

Abstract
Amide hydrogen exchange (HX) in combination with mass spectrometry (MS) is a powerful tool to analyze the folding and dynamics of proteins. In the traditional methodology the exchange time is controlled by manual pipetting, thereby limiting the time resolution to several seconds. Some conformational changes in proteins, however, occur in the subsecond time scale, making it desirable to perform HX at shorter time intervals down to the limit set by the intrinsic chemical exchange rate. We now report the development of the first completely on-line quenched-flow setup that allows the performance of HX experiments in the 100-sec to 30-sec time scale, on-line proteolytic digestion using immobilized proteases, rapid desalting, and MS analysis. We show that conformational fluctuations in the range of seconds can be detected and protection factors as small as 10 reproducibly determined. Using this setup we investigated the conformational properties of Escherichia coli heat-shock transcription factor σ32 free in solution. Our results indicate that the C-terminal σ4 domain of σ32, which is responsible for the recognition of the −35 region of heat shock promoters, contains more extensive secondary structure than expected when compared with the structure of the homologous σ-factor σA in complex with the RNA-polymerase. This setup should be very useful for a more accurate analysis of structural motions in proteins in the subsecond to second time scale relevant to allostery and enzyme function.
Keywords: quenched flow, amide hydrogen exchange, protein conformation, protein folding, mass spectrometry
As more and more structural information on proteins becomes available, the focus of research interests shifts to the dynamics of conformational changes within proteins. Time-resolved X-ray crystallography and NMR, which are used to address these questions, have their own specific limitations. Very often flexible parts of proteins do not crystallize or are not visible in the electron density maps, and rapid conformational changes of protein domains are very difficult to visualize in crystallography (Hajdu et al. 2000; Schlichting 2000; Moffat 2001). Analysis by NMR is generally limited to proteins not larger than 50 kDa; it requires relatively high protein concentrations, and NMR cannot resolve coexisting conformations. An alternative approach is amide hydrogen exchange (HX) in combination with mass spectrometry (MS) (Engen and Smith 2000; Hoofnagle et al. 2003). In recent years this approach has become increasingly popular because of the high sensitivity allowing the investigation of proteins which tend to aggregate at high concentrations, the accessibility of flexible structures, the greatly extended size range compared with NMR, and the possibility to detect coexisting conformations. The spatial resolution of this method is limited by the size (on average 10–15 residues) of peptides generated after the exchange reaction by proteolytic cleavage under quench conditions, but is currently improved by different methods (Kaltashov and Eyles 2002).
The conformational properties of proteins in their native state are typically investigated by HX by incubating the proteins in deuterated buffer at neutral pH for different time intervals with subsequent acidification, desalting, and MS analysis (Engen and Smith 2001). Since the samples are handled manually, exchange times < 10 sec are from a practical point of view very difficult to obtain accurately. Conformational changes and transient structural fluctuations occurring on this time scale can therefore not be resolved. Protein folding studies are typically carried out in a quenched-flow instrument with pulse-labeling at high pH (≥ 9.5) for short time intervals (typically 10 msec) and automated quenching of the reaction. The subsequent steps, i.e., peptic digestion, desalting, and MS analysis, are performed off-line (Konermann and Simmons 2003). Proteins in a denaturing buffer are diluted into a refolding buffer and allowed to refold for various periods of time whereafter a short pulse at high pH labels regions still unfolded. The off-line desalting and proteolytic digestion required in this method make sample handling more labor intensive and limit reproducibility. Quenched-flow pulse-labeling with on-line electrospray ionization (ESI) MS analysis, which is so far only used to investigate full-length proteins, was hampered by the salt sensitivity of the ESI-MS analysis, and therefore restricted to proteins dissolved in buffers with low ionic strength (i.e., at nonphysiological conditions) and without metal ions essential for many nucleotide binding proteins.
We report here a quenched-flow rapid-desalting MS setup for on-line HX with exchange times of 0.1–30 sec, on-line proteolytic digestion, desalting and ESI-MS analysis.
Results and Discussion
Quenched-flow setup
To analyze fast conformational changes in proteins we developed a quenched-flow system that allows the monitoring of amide hydrogen exchange in time intervals from 100 msec to 30 sec. Shorter exchange times, though possible with our setup, are not amenable because amide hydrogen exchange at neutral to moderately basic pH and physiological temperatures is then limited by the intrinsic chemical exchange rate (kch) and not only by accessibility of the amide hydrogen. Our setup consists of five HPLC pumps, two 10-port valves, an injection valve, a trap column for rapid desalting, an optional column with immobilized pepsin for on-line proteolytic digestion, and an optional analytical column for peptide separation before analysis by ESI-MS as shown in Figure 1 ▶. The sample, delivered by pump 1 from the injection valve, is diluted 1:25 in a mixing tee with D2O-buffer, delivered by pump 2, thereby starting the exchange reaction, which continues in the delay line. The exchange reaction is quenched by addition of quench buffer, delivered by pump 3, in a second mixing tee. The protein is subsequently trapped on a reversed-phase microtrap column and, after switching the first 10-port valve, desalted with 0.05% trifluoroacetic acid in water, delivered by pump A. After switching the second 10-port valve, the sample is eluted with an acetonitrile gradient, generated by binary pump B, over an analytical microbore reversed-phase column directly into the electrospray ion source of the mass spectrometer. For localization of the exchanging regions within the protein a column with an immobilized pro-tease, e.g., pepsin, is inserted into the system before the trap column. The duration of the exchange reaction can be adjusted to the desired times by the choice of the delay line length and inner diameter. To limit back-exchange during proteolytic cleavage and desalting, the entire setup downstream of the delay line is submerged in an ice bath.
Figure 1.
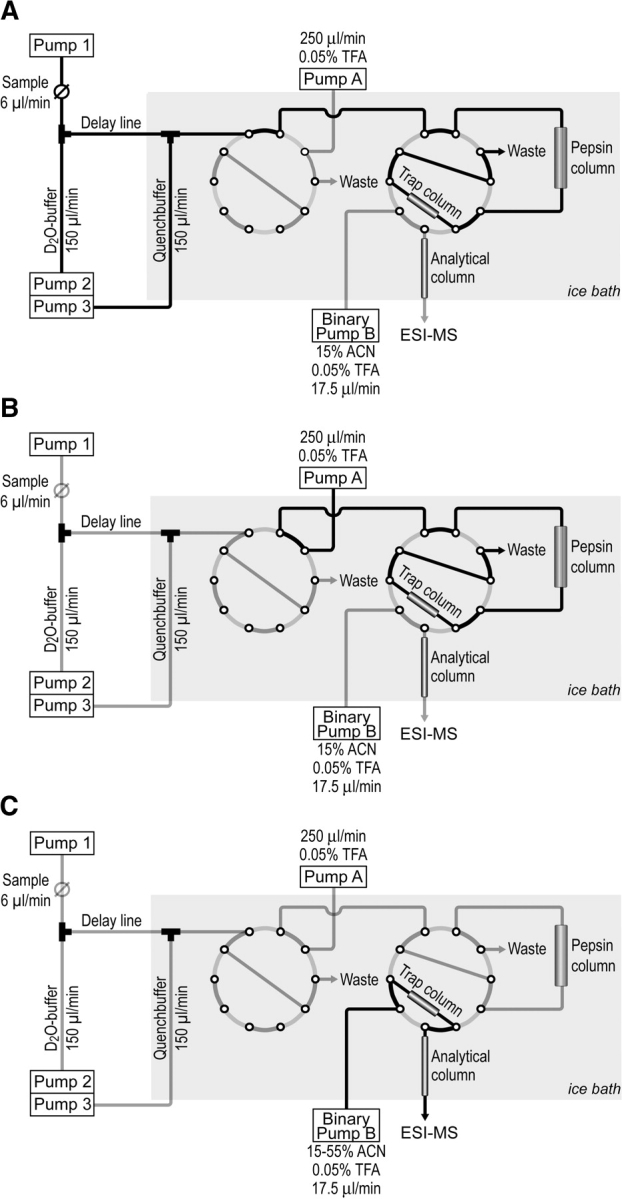
Schematic representation of the novel HPLC-MS quenched-flow setup. (A) HX position; (B) desalting position; and (C) MS analysis position.
When all pumps have reached the desired flow rates the exchange experiment is started by switching the injection valve to the inject position. This triggers a program that automatically switches the two 10-port valves at the preset time points. This full automation of the setup guarantees a high degree of reproducibility.
Performance of the quenched-flow system
To test our setup we analyzed deuteron incorporation into native apo-myoglobin at decreasing time intervals and compared the results with samples of manually performed HX reactions. Between 30 sec and 5 sec, both sample-handling procedures yielded identical results (Table 1). Below 5 sec, reproducible manual sample handling was very difficult, and below 2 sec impossible; however, gradual deuterium incorporation was still observed with the quenched-flow setup revealing the fluctuations within the protein in the millisecond-to-second time scale.
Table 1.
Reproducibility of manual and quenched-flow amide hydrogen exchange
| Exchange time (sec) | Off-line | On-line |
| 1 | — | 16,992.4 ± 0.7 |
| 2 | — | 16,997.8 ± 0.9 |
| 5 | 17,006.3 ± 0.5 | 17,005.4 ± 0.7 |
| 10 | 17,013.0 ± 0.7 | 17,011.0 ± 0.7 |
| 30 | 17,023.9 ± 0.7 | 17,022.2 ± 0.1 |
Apo-myoglobin was diluted 1:25 in D2O buffer by manual mixing or using
the quenched-flow setup and deuteron incorporation determined after different time intervals. Desalting and analysis was identical for both methods.
Since sample loading into the quenched flow system took about 50 sec (5 μL at 6 μL/min flow-rate at pump 1), part of the sample remains slightly longer on the trap column compared with the sample obtained from the manually performed initiation and quenching of isotopic exchange. This results in a somewhat higher deuterium loss in the quenched flow system because of the prolonged exposure to protiated solvent. However, due to the short time interval between the quench of the exchange reaction and the transition of the sample into the vacuum, the overall retention of incorporated deuterons was close to 90%. Supplementary Figure 1 ▶ shows some representative mass spectra of full-length myoglobin.
Figure 2 ▶ shows the time course of deuteron incorporation into apo-myoglobin at pH 7.6 (left panels) and 8.5 (right panels) in comparison with the intrinsic chemical amide hydrogen exchange (dashed lines) as calculated for each amide hydrogen of the entire protein using the HXPep program (courtesy Z. Zhang) (Bai et al. 1993). A triple exponential rate equation was fitted to the measured and calculated data. The shortest time constant for the measured deuteron incorporation could not be fitted and was set to the value determined for the intrinsic chemical exchange time constant.
Figure 2.

Quenched-flow amide hydrogen exchange of full-length apo-myoglobin. Amide hydrogen exchange of apo-myoglobin (100 pmol) was measured at pH 7.6 (left) and 8.5 (right) for 0.1 sec to 30 sec and compared to the overall intrinsic chemical exchange (dashed curves) as determined for each amide using the HXPep program (courtesy Z. Zhang). The solid line represents the fit of a triple exponential rate equation (y = A∞− A1•exp(−k1•t) − A2•exp(−k2•t) − A3•exp(−k3•t)) to the data whereby for the fastest rate k1 the value derived from a fit to the intrinsic chemical exchange data was used. The lower panels show a zoom of the first 2 sec of the exchange reaction.
The fit parameters indicated that at pH 7.6 31 deuterons were incorporated into apo-myoglobin with a time constant shorter than 0.1 sec marking the number of completely exposed amide hydrogens. Seventeen deuterons were incorporated with a time constant of 1.7 sec, revealing relatively fast fluctuations in amide hydrogen accessibility most likely in the form of opening and closing H-bonds close to the protein surface. An additional 29 deuterons were incorporated with a time constant of 22 sec. The total number of exchanging amide hydrogens at pH 7.6 as calculated from the fitting parameters was 77. Since apo-myoglobin has 147 exchangeable amide hydrogens about 50% of the amide hydrogens are completely protected at the time scale investigated. Our results for the longest incubation times are similar to published data (Johnson and Walsh 1994).
At pH 8.5, the intrinsic chemical exchange exceeds 98% already after 250 msec; any measured slower exchange therefore indicates protection by structural elements. As indicated by the fitting parameter, 47 deuterons were incorporated with a time constant < 0.1 sec, 28 amide hydrogens exchanged with a time constant of 0.8 sec and additional 75 protons with a time constant of ~70 sec. The total number of exchanging amide hydrogens at pH 8.5 as calculated from the fitting parameters was 150, which is very similar to the total number of exchangeable amide hydrogens in apo-myoglobin. At pH 8.5, deuteron incorporation was therefore faster and more extensive than at pH 7.6, indicating pH-dependent structural fluctuations in apo-myoglobin. The increased rate of deuteron incorporation at higher pH is caused by two effects. First, the accelerated intrinsic chemical exchange rate at higher pH increases the probability of exchange in any transient structural opening event. Second, the flexibility of apo-myoglobin increases at higher pH, consistent with published data (Haouz et al. 1998).
Taken together, these data demonstrate that our quenched-flow setup can accurately measure the deuterium incorporation into full-length proteins within the time interval limited by kch and the manual pipetting speed.
Detection of protected amides in the three-helix bundle of σ32
To test the performance of our quenched-flow setup including the on-line peptic digestion we analyzed the HX of the E. coli heat-shock transcription factor σ32 for which only a homology model onto the structure of Thermus thermophilus σA bound to RNA polymerase exists. In our recent study on temperature-dependent conformational changes of σ32 we found good overall consistency between the structural model of σ32 and HX data, except in the C terminus, where the structural model shows α-helices while the HX data did not show any protection (Rist et al. 2003). This inconsistency could be explained either by the incorrectness of the model or by a highly dynamic nature of the α-helices in this region with opening and closing kinetics in the second time range. The shortest exchange times used in the reported experiments were 12 sec, and therefore a dynamic structure of the α-helices would have remained undetected. Since this region is involved in the recognition of the −35 region of heat-shock promoters, its structural properties are important for the biological function of σ32. We were therefore interested in analyzing HX in σ32 in the 0.1-sec to 30-sec time scale.
Almost complete digestion of σ32 was achieved with a 16 μL column packed with immobilized pepsin (< 5% of the protein remained undigested). The small volume results in a very short residence time for the sample molecules in the pepsin column (~ 3.1 sec), and any deuterium loss or gain will be negligible relative to the deuterium loss during the chromatographic separation with the analytical column (retention times were between 8 and 12 min). Deuterium loss during desalting and analytical chromatography was between 13% and 18%. Supplementary Figure 2 ▶ shows some representative mass spectra of peptic fragments of σ32.
When HX of σ32 was analyzed in our setup and the measured deuteron incorporation compared with the kch of the individual peptide segments we found that a total of at least 30 amide protons in the C-terminal region are still protected after 1 sec and at least 25 after 5 sec. Figure 3 ▶ shows the kinetics of amide hydrogen exchange of individual peptide segments under native state conditions in comparison with the intrinsic chemical exchange kinetics of corresponding unstructured peptides. While in some peptide segments (amino acids 209–244,1 240–253, 255–263, 259–272, and 258–294) (Figs. 3 ▶ and 4 ▶, upper panel; data not shown) significant protection was observed over a time interval of 1–5 sec, other peptide segments (amino acids 184–199 and 201–208) exchanged almost all amide protons for deuterons within 0.5 sec. The determined protection factors of the intermediately exchanging amide hydrogens were 10.0 14.4, 33.9, 9.3, and 35.9 for the peptides 209–244, 240–253, 255–263, 259–272, and 258–294, respectively. Repetitive determination of the m/z values for the different peptides at the individual time points of HX was within 0.1 m/z units demonstrating the high reproducibility.
Figure 3.
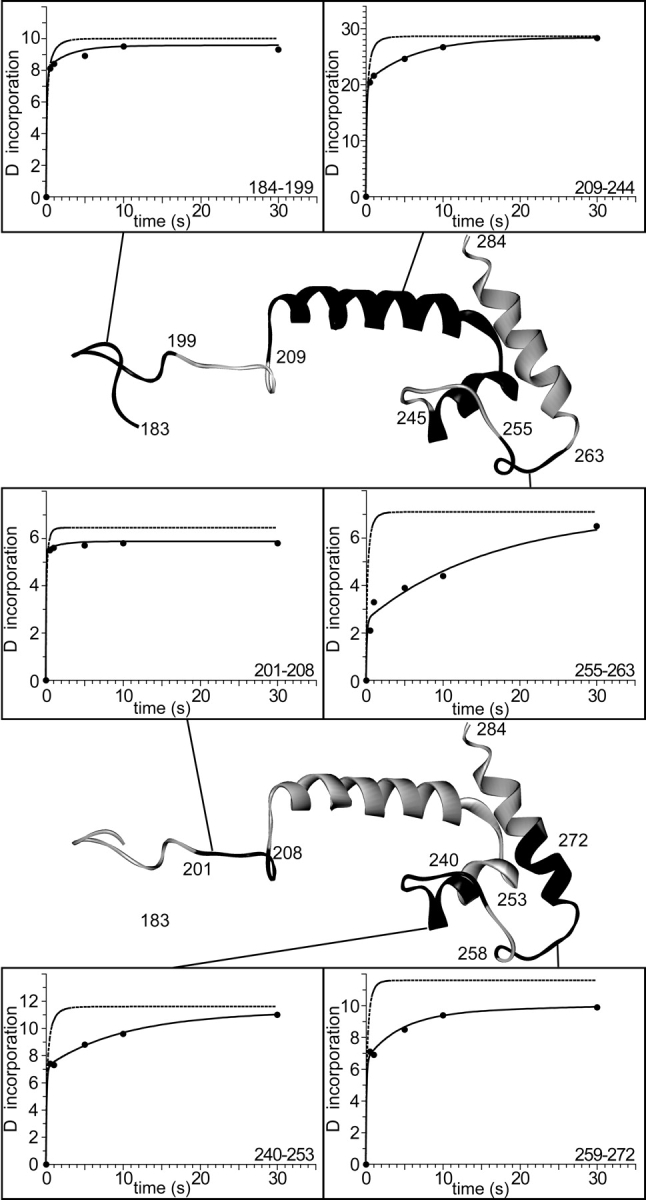
Amide hydrogen exchange kinetics of peptide segments within the E. coli heat-shock transcription factor σ32. Incorporation of deuterium into σ32 was measured using quenched-flow with on-line peptic digestion. The exchange kinetics is shown for six different peptides [indicated in the structural model of σ32(183–284) in black] and compared to their individual intrinsic chemical exchange kinetics (dashed line). The solid line represents the fit of a triple exponential rate equation (y = A∞ − A1•exp(−k1•t) − A2•exp(−k2•t) − A3•exp(−k3•t)) to the data whereby the values for the fastest rate, k1, and for the total number of exchangeable amides, A∞, were derived from a fit to the intrinsic chemical exchange data.
Figure 4.

Comparison of expected and observed amide hydrogen exchange in the C-terminal half of σ32. (Upper panel) Number of protected amide hydrogens ([number of hydrogens exchanging according to intrinsic chemical exchange rate after 30 sec] − [observed incorporated deuterons at the given time point]) after 0.5, 1, 5, 10, and 30 sec. The number over the columns indicates the number of observable protons, i.e., the number of protons exchanged to equilibrium in the corresponding unfolded peptide according to the intrinsic chemical exchange rate, taking into account back-exchange during desalting. The numbers under the abscissa indicate the amino acid numbers of the peptides analyzed minus the first residue, which does not have an amide group. (Lower panel) Number of potential hydrogen bonds in the individual peptide stretches as calculated from the distances between amide nitrogen and nearest carbonyl oxygen in the structural model. Gray bars give the number of distances smaller than 3.2 Å; white bars, distances smaller than 3.0 Å. Distances were calculated using the Insight II program (Accelrys).
Using our model we determined the number of possible hydrogen bonds involving backbone amides by measuring the distances between the amide nitrogens and corresponding carbonyl oxygens using the insight II program (Accelrys). The lower panel of Figure 4 ▶ gives the number of potential hydrogen bonds (nitrogen–oxygen distance < 3.0 Å and < 3.2 Å, respectively) and compares these with the number of protected amide hydrogens after different exchange times (Fig. 4 ▶, upper panel). In most peptides originating from the C-terminal part of σ32 we observed a higher degree of protection of amide hydrogens than expected from the structural model using 3 Å or 3.2 Å as cutoff criterion for a stable hydrogen bond, indicating more extensive secondary structure. In one peptide the opposite was observed; less amide hydrogens were protected than expected from the model. This discrepancy could be due to the fact that σ32 was modeled onto the RNA polymerase bound from of σA. Free in solution this region may exhibit more extensive secondary structure. Our quenched-flow setup therefore allowed a close test of the structural model of σ32 and it will be interesting to analyze σ32 in complex with RNA polymerase and DNA.
Conclusion
We present here a novel quenched-flow setup that allows the HX experiments with exchange times as short as 100 msec. Thereby incubation times between the limitations set by manual sample handling and the intrinsic chemical exchange rate at physiological pH are accessible in a highly reproducible manner. Compared to previous published systems (Dharmasiri and Smith 1996; Simmons and Konermann 2002; Simmons et al. 2003), our setup has the advantage that all steps are performed automatically and on-line from the start of the HX reaction, over the digestion with immobilized proteases and the desalting, to the mass spectrometer. The analysis time is thereby minimized to < 3 min if no analytical reversed-phase separation of the peptides is necessary, or to < 12 min including an analytical reversed-phase column. The back-exchange is thereby minimized and the results are highly reproducible.
This setup should find a wide range of applications. It is very suitable for native state HX allowing the accurate determination of protection factors (kch/kobs) as small as 10, corresponding to structural fluctuations with a free energy barrier of around −6 kJ/mol (Bai et al. 1995a,b; Clarke and Itzhaki 1998). This setup would also allow the detection of induced short-term conformational changes due to ligand binding according to an induced fit mechanism or to enzyme catalysis. For this purpose the ligand/substrate could be present in the D2O buffer and encounter the enzyme at the same moment as D2O. Similarly, ligand binding sites or dimer interfaces of interaction partners with dissociation rates as high as 1 sec−1 could be mapped (HD footprinting). Even protein folding studies could be performed with this quenched-flow setup to analyze early folding processes. Acid denatured proteins could be mixed with D2O buffer that at the same time initiate refolding and HX. Since the initial hydrophobic collapse occurs in the microsecond time scale, the amide protons that are protected in the “molten globule” state should not exchange to any significant degree because the collapse is much faster than kch. However, the subsequent folding processes were amenable for analysis without the problem that is imposed by the stabilization through the native state. The detection and characterization of off-path intermediates that are lost in the generally used pulse-labeling studies should be possible. The ability of the MS analysis to detect coexisting conformations at the same time without averaging is the prerequisite for such a study. Even later stages of the protein folding could be analyzed by small modifications of our setup. The use of an additional pump and a second delay line and mixing tee would expand the system to allow the separation of the initiation of re-folding from the start of the HX reaction.
Materials and methods
σ32 was prepared as described previously (Tomoyasu et al. 2001). D2O (99.9%) was from Cambridge Isotope Laboratories, Poroszyme Immobilized Pepsin and Poros R1 from Applied Biosystems, myoglobin and other chemicals from Sigma.
On-line quenched-flow amide hydrogen exchange setup
The setup employed to measure HX in a millisecond time scale consisted of five HPLC pumps (one Agilent 1100 Series Capillary pump [A], one Agilent 1100 Series Binary pump [B], one Rheos 2000 Micro pump [1], and two Shimadzu LC-10ADVP pumps [2 and 3]), a Rheodyne injection valve (Model 8125) with a 5-μL stainless steel sample loop, and two Valco 2-position/10-port valves with microelectric actuators (Model C2-1000EP6). A schematic drawing of the setup is shown in Figure 1 ▶. Protein samples (100–200 pmol in < 5 μL) were injected and pushed by pump 1 with a flow rate of 6 μL/min toward a mixing tee, which was also connected with pump 2 delivering D2O-buffer (25 mM HEPES, pD 7.6, 50 mM KCl, and 5 mM MgCl2) with a flow rate of 150 μL/min. Hence, the injected samples were diluted 1:25 in D2O uffer and pushed through the delay line where amide hydrogen exchange occurred. The exchange time was adjusted by the volume of the delay line (e.g., 2.6 μL ≡ 1 sec). The exchange reaction was quenched in a second mixing tee by 1:1 dilution of the sample with quench buffer (0.5 M KH2PO4/H3PO4, pH 2.2) delivered by pump 3 with a flow rate of 150 μL/min. The quenched sample was then pushed through a 16-μL pepsin column (1 mm ID × 20 mm) and immediately trapped on a reversed-phase column (0.8 mm ID × 3 mm, Poros 10 R1).
For the desalting of trapped peptic peptides, the left two-position/10-port valve was switched, and 0.05% TFA was delivered by pump A (250 μL/min). After 1 min, the right two-position/10-port valve was switched to elute the desalted peptic peptides from the trap column over a 0.75 mm ID × 6 cm analytical reversed-phase column packed with Zorbax 300SB-C8 (3.5-μm particles) directly into the electrospray source. The solvent for elution was delivered by pump B with a flow rate of 17.5 μL/min using the following short gradient: buffer A (0.05% TFA) to buffer B (90% ACN, 0.05% TFA), having the profile: %B: 15 → 55 (0 → 10 min), %B: 55 → 100 (10 → 11 min), %B: 100 → 15 (11 → 12 min). The last eluted peptide was detected 12 min after sample injection. All lines delivering quench buffer or quenched sample were immersed in an ice bath to minimize back-exchange. For kinetic measurements of the full-length proteins the pepsin column was omitted.
Off-line amide hydrogen exchange experiments
Amide hydrogen exchange was initiated by a 25-fold dilution of 100 pmol apo-myoglobin into D2O containing 25 mM HEPES, pD 7.6, 50 mM KCl, and 5 mM MgCl2 at room temperature. After different elapsed times (from 1 to 30 sec), the exchange reaction was quenched by decreasing the temperature to 0°C and the pH to 2.2 with 500 mM phosphate buffer. Quenched samples were loaded via the injection valve and pushed through the pepsin column by pump A. Hereafter, the experimental setup was as described above.
Mass spectrometry and data treatment
Electrospray ionization mass spectra were acquired on a quadrupole time-of-flight instrument (QSTAR Pulsar, ABI SCIEX). Molecular masses of the proteins were calculated from the ESI mass spectra using the Bayesian Reconstruct tool of the BioAnalyst software (ABI SCIEX). For full-length proteins, spectra were externally calibrated using apo-myoglobin as standard.
Peptic peptides of σ32 were identified on the basis of their MS/MS spectra. The deuterium content of the peptides was calculated by using the average mass difference between the isotopic envelopes of deuterated and undeuterated peptides. The average masses were determined with the MagTran software (Zhang and Marshall 1998).
Acknowledgments
We thank B. Bukau for continuous support of the project. This work was supported by grants of the Deutsche Forschungsgemeinschaft (SFB638 to M.P.M., Leibnizprogram to B. Bukau) and by the Fonds der Chemischen Industrie.
Abbreviations
ESI, electrospray ionization
HPLC, high-pressure liquid chromatography
HX, amide hydrogen-(1H/2H)-exchange
kch, intrinsic chemical exchange rate
kobs, observed exchange rate
MS, mass spectrometry
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041098305.
Supplemental material: see www.proteinscience.org
Footnotes
All numbers of peptides are based on the observable exchange and therefore exclude the N-terminal amino acid, which does not have a backbone amide.
References
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, Y., Englander, J.J., Mayne, L., Milne, J.S., and Englander, S.W. 1995a. Thermodynamic parameters from hydrogen exchange measurements. Methods Enzymol. 259 344–356. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995b. Protein folding intermediates: Native-state hydrogen exchange. Science 269 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, J. and Itzhaki, L.S. 1998. Hydrogen exchange and protein folding. Curr. Opin. Struct. Biol. 8 112–118. [DOI] [PubMed] [Google Scholar]
- Dharmasiri, K. and Smith, D.L. 1996. Mass spectrometric determination of isotopic exchange rates of amide hydrogens located on the surfaces of proteins. Anal. Chem. 68 2340–2344. [DOI] [PubMed] [Google Scholar]
- Engen, J.R. and Smith, D.L. 2000. Investigating the higher order structure of proteins. Hydrogen exchange, proteolytic fragmentation, and mass spectrometry. Methods Mol. Biol. 146 95–112. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Investigating protein structure and dynamics by hydrogen exchange MS. Anal. Chem. 73 256A–265A. [DOI] [PubMed] [Google Scholar]
- Hajdu, J., Neutze, R., Sjogren, T., Edman, K., Szoke, A., Wilmouth, R.C., and Wilmot, C.M. 2000. Analyzing protein functions in four dimensions. Nat. Struct. Biol. 7 1006–1012. [DOI] [PubMed] [Google Scholar]
- Haouz, A., Glandieres, J.M., Zentz, C., Pin, S., Ramstein, J., Tauc, P., Brochon, J.C., and Alpert, B. 1998. Solvent effects on horse apomyoglobin dynamics. Biochemistry 37 3013–3019. [DOI] [PubMed] [Google Scholar]
- Hoofnagle, A.N., Resing, K.A., and Ahn, N.G. 2003. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32 1–25. [DOI] [PubMed] [Google Scholar]
- Johnson, R.S. and Walsh, K.A. 1994. Mass spectrometric measurement of protein amide hydrogen exchange rates of apo- and holo-myoglobin. Protein Sci. 3 2411–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltashov, I.A. and Eyles, S.J. 2002. Crossing the phase boundary to study protein dynamics and function: Combination of amide hydrogen exchange in solution and ion fragmentation in the gas phase. J. Mass Spectrom. 37 557–565. [DOI] [PubMed] [Google Scholar]
- Konermann, L. and Simmons, D.A. 2003. Protein-folding kinetics and mechanisms studied by pulse-labeling and mass spectrometry. Mass Spectrom. Rev. 22 1–26. [DOI] [PubMed] [Google Scholar]
- Moffat, K. 2001. Time-resolved biochemical crystallography: A mechanistic perspective. Chem. Rev. 101 1569–1581. [DOI] [PubMed] [Google Scholar]
- Rist, W., Jørgensen, T.J.D., Roepstorff, P., Bukau, B., and Mayer, M.P. 2003. Mapping temperature-induced conformational changes in the Escherichia coli heat shock transcription factor σ32 by amide hydrogen exchange. J. Biol. Chem. 278 51415–51421. [DOI] [PubMed] [Google Scholar]
- Schlichting, I. 2000. Crystallographic structure determination of unstable species. Acc. Chem. Res. 33 532–538. [DOI] [PubMed] [Google Scholar]
- Simmons, D.A. and Konermann, L. 2002. Characterization of transient protein folding intermediates during myoglobin reconstitution by time-resolved electrospray mass spectrometry with on-line isotopic pulse labeling. Biochemistry 41 1906–1914. [DOI] [PubMed] [Google Scholar]
- Simmons, D.A., Dunn, S.D., and Konermann, L. 2003. Conformational dynamics of partially denatured myoglobin studied by time-resolved electrospray mass spectrometry with on-line hydrogen-deuterium exchange. Biochemistry 42 5896–5905. [DOI] [PubMed] [Google Scholar]
- Tomoyasu, T., Arsene, F., Ogura, T., and Bukau, B. 2001. The C terminus of σ32 is not essential for degradation by FtsH. J. Bacteriol. 183 5911–5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. and Marshall, A.G. 1998. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 9 225–233. [DOI] [PubMed] [Google Scholar]