 2D plot (Figs. 1 ▶, 2 ▶, and so on).
Figure 1.

Threshold lines with 50%, 70%, and 90% true positive constraints. Red crosses indicate the correct answers, and incorrect answers are plotted in the form of density plot.
Figure 2.

Search result with the 2D plots for the coverage and the Z-score for 10 hypothetical proteins. Red, green, and white circles indicate the “similar” binding sites that exceed the 50% TP, 70% TP, and 90% TP lines, respectively. The white line is the 90% TP line, for which we used the loosest threshold line. Each 2D plot conceptually consists of the 26,359 (the number of entries in the search database) dots for each plot, but here the dots below the 90% TP line (a white line) are represented as a contour map of the dots density for clarity. The plots are aligned from the top left to the right in the order that appeared in the text with the PDB identifications. The PDB identifications with yellow color indicate the entries discussed in the text.
Threshold determination
The next problem is to determine the threshold line in the 2D plot, to judge whether the binding sites in the database to be searched are similar. For this purpose, we prepared a learning data set including the 10 representative protein pairs whose free and complex structures were determined. These 10 proteins were selected randomly, so that various ligand sizes were included in the learning data set (see Materials and Methods).
For each entry for the free form of the 10 proteins, we carried out similarity searches against the search database and determined the correct and incorrect answers by analyzing the results as described in Materials and Methods section. In short, the judgment of whether the prediction was correct or not was done by using the following criteria: (1) the distance between the center of gravity of the predicted ligand and that of the ligand in the complex form is <5 Å, and (2) the predicted ligand is “similar” to the known ligand. The definition of the similarity of the ligand was judged manually by inspecting to the heteroatom dictionary in the PDB. (All the correct answers in our learning data set are listed in Supplemental Table S1.)
Figure 1 ▶ shows the distribution of correct and incorrect answers that we determined. To establish the threshold line, we used Matthews’ correlation coefficient (MCC) as an evaluation indicator. The MCC can be calculated by
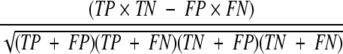 |
where TP, TN, FP, and FN are the number of true positives, true negatives, false positives, and false negatives, respectively. These numbers can be calculated when a threshold line is given. If a correct answer is placed above the given threshold line, it is considered as a true positive, otherwise it is a false negative. There seem to be no guiding principles for determining a particular mathematical function for the threshold line. Thus, we tried several types of threshold lines, and the coverage = a/(Z + b) + c was found to give a good MCC value, where a, b, and c are parameters and Z is the Z-score. The parameters were determined by maximizing the MCC by calculating all possible combinations of the parameters in the range of 0.1–5.0, −2.0–2.0, and −0.3–0.3 for a, b, and c, respectively, with 50 equal intervals, thus 503 times calculations were carried out. However, it should be noted that the number of correct examples (TP + FN) was much smaller than that of incorrect examples (TN + FP) in this case. Therefore, the maximum MCC would be achieved by reducing FP at the expense of decreasing TP. However, this would not be desirable, because our aim was to find as many similar binding sites as possible, even though some false positives would be included in the results. In other words, sufficient numbers of TP should be retained. Therefore, we introduced another constraint in the maximization of the MCC, so that the TP percentage should exceed a given value. Here, we show three results, with 90%, 70%, and 50% TP constraints. Each TP percentage must be larger than each value in the maximization processes. With the constraints, we obtained 0.68, 0.46, and 0.34 MCC values with 50%, 70%, and 90% constraints, respectively. The loosest threshold, the 90% TP line, was used for the following applications, but to evaluate the confidence of the prediction, we used all of the threshold lines.
Newly determined hypothetical proteins
We applied our methods to 18 newly determined, hypothetical proteins. They were selected as described in the Materials and Methods section. The results are summarized in Figure 2A ▶ and Supplemental Figure S1, where similar binding sites above the 50% TP line, 70% TP line, and 90% TP line are indicated by red, green, and white circles, respectively. In addition, all of the detected binding sites are listed in Table 1 for the first four entries and Supplemental Table S2 for the other 14 entries.
Table 1.
Detected similar binding sites with the 90% TP line for four hypothetical proteins
| 1j85 (HI0766) |
| 1: OLA_1j78_0_303 (2.0, 0.578) |
| 2: MAL_1cgx_0_689 (2.1, 0.648) GOL_1f0v_0_906 (1.6, 0.710) BOG_lecz_0_1015 (2.4, 0.626) |
| 3: E09_1gg5_0_702 (4.4, 0.440) ANP_1kor_0_3510 (3.5, 0.364) RH1_1h66_D_1274 (3.1, 0.377) LMT_1qla_Y_8 (3.6, 0.319) UDP_1f6d_0_3377 (3.4, 0.331) GLC_1hgg_I_203 (3.2, 0.434) MIN_1tom_0_1 (3.8, 0.329) NAD_1guz_C_1306 (4.4, 0.271) E09_1gg5_0_701 (3.5, 0.383) |
| 4: MAL_1qhp_0_1291 (2.6, 0.688) GLC_2dij_0_694 (1.8, 0.725) GLF_1cxl_C_695 (1.8, 0.809) GLC_1kcl_C_1698 (2.0, 0.781) GLC_5cgt_0_708 (2.0, 0.800) GLC_1cxk_D_702 (1.9 0.852) MAL_1cdg_0_689 (2.3, 0.689) MAL_1b9z_0_903 (1.9, 0.631) GLC_1eo5_C_700 (1.7, 0.786) GLC_1eo7_C_699 (1.5, 0.764) GLC_1dtu_E_705 (1.6, 0.822) MAL_1qho_0_1201 (2.6, 0.679) ACX_1cxf_0_688 (2.3, 0.632) PG4_1h17_A_9012 (1.8, 0.645) MAL_1cxi_0_688 (1.9, 0.622) GLC_5cgt_0_707 (1.8, 0.745) BOG_1i78_0_700 (1.9, 0.621) |
| 5: FD1_1k1i_0_999 (2.8, 0.436) AMN_1hgj_H_101 (2.7, 0.464) |
| 6: NG6_1hmw_D_710 (2.5, 0.482) FAD_1j9z_0_850 (4.2, 0.319) NAG_1k7t_C_1 (2.2, 0.580) HEM_1mdv_B_110 (3.9, 0.271) |
| 7: CIT_1tet_0_1 (1.9, 0.628) C15-1gzz_B_1067 (1.8, 0.627) |
| 1mp2 (RV1700) |
| 1: BOG_1lpb_0_1 (2.2, 0.528) |
| 2: LI1_1kgb_0_610 (2.8, 0.427) |
| 3: NAG_1at6_0_132 (2.6, 0.475) |
| 4: MAL_1cxi_0_688 (2.0, 0.616) GLC_1cxl_C_696 (1.8, 0.679) GLC_5cgt_0_707 (1.8, 0.725) SBA_1bqi_0_300 (3.0, 0.444) MAL_1cxh_0_688 (2.3, 0.616) MAL_1cgy_0_689 (2.2, 0.611) |
| 1qwk (XH961) |
| 1: FAD_1jra_0_335 (4.3, 0.246) |
| 2: MAN_1kj1_0_309 (1.6, 0.720) |
| 3: NAG_1abr_0_1C (2.1, 0.702) |
| 4: NGA_1lu1_0_1 (2.1, 0.559) |
| 5: XYP_1goq_A_404 (2.1, 0.613) |
| 6: XYS_1b3x_0_607 (1.7, 0.653) |
| 7: Z34_1fjs_0_500 (3.7, 0.300) |
| 8: MAL_1cdg_0_689 (2.1, 0.670) CIT_1erx_0_347 (2.1, 0.692) MAL_1cgy_0_689 (2.3, 0.652) LAT_1it0_0_471 (1.6, 0.693) |
| 9: MES_1aqw_0_2600 (2.2, 0.537) EPE_1pgt_A_1 (2.6, 0.487) |
| 10: MO6_1qh1_0_3007 (2.5, 0.655) MES_19gs_B_4 (2.2, 0.544) |
| 11: NAP_11wi_B_350 (4.4, 0.310) NAP_1lwi_A_350 (4.8, 0.331) NAP_1ah0_0_318 (4.1, 0.296) FAD_1jal_0_1850 (3.9, 0.282) NAP_1k8c_0_3350 (4.9, 0.266) NAP_1afs_B_320 (5.1, 0.323) NAP_1k8c_0_2350 (4.4, 0.247) |
| 12: NOJ_1i75_0_2694 (1.8, 0.724) GLC_1eo7_C_699 (1.4, 0.760) MAL_1cgx_0_689 (2.2, 0.676) PTY_1l8j_0_1 (5.3, 0.186) NAG_1bcs_0_1131 (1.3, 0.811) |
| 1nc5 (yteR) |
| 1: CBI_3eng_0_1 (2.6, 0506) |
| 2: GLC_1d3c_D_707 (1.8, 0.736) |
| 3: GMP_2sar_A_98 (2.7, 0.456) |
| 4: HEM_1d4c_B_603 (3.9, 0.277) |
| 5: PUB_1eyx_L_179 (4.0, 0.353) |
| 6: SUC_1ld8_0_901 (3.0, 0.403) |
| 7: VS2_1f2a_A_300 (3.9, 0.345) |
| 8: BME_3pcn_O_429 (1.5, 0.747) BME_3pcf_N_429 (1.5, 0.733) |
| 9: GLC_1kcl_C_1699 (1.6, 0.709) ACX_1cxf_0_688 (2.1, 0.606) |
| 10: POP_1h52_A_1124 (2.0, 0.607) GNP_1g7t_0_601 (3.3, 0.373) SIA_1qfo_E_201 (2.2, 0.565) |
| 11: ANA_1hgi_I_101 (2.8, 0.457) 2GP_1gmp_B_97 (2.8, 0.443) AI1_1fd7_P_104 (3.2, 0.441) A32_1jqy_L_104 (4.0, 0.420) |
| 12: XYS_1isx_0_961 (2.2, 0.689) TDG_1lt5_G_104 (2.7, 0.430) DMS_1jz7_B_8504 (1.5, 0.720) DMS_1jz2_A_8504 (1.9, 0.609) BOG_1ecz_0_1039 (2.6, 0.629) |
| 13: GDP_1mre_0_901 (2.8, 0.472) ACT_1fs7_0_655 (1.6, 0.683) |
| 14: PT1_1br6_0_301 (2.7, 0.459) A32_1jqy_E_104 (3.2, 0.377) A32_1jqy_V_104 (4.4, 0.461) PEH_1m56_0_3010 (5.4, 0.267) GCR_1efd_0_503 (3.0, 0.401) |
| 15: GLC_5cgt_0_708 (1.6, 0.714) BOG_1ecz_0_1015 (2.0, 0.573) AG7_1cqq_0_501 (3.9, 0.271) 1PG_1i4f_0_502 (2.6, 0.501) ACR_1ded_0_2001 (3.2, 0.520) |
The binding sites found to be similar with the 90% TP line are listed. Each binding site has an ID code with the form of “ligand name,” “pdb-idl,” “chain identifier,” and “residue number” joined with “_.” When the chain identifier is null, it was indicated with “0.” The detected binding sites are clustered according to the position on the molecular surface for convenience. The first column is the cluster number and the numbers in parentheses after each ID code are the Z-score and the coverage, respectively.
All of the proteins are hypothetical, and thus a quantitative assessment of the quality of our prediction is not straightforward. However, in three cases, HI0766 (PDB: 1j85), RV1700 (PDB: 1mp2), and XH961 (PDB: 1qwk), the complex form of the query proteins or that of homologous proteins was also available, which allowed us to evaluate the prediction result. Among 15 other hypothetical proteins, seven (1nc5, 1o6d, 1mw7, 1nog, 1j7g, 1lpl, and 1oz9) had a sufficient number of homologs in the SwissProt Database to infer the functional sites from the sequence conservation, and thus we were able to compare the conserved and predicted sites. However, the remaining eight entries are only open for discussion, and the results are shown as the supplementary materials (Supplemental Table S2; Supplemental Fig. S1).
One of the three complex forms is the protein HI0766 (1j85), which is a hypothetical protein from Haemophilus influenzae. The protein shows sequence similarity to the spoU family, which catalyzes the reaction of S-adenosylmethionine (AdoMet)–dependent tRNA/rRNA methyltransferase, with 20%–30% sequence identity, but it has a shorter polypeptide chain than spoU by ~70 amino acids. Because of the low sequence similarity, HI0776 was considered as a putative relative to spoU or as a hypothetical protein. However, Lim et al. (2003) suggested that this protein is likely to be a member of the spoU family by their X-ray crystallography analyses with and without S-adenosylhomocysteine (AdoHcy), which is a product of the methytransfer reaction using AdoMet. Their structural determination unexpectedly revealed that HI0766 (1j85) has a novel fold and AdoHcy assumes an unusual conformation.
As a result of our similarity search, 38 similar binding sites were found, and they could be classified into seven putative binding sites according to their position on the query protein (Table 1). Among them, several strong similarities (red or green circles in Fig. 2 ▶ of 1j85) were found, but no AdoHcy binding sites were detected. However, the E09 (indolequinone derivatives, 3-hydroxymethyl-5-aziridi-nyl-1-methyl-2-(H-indole-4,7-indione)-propenol) binding site found in NAD(P)H:Quinone oxidoreductase (PDB: 1gg5) showed relatively high similarity (Z = 4.4, coverage = 0.44), as seen in Figure 2 ▶, 1j85, and it was the SAH binding site revealed by Lim and colleagues (Lim et al. 2003). In the same binding site, the possibility of mono-nucleotide binding was also predicted by our methods, from the similarity to the ANP binding site in 1kor (Z = 3.5, coverage = 0.36), the UDP binding site in 1f6d (Z = 3.4, coverage = 0.33), and the NAD binding site in 1guz (Z= 4.4, coverage = 0.27) (Table 1).
As shown in the virtual complex in Figure 3 ▶, our method could successfully predict the position of the binding site but failed to predict the kind of compound that could be bound. To understand why our methods could not find the AdoHcy binding site in the query protein, we examined all of the AdoHcy structures in the PDB, and found that the AdoHcy in HI0766 (1j85) has a unique conformation, as pointed out by Lim et al. (2003). Usually, AdoHcy has an extended conformation, but in HI0766 (1j85) it has a compact conformation. This difference seemed to prevent our methods from predicting the AdoHcy binding site of this protein.
Figure 3.

A real complex form with AdoHcy, a virtual complex predicted with E09, and a similar binding site to the query of HI0776 with E09. (A) Molecular surfaces of the entire structures are shown. Red and blue colors indicate that the electrostatic fields at the molecular surface are negative and positive, respectively. (B) Close-up views of real, putative, and similar binding. The ligands (an AdoHcy, and two E09s) are shown in ball-and-stick models. These figures were generated with MOLSCRIPT (Kraulis 1991).
Another case is for RV1700 (PDB: 1mp2), from Mycobacterium tuberculosis, which is now characterized as an ADP-ribose hydrolase even though the annotation in PDB is still a hypothetical protein. The complex form of this protein with ADP-ribose is available (PDB: 1mqw). Our calculation was done for the free form of this protein, but no significant similarities were observed (Fig. 2 ▶, 1mp2). From a comparison between the free and complex forms, we noticed that in the free form of this protein, one flexible loop is missing due to its high mobility, as indicated by the green fragment in Figure 4 ▶. As we reported previously (Kinoshita et al. 2002), our method might be able to detect the similarities among proteins with localized structural changes showing only a little change of electrostatic potential distribution; however, a large structural change such as a loop disappearing is beyond the scope of our approach. This is a limitation of our method, and the missing loop issue would be a serious problem in this kind of approach. To overcome this problem, some good loop modeling methods will be required for more robust function identification based on the structural information.
Figure 4.

The molecular surfaces and the ribbon models of the complex form (left) and free form (right) of RV1700 (1mp2). The color of the molecular surface is assigned in the same way as in Figure 3 ▶. The ligands with ball-and-stick models on the molecular surfaces are the real ones in the complex form (left) and the predicted ones for the free form (right). Green circles in the surface models and green parts in the left ribbon model are the missing regions in the free form. These figures were generated with MOLSCRIPT (Kraulis 1991).
In the case of XH961 (PDB: 1qwk), a FASTA search (Pearson 1994) with a 10−5 E-value threshold have yielded 27 homologous proteins in the PDB as the complex forms with NADP (nicotinamideadenine-dinucleotide phosphate), with sequence identities of ~37%–40%. In these ranges of sequence similarity, the folds of the protein are very similar, but the side-chain conformations are variable and thus molecular surface can be changed. As a result of our similarity search, four binding entries among the 27 candidates were detected as having similar binding sites. Furthermore, our methods detected a similarity (Z = 3.9, coverage = 0.28) to the FAD binding site within NADPH-cytochrome P450 oxidoreductase (PDB: 1ja1), which has a completely different fold according to the SCOP classification (SCCS: c.1.7 for 1qwk homolog and b.43.1 for 1ja1) (Lo Conte et al. 2002). In 1ja1, FAD is bound to the position adjacent to NADP, but our method detected a similarity to FAD binding site rather than the site for NADP, which is the putative ligand inferred from the homology. It might be possible that this similarity is false, because large ligands such as FAD and HEM tend to appear in the region with a high Z-score and low coverage, as discussed later. However, it may also be possible that there is a FAD binding site near the NADP binding site in this hypothetical protein, because FAD often works with NADP in oxidoreductase reactions.
Among the other seven hypothetical proteins, three proteins (1nc5, 1o6d, and 1mw7) have distinctive homologs in the sequence database.
The conserved hypothetical protein yteR is taken from Bacillus subtilis (PDB: 1nc5), and is annotated as an unknown protein in the NCBI Entrez/Protein database (http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=protein&val=16080064). However, from the structural viewpoint, this protein is considered to be a member of the Six-hairpin glycosyltransferase superfamily, and a weak sequence similarity to the catalytic domain of cellulases was reported in the SCOP database (Lo Conte et al. 2002). Furthermore, our sequence analysis of this protein identified some conserved residues. According to the similarity search with our methods, there were 35 significant similarities that could be classified into 15 clusters in their positions on the molecular surface. Among them, the one with high similarity (Z = 4.4, coverage = 0.46) to the BMSC-10 ((3-nitro-5-(3-morpholin-4-yl-propylamino-carbonyl)-phenyl)- galactopyranoside) binding site on heat-labile enterotoxin (LT; PDB: 1jqy) may be promising, because LT (1jqy) is known to interact with ganglioside, which is a similar compound to similar cellulose and is located on the surface of human intestinal epithelial cells (BMSC-10 is an inhibitor of this interaction), and because the conserved residues found in yteR (1nc5; 88D, 132H, 136Y, 141W, 143D, 189H, 211W, 213R, 217W, 340Y) are located in the vicinity of the putative BMSC-10 binding site.
In the case of 1o6d, the BCX-1812 (3-(1-acetylamino-2-ehyl-butyl)-4-guanidino-2-hydroxy-cyclopentanecarboxylic acid) binding site in 1l7g was found to be similar (Z = 4.5, coverage = 0.44) to the surface of 1o6d in the vicinity of the conserved site constructed by Gly100, Gly104, and Ser120. In 1mw7, the N-benzyl-e-(α-d-galactos-1-yl)-benzamide binding site in 1fd7 was found to be similar (Z = 3.5, coverage = 0.47) to the surface of 1mw7 near the conserved site consisting of Gly125, Leu127, and Phe131.
Four other cases (1nog, 1j7g, 1lpl, 1oz9) are predicted as sugar binding sites by our methods and the conservation analysis. The significance of sugar binding sites will be discussed later. The other eight entries are orphan hypothetical proteins, and thus an evaluation of our prediction is difficult. Therefore, the results of the remaining eight entries are attached as supplementary materials, Supplemental Figure S1 and Supplemental Table S2, to be open for discussion.
As seen in these examples, our prediction tended to find several putative binding sites on the molecular surfaces of the query proteins. Usually some of them can be judged to be correct in the cases where the complex form of the query protein or a protein homologous to the query protein is known, but the other cases are ambiguous. This ambiguity comes from the lack of biochemical function information. It should be noted that the biochemical function of a protein is such a function for which an experimental assay has been carried out. In other words, there is no evidence that the protein has no other functions. For example, our search methods often detected sugar binding sites in many proteins. They are usually found in the region with the low Z-score and high coverage value. It is true that no experimental support is available for most of the sugar binding sites, but the situation is the same in the case that sugars could not bind to the proteins. In contrast, the heme binding sites tend to be found in the region with the high Z-score and low coverage, and are believed to bind specifically. Therefore, our threshold line in that region may need to be improved with a larger learning data set, because there are only a few true answers in the regions, as seen in Figure 1 ▶.
In summary, the application of our method to the newly determined hypothetical proteins worked well in five cases (1j85, 1qwk, 1nc5, 1o6d, and 1mw6), failed in one case (1mp2), and yielded unknowns in 12 other cases. Besides these applications, as reported previously (Handa et al. 2003; Kinoshita and Nakamura 2003), our methods have some potential to make promising predictions for hypothetical proteins. However, several problems still exist. The large structural change and the missing loop issues were pointed out above. The other problem is the biased variety of ligands in the structural database, which has too many sugar and sugar derivatives. It will be necessary to construct a database that contains representative binding sites not from the viewpoint of the sequence homology but from that of the tertiary structures of proteins.
Materials and methods
Search database
The binding sites of all heteroatoms, except for metal, PO4, SO4, and modified residues (or covalently attached heterogens such as sugars), are considered as the database to be searched. It is because the small molecules are often used in the crystallization buffer whose interactions with the proteins may not be realistic, and because the covalent bond between covalently linked molecules and proteins would disturb the other interactions between the molecule and proteins. The database was prepared from the X-ray crystallographic entries with ≥2.5 Å resolution in the PDB (January 2003 release), and 26,359 binding sites appeared in the data set, which is available through the eF-site database (Kinoshita and Nakamura 2004).
Learning data set
To construct the learning data set, we first picked all the protein pairs with free and complex structures determined by X-ray crystallography with ≥2.5Å resolution, where the free structures are those without any heterogeneous atoms other than water, SO4, PO4, Cl, Na, and modified residues such as selenomethionine. Furthermore, we picked up the proteins registered as single chain to avoid the problem of finding the correspondence between several chains in the calculation of the sequence identity. The correspondence between the free and complex structures was determined with the 100% sequence identity and ≥95% alignment coverage. The representative entries were then selected according to the sequence comparison, with the threshold of 30% sequence identity and 80% alignment coverage. As a result, we identified 192 representative pairs in January 2004 from the PDB. In order to select the various ligand sizes in the learning data set, we sorted the 192 representatives pairs according to the number of atoms in the ligand and selected one in every 20 from the list. Finally, we obtained 10 entries as the learning data set, whose PDB IDs are 1af9-1d0h (NGA), 1ah6-1am1 (ADP), 1cz1-1eqc (CTS), 1gta-1gtb (PZQ), 1qj9-1qj8 (C8E), 1qlq-1g6x (EDO), 1xaa-1hex (NAD), 2plc-1aod (INS), 3app-1bxo (GOL), and 3thi-4thi (PYD). The three-letter code in the parentheses is the heteroatom name assigned in each PDB entry.
Clustering according to the position of the predicted ligand
Usually, several tens of similarities to the known binding sites were detected with our method. To reduce the redundancy, we carried out a cluster analysis according to the position of the predicted ligand. The position of the putative ligand is represented by the center of gravity of the ligand after the superimposition, according to the correspondence of the molecular surface. The clustering analysis was carried out by the single linkage clustering algorithm, and the final clusters were identified with a 5 Å threshold; that is, no further cluster joints would be carried out once all of the distances among the existing clusters exceeded 5 Å. The distance between a pair of clusters was measured by the minimum distance between the members of the two clusters.
Assignments of correct answers in the learning data set
To assign correct answers for each free structure in the search database, we first carried out the similarity searches by surface similarity for each free structure followed by the clustering as described above with the temporary threshold line with the step function form:
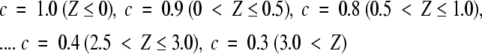 |
At the same time, the sequence similarity searches against the same search database using FASTA (Pearson 1994) were carried out. Then we identified such clusters that include the homologous proteins with similar ligands to that appeared in the complex form. The similarity of the ligand was judged according to the ligand name that appeared in the heteroatom dictionary in the PDB. For the entries in the clusters, we manually checked the similarity and determined if the entries are correct or not. All entries that we assigned as correct are shown in the Supplemental Table S1.
Newly determined hypothetical proteins
We have selected the newly determined hypothetical proteins according to the following criteria; that is, the proteins were (1) released after 2003, (2) resolved by X-ray crystallography with ≥2.5 Å resolution, (3) free from ligand, and (4) had the monomeric structure available in January 2004. We obtained 23 entries. Four of them were a membrane protein, RNA binding proteins (two cases), and a lipid binding protein, which were excluded. One pair, 1iuk and 1iul, is identical, so 1iul was excluded from the final list due to its poorer resolution than that of 1iuk. Finally, we picked up 18 hypothetical proteins from the PDB: 1iuk, 1j27, 1j7g, 1j85, 1jhs, 1lpl, 1mp2, 1mw7, 1nc5, 1nfj, 1ng6, 1nig, 1nij, 1nog, 1o50, 1o6d, 1oz9, and 1qwk. It should be noted that these hypothetical proteins were selected by just a keyword search, so some proteins have been annotated, as in the case of 1mp2, as discussed in the Results and Discussion.
Conservation analysis
The sequence conservation analysis was done by the recent version of an evolutionary trace method (Mihalek et al. 2004). Similar sequences were searched with BLAST (Altschul et al. 1997) with the E-value threshold of 10−5. Multiple sequence alignments were constructed with the clustalW (Higgins et al. 1996). The top 20% of residues with a high degree of importance in terms of entropy measurement were selected as the conserved residues (Mihalek et al. 2004).
Supplementary material
Supplemental materials are a table of entries in the learning data set as “correct” answer (Supplemental Table S1), a table of search results for orphan hypothetical proteins (Supplemental Table S2), and a 2D plot for orphan hypothetical proteins (Supplemental Fig. S1).
Acknowledgments
We thank Eiji Kanamori for the implementation of the recent version of the evolutionary trace method. This work was supported by a grant from MEXT to K.K. Development of the binding sites databases as a part of the eF-site database has been supported by a grant-in-aid from the Institute for Bioinformatics Research and Development, Japan Science and Technology Agency (BIRD-JST) to H.N.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041080105.
Supplemental material: see www.proteinscience.org
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artymiuk, P.J., Poirrette, A.R., Grindley, H.M., Rice, D.W., and Willett, P. 1994. A graph-theoretic approach to the identification of three-dimensional patterns of amino acid side-chains in protein structures. J. Mol. Biol. 243 327–344. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H., Henrick, K., and Nakamura, H. 2003. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 10 980. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E. 2001. A tour of structural genomics. Nat. Rev. Genet. 2 801–809. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Chothia, C., and Hubbard, T.J. 1997. Population statistics of protein structures: Lessons from structural classifications. Curr. Opin. Struct. Biol. 7 369–376. [DOI] [PubMed] [Google Scholar]
- Connolly, M.L. 1983. Solvent-accessible surfaces of proteins and nucleic acids. Science 221 709–713. [DOI] [PubMed] [Google Scholar]
- Dawe, J.H., Porter, C.T., Thornton, J.M., and Tabor, A.B. 2003. A template search reveals mechanistic similarities and differences in β-ketoacyl synthases (KAS) and related enzymes. Proteins 52 427–435. [DOI] [PubMed] [Google Scholar]
- Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. 1998. Biological sequence analysis: models of proteins and nucleic acids. Cambridge University Press, Cambridge, United Kingdom.
- Handa, N., Terada, T., Kamewari, Y., Hamana, H., Tame, J.R., Park, S.Y., Kinoshita, K., Ota, M., Nakamura, H., Kuramitsu, S., et al. 2003. Crystal structure of the conserved protein TT1542 from Thermus thermophilus HB8. Protein Sci. 12 1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins, D.G., Thompson, J.D., and Gibson, T.J. 1996. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 266 383–402. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Park, J. 2000. DaliLite workbench for protein structure comparison. Bioinformatics 16 566–567. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. Mapping the protein universe. Science 273 595–603. [DOI] [PubMed] [Google Scholar]
- Kinoshita, K., and Nakamura, H. 2003. Identification of protein biochemical functions by similarity search using the molecular surface database eF-site. Protein Sci. 12 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2004. eF-site and PDBjViewer: database and viewer for protein functional sites. Bioinformatics 20 1329–1330. [DOI] [PubMed] [Google Scholar]
- Kinoshita, K., Sadanami, K., Kidera, A., and Go, N. 1999. Structural motif of phosphate-binding site common to various protein superfamilies: All-against-all structural comparison of protein-mononucleotide complexes. Protein Eng. 12 11–14. [DOI] [PubMed] [Google Scholar]
- Kinoshita, K., Furui, J., and Nakamura, H. 2002. Identification of protein functions from a molecular surface database, eF-site. J. Struct. Funct. Genomics 2 9–22. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. 1999. Recognition of spatial motifs in protein structures. J. Mol. Biol. 285 1887–1897. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of proteins structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Lim, K., Zhang, H., Tempczyk, A., Krajewski, W., Bonander, N., Toedt, J., Howard, A., Eisenstein, E., and Herzberg, O. 2003. Structure of the YibK methyltransferase from Haemophilus influenzae (HI0766 (1J85)): A cofactor bound at a site formed by a knot. Proteins 51 56–67. [DOI] [PubMed] [Google Scholar]
- Lin, S.L. and Nussinov, R. 1996. Molecular recognition via face center representation of a molecular surface. J. Mol. Graph. 14 78–90, 95–77. [DOI] [PubMed] [Google Scholar]
- Lin, S.L., Nussinov, R., Fischer, D., and Wolfson, H.J. 1994. Molecular surface representations by sparse critical points. Proteins 18 94–101. [DOI] [PubMed] [Google Scholar]
- Lo Conte, L., Brenner, S.E., Hubbard, T.J., Chothia, C., and Murzin, A.G. 2002. SCOP database in 2002: Refinements accommodate structural genomics. Nucleic Acids Res. 30 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalek, I., Res, I., and Lichtarge, O. 2004. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J. Mol. Biol. 336 1265–1282. [DOI] [PubMed] [Google Scholar]
- Moodie, S.L., Mitchell, J.B., and Thornton, J.M. 1996. Protein recognition of adenylate: An example of a fuzzy recognition template. J. Mol. Biol. 263 486–500. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Jones, D.T., and Thornton, J.M. 1994. Protein superfamilies and domain superfolds. Nature 372 631–634. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. 1994. Using the FASTA program to search protein and DNA sequence databases. Methods Mol. Biol. 24 307–331. [DOI] [PubMed] [Google Scholar]
- Rosen, M., Lin, S.L., Wolfson, H., and Nussinov, R. 1998. Molecular shape comparisons in searches for active sites and functional similarity. Protein Eng. 11 263–277. [DOI] [PubMed] [Google Scholar]
- Shulman-Peleg, A., Nussinov, R., and Wolfson, H.J. 2004. Recognition of functional sites in protein structures. J. Mol. Biol. 339 607–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, J.M., Todd, A.E., Milburn, D., Borkakoti, N., and Orengo, C.A. 2000. From structure to function: Approaches and limitations. Nat. Struct. Biol. 7(Suppl) 991–994. [DOI] [PubMed] [Google Scholar]
- Todd, A.E., Orengo, C.A., and Thornton, J.M. 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307 1113–1143. [DOI] [PubMed] [Google Scholar]
- Wallace, A.C., Laskowski, R.A., and Thornton, J.M. 1996. Derivation of 3D coordinate templates for searching structural databases: Application to Ser-His-Asp catalytic triads in the serine proteinases and lipases. Protein Sci. 5 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, A.C., Borkakoti, N., and Thornton, J.M. 1997. TESS: A geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases: Application to enzyme active sites. Protein Sci. 6 2308–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]