

Abstract
Tumors induced in the rat by 1,2-dimethylhydrazine (DMH) contain mutations in β-catenin, but the spectrum of such mutations can be influenced by phytochemicals such as chlorophyllin (CHL) and indole-3-carbinol (I3C). In the present study, we determined the mutation status of β-catenin in more than 50 DMH-induced colon tumors and small intestine tumors, and compared this with the concomitant expression of β-catenin mRNA using quantitative real-time RT-PCR analysis. In total, 19/57 (33%) of the tumors harbored mutations in β-catenin, and 14/19 (74%) of the genetic changes substituted amino acids adjacent to Ser33, a key site for phosphorylation and β-catenin degradation. These tumors were found to express a 10-fold range of β-catenin mRNA levels, independent of the β-catenin mutation status and phytochemical exposure, i.e. CHL or I3C given post-initiation. However, β-catenin mRNA levels were strongly correlated with mRNA levels of c-myc, c-jun and cyclin D1, which are targets of β-catenin/Tcf signaling. Tumors with the highest levels of β-catenin mRNA often had over-expressed β-catenin protein, and those with lower β-catenin mRNA typically had low β-catenin protein expression, but there were exceptions (high β-catenin mRNA/low β-catenin protein, or vice versa). We conclude that DMH-induced mutations stabilize β-catenin protein in tumors, which increase c-myc, c-jun and cyclin D1, but there also can be over-expression of β-catenin itself at the mRNA level, contributing to high β-catenin protein levels. Similar findings have been reported in primary human colon cancers and their liver metastases, compared with matched normal-looking tissue. Thus, further studies are warranted on the mechanisms that upregulate β-catenin at the transcriptional level in human and rodent colon cancers.
Keywords: β-Catenin/Wnt signaling, DMH, Colon cancer, Chlorophyllin, Indole-3-carbinol
1. Introduction
There is growing interest in the β-catenin/T-cell factor (TCF)/lymphoid enhancer factor (LEF) signaling pathway and its role in human cancer development [1]. β-Catenin is a cadherin-binding protein that also functions as a transcriptional activator when complexed in the nucleus with members of the TCF/LEF family [2]. Cytosolic β-catenin interacts with APC, Axin, glycogen synthase kinase-3β (GSK-3β) and other protein partners, leading to phosphorylation of Ser33, Ser37, Thr41 and Ser45 residues in the N-terminal region of β-catenin, followed by ubiquitination and proteosomal degradation [1–3]. In primary human colon tumors and colorectal cancer cell lines, mutations in CTNNB1 substitute one of the four critical Ser/Thr residues and stabilize β-catenin, leading to accumulation of β-catenin/TCF complexes in the nucleus, and activation of downstream target genes [4–6].
Mutations in β-catenin also have been detected in colon tumors of animals treated with chemical carcinogens, such as 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), 2-amino-3-methylimidazo[4,5-f] quinoline (IQ), azoxymethane, 1,2-dimethylhydrazine (DMH), and methylazoxymethanol acetate plus 1-hydroxyanthraquinone [7–11]. In fact, there are a number of similarities between human and rat colon tumors with respect to the β-catenin pathway. First, as in the human situation, colon tumors in the rat contain mutations in Apc or Ctnnb1, but not in both of these genes [7]. Second, β-catenin/Tcf downstream targets frequently are over-expressed, including c-Myc, c-Jun and cyclin D1 [9,11]. Third, genetic changes in human CTNNB1 or murine Ctnnb1 substitute amino acids within the GSK-3β regulatory domain of β-catenin. However, whereas the vast majority of β-catenin mutations in human colon cancers substitute critical Ser/Thr residues directly, in rat colon tumors the mutations often localize to two CTGGA ‘hotspot’ sequences and substitute amino acids adjacent to Ser33 [7–11].
To complicate matters, the spectrum of β-catenin mutations can be influenced by exposure to dietary phytochemicals, such as chlorophyllin (CHL) and indole-3-carbinol (I3C). The latter compound is found in cruciferous vegetables [12–14], whereas CHL is a water soluble derivative of chlorophyll, the ubiquitous pigment in green, leafy vegetables [15–17]. In the tumors from rats given DMH or IQ alone, virtually all of the β-catenin mutations substituted amino acid residues adjacent to Ser33, whereas in animals given carcinogen followed by I3C or CHL, β-catenin mutations more often substituted one of the critical Ser/Thr residues [9]. Subsequent work [18] showed that amino acid substitutions adjacent to Ser33 retard, rather than completely block, the proteasome degradation pathway, leading to a range of β-catenin protein expression levels in colon tumors. High levels of β-catenin and c-Jun were detected in tumors that contained mutations affecting Ser45 or Thr41 of β-catenin, tumors with genetic changes substituting Gly34 and Asp32 had intermediate levels of β-catenin and c-Jun, and the lowest levels of β-catenin and c-Jun were observed in tumors with wild type β-catenin [11].
In the latter experiments, it was assumed that each specific mutation in β-catenin affected its relative stability and turnover at the protein level, the degree of nuclear trafficking and interaction with Tcf/Lef transcription factors, and thus the extent to which downstream target genes became activated. However, in the present investigation of more than 50 DMH-induced tumors in the rat, we observed that β-catenin frequently was over-expressed at the mRNA level. This suggested transcriptional dysregulation of β-catenin, as distinct from the β-catenin protein stabilization reported before [11]. Expression of β-catenin mRNA was correlated with cyclin D1, c-myc and c-jun mRNA levels, but not with the mutation status of β-catenin, or treatment with I3C or CHL.
2. Materials and methods
2.1. Source of tumors
Tumors were from a study in which male F344 rats were initiated during the first 5 weeks with DMH (20 mg/kg body weight, by subcutaneous injection), and treated post-initiation with 0.1, 0.01 or 0.001% CHL in the drinking water, or with 0.1, 0.01 or 0.001% I3C in the diet. Full details were provided in the original report [19].
2.2. Mutation screening
A subset of 57 DMH-induced small intestine and colon tumors, from each of the treatment groups in the original study, was screened for mutations in β-catenin. The methodology for PCR-based single strand conformation polymorphism (PCR-SSCP) analysis coupled with direct sequencing was reported elsewhere [9].
2.3. Quantitative real-time RT-PCR (qPCR)
Frozen samples of tumor and normal tissue were thawed and the mRNA was extracted using the RNeasy kit (Qiagen, Valencia, CA). RNA (2 μg) was reverse-transcribed in 20 μl of 1 × RT buffer, containing 10 U RNase inhibitor (Invitrogen, Carlsbad, CA), 0.5 mM each dNTP, 4 U Omniscript Reverse Transcriptase (Qiagen) and 50 ng random hexamers (Invitrogen). β-Catenin, cyclin D1, c-myc, c-jun and activator protein-2α (AP-2α) mRNA levels were measured by qPCR and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primer sequences were as shown in Table 1. Forty cycles of PCR (95 °C/10 s, 58 °C/20 s, 72 °C/20 s) were run on an Opticon Monitor 2 system (Finnzymes, Finland), in 20 μl total reaction volume containing cDNAs, SYBR Green I dye (DyNAmo master solution, Finnzymes) and primer set. The amount of specific mRNA was quantified by determining the point at which the fluorescence accumulation entered the exponential phase (Ct), and the Ct ratio of the target gene to GAPDH was calculated for each sample. Three separate experiments were performed for each tumor sample, and the corresponding results were expressed as mean ± S.E. in the figures.
Table 1.
Primer sequences for qPCR
| Primers | Nucleotide sequence | Size (bp) |
|---|---|---|
| β-Catenin F | 5′-CGA GGA CTC AAT ACC ATT CC-3′ | 250 |
| β-Catenin R | 5′-AGC CGT TTC TTG TAG TCC TG-3′ | |
| Cyclin D1 F | 5′-CGC CTT CCG TTT CTT ACT TCA-3′ | 251 |
| Cyclin D1 R | 5′-AAC TTC TCG GCA GTC AGG GGA-3′ | |
| c-myc F | 5′-TCAA GAG GCC ACA GCA AAC-3′ | 274 |
| c-myc R | 5′-AAA AGC TAC GCT TCA GCT CG3-′ | |
| c-jun F | 5′-CCG GCT AGA GGA AAA AGT GA-3′ | 135 |
| c-jun R | 5′-TGA GTT GGC ACC CAC TGT TA-3′ | |
| AP-2α F | 5′-TCC GAT CCC AAC GAG GAA GT-3′ | 170 |
| AP-2α R | 5′-GAA GTG GGT CAA GCA ACT CT-3′ | |
| GAPDH F | 5′-ATG GGA GTT GCT GTT GAA GTC A-3′ | 77 |
| GAPDH R | 5′-CCG AGG GCC CAC TAA AGG-3′ |
2.4. Immunoblotting
β-Catenin and β-actin protein expression levels in tumors were examined using the immunoblot methodology described in detail elsewhere [11].
2.5. Statistics
For multiple group comparisons the data (mean ± S.E.) were compared by ANOVA (Waller–Duncan K-ratio t-test), and Student’s t-test was used for paired comparisons. Linear regression analyses were performed using SigmaPlot 8.0.
3. Results
3.1. Confirmation of a mutational ‘hotspot’ involving codons 32 and 34
In the present investigation, 16 tumors from the DMH control group, 25 tumors from the DMH + CHL group, and 22 tumors from the DMH + I3C group were screened using PCR-SSCP analysis (Fig. 1). One of the samples lacked any of the bands for the wild type, indicating that the tumor was likely to be homozygous for the corresponding β-catenin mutation. Sequencing of this tumor identified a mutation in codon 32 of β-catenin (GAT → AAT, D32N). Table 2 shows the complete list of β-catenin mutations, confirmed by sequencing. One-third of the tumors (19/57 = 33%) contained a β-catenin mutation, and 14/19 (74%) were localized to the ‘hotspot’ region involving codons 32 and 34 (Fig. 2). The remaining mutations (5/19 = 26%) substituted one of the critical Ser/Thr residues in β-catenin directly, namely codon 33 TCT → CCT (S33P), codon 37 TCT → TTT (S37F), codon 41 ACC → ATC (T41I, two cases), and codon 45 TCC → TTC (S45F). The pattern of β-catenin mutations in this subset of DMH-induced tumors was consistent with that reported previously for the larger group of tumors, which included colon, small intestine, and liver tumors induced by IQ [9,11].
Fig. 1.

DMH-induced tumors in the rat were screened for β-catenin mutations using PCR-based single strand conformation polymorphism (PCR-SSCP) analysis. Each lane marked with an arrow had an altered band pattern compared with wild type (WT) β-catenin, and sequence analysis confirmed the mutation in β-catenin as affecting codon: (a) 32, (b) 33, (c) 34, (d) 37, (e) 41 or (f) 45. One sample (asterisk, *) lacked any of the bands from the corresponding WT control, and was homozygous for D32N mutant β-catenin (see Table 2 for mutations confirmed by sequencing).
Table 2.
Summary of β-catenin mutations in DMH-induced tumors
| Codon | Mutations in β-catenin | Number of Mutations | Amino acid substitutions |
|---|---|---|---|
| 32 | GAT → AAT | 4 | D32N |
| → GTT | 1 | D32V | |
| 33 | TCT → CCT | 1 | S33P |
| 34 | GGA → GAA | 6 | G34E |
| → AGA | 2 | G34R | |
| → GTA | 1 | G34V | |
| 37 | TCT → TTT | 1 | S37F |
| 41 | ACC → ATC | 2 | T41I |
| 45 | TCC → TTC | 1 | S45F |
| 19/57 (33.3%) |
Fig. 2.
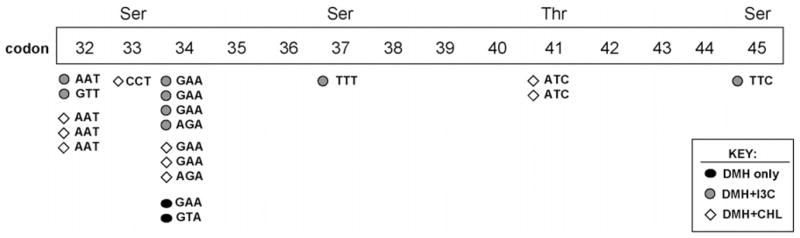
Summary of β-catenin mutations in DMH-induced tumors. An apparent mutational ‘hotspot’ around codons 32 and 34 has been reported in prior studies with colon carcinogens in the rat [7–11], substituting amino acids adjacent to Ser33, which is a critical target for phosphorylation and β-catenin protein degradation.
3.2. β-Catenin mRNA expression varies markedly among DMH-induced tumors
Tumors that had been screened for β-catenin mutation status were further examined for β-catenin mRNA expression. After normalizing to GAPDH, there was a ~ 10-fold range of β-catenin mRNA expression in tumors that were WT or mutant for β-catenin (Fig. 3, upper and lower panels). Collectively, the β-catenin/GAPDH mRNA expression ratios were 2.14 ± 0.17 versus 2.00 ± 0.16 in tumors WT and mutant for β-catenin, respectively (mean ± S.E., P > 0.05, not statistically significant).
Fig. 3.
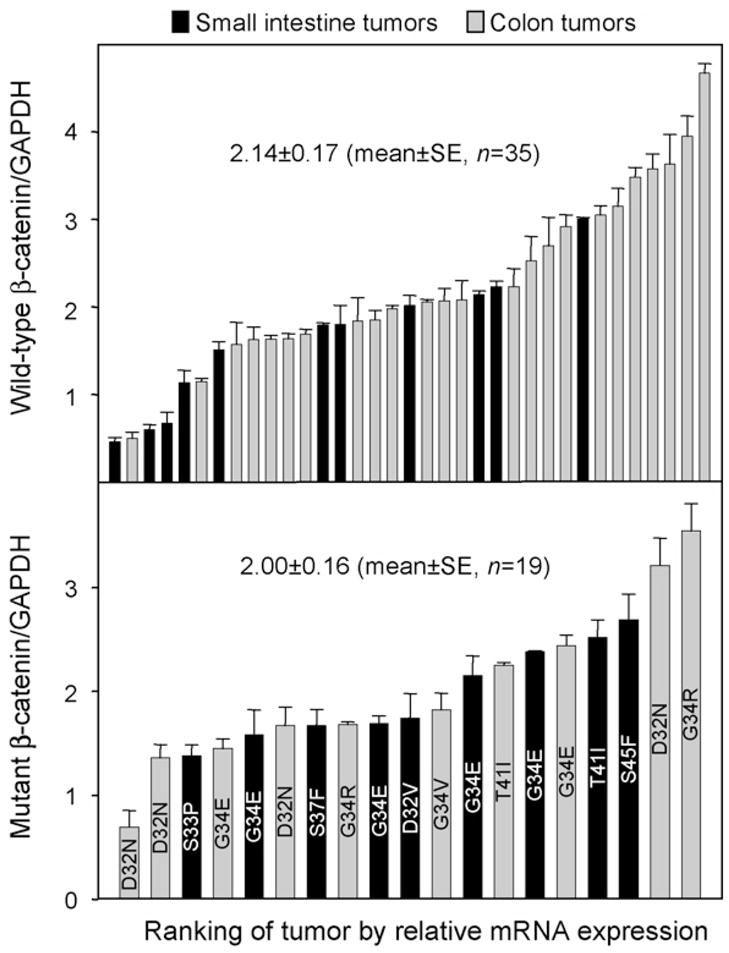
β-Catenin mRNA expression varies markedly in DMH-induced tumors. Each tumor was screened by qPCR, and β-catenin mRNA expression was normalized to the corresponding levels for GAPDH. Data bars (mean ± S.E., n = 3) were arranged from lowest to highest mRNA expression for small intestine tumors (black bars) and colon tumors (gray bars), and according to whether β-catenin was wild type (upper panel, n = 35) or mutant (lower panel, n = 19). The lower panel also shows each amino acid substitution identified in β-catenin (e.g. D32N).
3.3. β-Catenin mRNA was correlated with cyclin D1, c-myc and c-jun mRNA levels
We next examined the mRNA expression levels for three reported β-catenin/Tcf downstream targets (Fig. 4). A highly significant correlation was observed for cyclin D1, c-myc, and c-jun mRNA expression when plotted as a function of the corresponding β-catenin mRNA levels, and this was independent of the β-catenin mutation status (compare open and closed symbols in each panel of Fig. 4). No correlation was seen for AP-2α and β-catenin at the mRNA level (Fig. 4, lower right panel); AP-2α recently was shown to inhibit β-catenin/Tcf signaling via the formation of a nuclear AP-2α/Apc/β-catenin protein complex [20].
Fig. 4.

β-Catenin mRNA expression was correlated with cyclin D1, c-myc and c-jun mRNA levels in DMH-induced tumors. qPCR was used to examine DMH-induced tumors and normal mucosa for expression of three reported β-catenin/Tcf downstream targets, as well as AP-2α, a negative regulator of β-catenin/Wnt signaling [20]. In each case, mRNA expression was first normalized to the housekeeping gene, GAPDH. Open symbols (○), tumors containing WT β-catenin; closed symbols (●), tumors with mutant β-catenin.
3.4. β-Catenin mRNA expression was unrelated to CHL or I3C treatment
The β-catenin mRNA results were examined as a function of carcinogen and phytochemical exposure (Fig. 5). Low levels of β-catenin mRNA were detected in normal mucosa from untreated control rats, whereas in tumors from rats treated with DMH, DMH + I3C, or DMH + CHL the average β-catenin mRNA expression levels were on the order of 1.5–2.5-fold higher than the housekeeping gene, GAPDH. No significant differences were seen among the various groups with respect to concentration of CHL or I3C administered. Similar findings were obtained for c-myc, c-jun and cyclin D1 mRNA expression when plotted as a function of carcinogen and phytochemical treatment (data not shown).
Fig. 5.
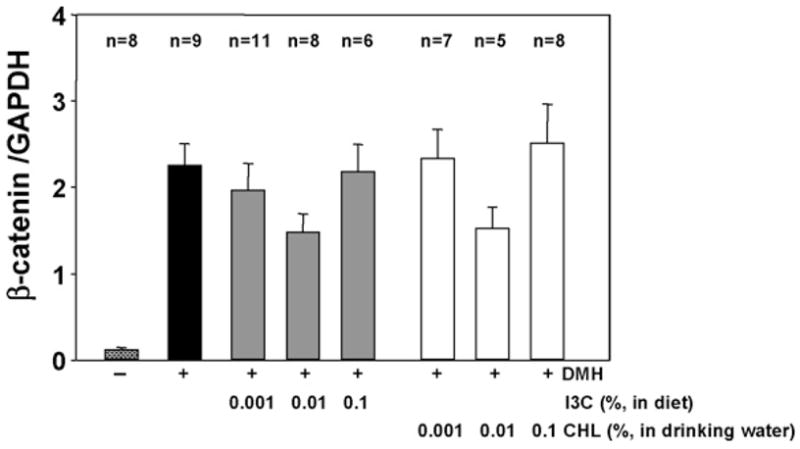
β-Catenin mRNA expression in DMH-induced tumors was unrelated to chlorophyllin (CHL) or indole-3-carbinol (I3C) treatment. qPCR was used to determine β-catenin mRNA expression relative to GADPH, as described in Fig. 4. Data = mean ± S.E., for the number of tumors (n) shown above each bar, and for the number of normal mucosal samples from control rats given no DMH. Tumors analyzed here were from a larger study [19] in which rats were treated with DMH and then exposed post-initiation to one of three different concentrations of CHL in the drinking water, or I3C in the diet, as indicated in the figure.
3.5. Concordance between β-catenin mRNA and protein levels in tumors
We reported [9,11] that colon tumors harboring mutant β-catenins had 4–10-fold higher β-catenin protein levels than those containing WT β-catenin, but we did not examine thoroughly the concordance between β-catenin mRNA and protein levels. Therefore, tumors from the present investigation were subjected to Western analyses (Fig. 6). β-Catenin protein was highly expressed in several tumors in which the β-catenin/GAPDH mRNA ratio was ≥2.5 (lanes 1, 2, 13 and 14), and the converse was true in tumors with lower β-catenin protein levels and β-catenin/GAPDH mRNA ratios ≤2.0 (lanes 4–7 and 9–12). However, there were exceptions, such as a tumor with high β-catenin mRNA but low β-catenin protein (lane 3), and low β-catenin mRNA with strongly over-expressed β-catenin protein (lane 8).
Fig. 6.
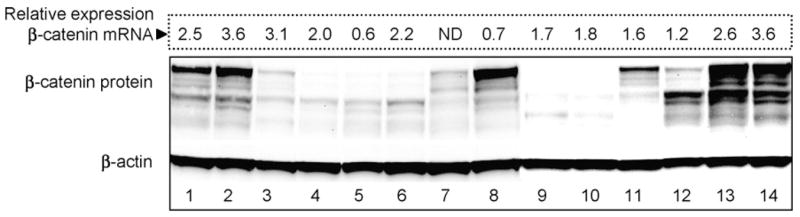
Concordance between β-catenin mRNA and protein levels in DMH-induced tumors. Immunoblotting was performed for β-catenin, with β-actin as loading control, and protein expression was compared with the corresponding levels of β-catenin mRNA. Values shown above each lane were obtained from Fig. 3, and represent β-catenin mRNA levels normalized to GAPDH. ND, not detected.
4. Discussion
The present investigation builds upon prior work showing that phytochemicals such as CHL and I3C can alter the spectrum of β-catenin mutations in DMH- and IQ-induced tumors [9,11]. Under conditions of tumor promotion, there was an increase in mutations affecting Ser37, Thr41 and Ser45 of β-catenin [11]. The latter mutations have been reported in human cancers, but they occur less frequently in the colon tumors from rats exposed to chemical carcinogens in the absence of a tumor promoter [7–9]. It is tempting to speculate that Ser37, Thr41 and Ser45 β-catenin mutations represent a genetic ‘fingerprint’ for cancers that have arisen due to initiating events coupled with a promotional stimulus, when cells normally deleted by apoptosis might survive to frank tumors. However, this would be an over-simplification. In PhIP-treated rats, promotion by a high-fat diet led to an increase in dysplastic aberrant crypt foci and colon tumors, but none of the mutations identified affected critical Ser/Thr residues in β-catenin [21,22]. Conversely, a high frequency of codon 41 β-catenin mutation (Thr41Ile) was found in rats given DMH over a period of 20 weeks as part of a ‘complete carcinogen’ protocol, without a separate tumor promoting agent [10]. Nonetheless, it is clear from published reports [9–11,21,22] that the carcinogen and/or phytochemical exposure protocol can influence the final pattern of β-catenin mutations in colon tumors. Understanding how such patterns arise might provide better insight into the risk factors for human colorectal cancer development.
Previously, rat colon tumors with the highest levels of β-catenin, c-Myc and c-Jun proteins were found to contain codon 41 and 45 mutant β-catenins [9,11]. These specific mutations in β-catenin were thought to more effectively stabilize β-catenin at the protein level, and thus provide for greater activation of β-catenin/Tcf target genes. In the present study, however, DMH-induced colon tumors and small intestine tumors had a wide range of β-catenin mRNA expression, and this was essentially independent of the β-catenin mutation status (Fig. 4) or phytochemical exposure (Fig. 5 and data not shown). Transcript levels for three β-catenin/Tcf ‘downstream’ targets, namely c-myc, c-jun, and cyclin D1, were highly correlated with each other and with the β-catenin mRNA expression levels. The latter findings suggest a global dysregulation of gene expression, including transcriptional changes in the gene for β-catenin (Ctnnb1).
It is well established that the progression to colorectal adenoma and carcinoma involves multiple genetic events and transcription changes in a broad array of genes [23,24]. Indeed, β-catenin mRNA levels are known to be increased in human primary colorectal cancers and their liver metastases, compared with matched normal-looking tissue [5]. A major regulator of β-catenin expression in human colon cancers is APC, but this is usually viewed from the perspective of β-catenin protein stabilization rather than transcriptional control of β-catenin mRNA levels. It is unclear what relationship, if any, exists between Apc mutation status and β-catenin mRNA expression in tumors. We did not examine Apc in the present investigation, because previous work showed that DMH- and IQ-induced tumors in the rat had a low frequency (<10%) of Apc mutations [11]. Apc might be a candidate for further study, especially in DMH-induced tumors that were wild type for β-catenin and that expressed high levels of β-catenin mRNA (Fig. 3). It also remains to be determined whether β-catenin mRNA levels are altered in the tumors induced by other carcinogens, such as IQ and PhIP, and the mechanisms involved.
As indicated above, the regulation of β-catenin mRNA levels in tumors has not been well studied, and establishing what relationship might exist between β-catenin transcript and β-catenin protein expression may be critical to understanding its relevance to tumor development. In the present investigation, many of the tumors with the highest β-catenin mRNA levels had highly over-expressed β-catenin protein, and others with low or undetectable β-catenin mRNA also had low β-catenin protein expression, but there were exceptions (e.g. lanes 3 and 8 in Fig. 6). We conclude that there is a general concordance between β-catenin mRNA and protein expression levels in DMH-induced tumors, but the correlation is not perfect. Other variables may be correlated with β-catenin transcript levels, such as tumor size, histopathology, or proliferative index, and additional work in this area appears to be warranted.
In summary, the present investigation has shown that small intestine tumors and colon tumors induced by DMH in the rat contain a wide range of expression of β-catenin mRNA. This was independent of β-catenin mutation status and treatment with CHL or I3C, but was highly correlated with c-myc, c-jun and cyclin D1 mRNA expression levels. These findings suggest the need for future studies on the mechanisms that regulate β-catenin at the transcriptional level, including further characterization of the transcription factors that bind to the promoter regions of human CTNNB1, rat Ctnnb1 and mouse Catbn1 genes [25].
Acknowledgments
Thanks are extended to Meirong Xu, who was responsible for overseeing the original carcinogenicity bioassay. We also thank Qingjie Li for help with primer design, and members of the Tanguay and Hagen laboratories for access to qPCR equipment. Sequencing was performed in the Center for Gene Research and Biotechnology at Oregon State University. This work was supported in part by NIH grants CA65525, CA80176, and CA90890.
Abbreviations
- APC
adenomatous polyposis coli
- AP-2α
activator protein-2α
- CHL
chlorophyllin
- DMH
1,2-dimethylhydrazine
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSK-3β
glycogen synthase kinase-3β
- I3C
indole-3-carbinol
- LEF
lymphoid enhancer factor
- PCR-SSCP
polymerase chain reaction-based single strand conformation polymorphism
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- qPCR
quantitative real-time RT-PCR
- TCF
T-cell factor
- WT
wild type
References
- 1.Katoh M. WNT/PCP signaling pathway and human cancer. Oncol Rep. 2005;14:1583–1588. [PubMed] [Google Scholar]
- 2.Fuchs SY, Ougolkov AV, Spiegelman VS, Minamoto T. Oncogenic β-catenin signaling networks in colorectal cancer. Cell Cycle. 2005;4:1522–1539. doi: 10.4161/cc.4.11.2129. [DOI] [PubMed] [Google Scholar]
- 3.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-Catenin is a target for the ubiquitin-proteosome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 5.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moey MP, Reicken EO, Buhr HJ, Hanski C. Target genes of β-catenin-T-cell factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA. 1999;96:1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tetsu O, McCormick F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 7.Dashwood RH, Suzui M, Nakagama H, Sugimura T, Nagao M. High frequency of β-catenin (Ctnnb1) mutations in the colon tumors induced by two heterocyclic amines in the F344 rat. Cancer Res. 1998;58:1127–1129. [PubMed] [Google Scholar]
- 8.Suzui M, Ushijima T, Dashwood RH, Yoshimi N, Sugimura T, Mori H, Nagao M. Frequent mutations of the rat β-catenin gene in colon cancers induced by methylazoxymethanol acetate plus 1-hydroxyanthraquinone. Mol Carcinog. 1999;24:232–237. doi: 10.1002/(sici)1098-2744(199903)24:3<232::aid-mc10>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 9.Blum CA, Xu M, Orner GA, Fong AT, Bailey GS, Stoner GD, Horio DT, Dashwood RH. β-Catenin mutation in rat colon tumors initiated by 1,2-dimethylhydrazine and 2-amino-3-methylimidazo[4,5-f]quinoline, and the effect of post-initiation treatment with chlorophyllin and indole-3-carbinol. Carcinogenesis. 2001;22:315–320. doi: 10.1093/carcin/22.2.315. [DOI] [PubMed] [Google Scholar]
- 10.Koesters R, Hans MA, Benner A, Prosst R, Boehm J, Gahlen J, Doeberitz MVK. Predominant mutation of codon 41 of the β-catenin proto-oncogene in rat colon tumors induced by 1,2-dimethylhydrazine using a complete carcinogenic protocol. Carcinogenesis. 2001;22:1885–1890. doi: 10.1093/carcin/22.11.1885. [DOI] [PubMed] [Google Scholar]
- 11.Blum CA, Tanaka T, Zhong X, Li Q, Dashwood WM, Pereira C, Xu M, Dashwood RH. Mutational analysis of Ctnnb1 and Apc in tumors from rats given 1,2-dimethylhydrazine or 2-amino-3-methylimidazo[4,5-f]quinoline: mutational ‘hotspots’ and the relative expression of β-catenin and c-jun. Mol Carcinog. 2003;36:195–203. doi: 10.1002/mc.10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leibelt DA, Hedstrom OR, Fischer KA, Pereira CB, Williams DE. Evaluation of chronic dietary exposure to indole-3-carbinol and absorption-enhanced 3,3′-diindolylmethane in Sprague–Dawley rats. Toxicol Sci. 2003;74:10–21. doi: 10.1093/toxsci/kfg103. [DOI] [PubMed] [Google Scholar]
- 13.Anderton MJ, Manson MM, Verschoyle RD, Gescher A, Lamb JH, Farmer PB, Steward WP, Williams ML. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin Cancer Res. 2004;10:5233–5241. doi: 10.1158/1078-0432.CCR-04-0163. [DOI] [PubMed] [Google Scholar]
- 14.Dashwood RH. Indole-3-carbinol: anticarcinogen or tumor promoter in brassica vegetables? Chem Biol Interact. 1998;110:1–5. doi: 10.1016/s0009-2797(97)00115-4. [DOI] [PubMed] [Google Scholar]
- 15.Ferguson LR, Philpott M, Karunasinghe N. Dietary cancer and prevention using antimutagens. Toxicology. 2004;198:147–159. doi: 10.1016/j.tox.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 16.Diaz GD, Li Q, Dashwood RH. Caspase-8 and apoptosis-inducing factor mediate a cytochrome c-independent pathway of apoptosis in human colon cancer cells induced by the dietary phytochemical chlorophyllin. Cancer Res. 2003;63:1254–1261. [PubMed] [Google Scholar]
- 17.Harttig U, Bailey GS. Chemoprotection by natural chlorophylls in vivo: inhibition of dibenzo[a,l]pyrene-DNA adducts in rainbow trout liver. Carcinogenesis. 1998;19:1323–1326. doi: 10.1093/carcin/19.7.1323. [DOI] [PubMed] [Google Scholar]
- 18.Al-Fageeh M, Li Q, Dashwood WM, Myzak MC, Dashwood RH. Phosphorylation and ubiquitination of oncogenic mutants of β-catenin containing substitutions at Asp32. Oncogene. 2004;23:4839–4846. doi: 10.1038/sj.onc.1207634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu M, Orner GA, Bailey GS, Stoner GD, Horio DT, Dashwood RH. Post-initiation effects of chlorophyllin and indole-3-carbinol in rats given 1,2-dimethylhydrazine or 2-amino-3-methylimidazo[4,5-f]quinoline. Carcinogenesis. 2001;22:309–314. doi: 10.1093/carcin/22.2.309. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Dashwood RH. Activator protein 2α associates with adenomatous polyposis coli/β-catenin and inhibits β-catenin/T-cell factor transcriptional activity in colorectal cancer cells. J Biol Chem. 2004;279:45669–45675. doi: 10.1074/jbc.M405025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ubagai T, Ochiai M, Kawamori T, Imai H, Sugimura T, Nagao M, Nakagama H. Efficient induction of rat large intestinal tumors with a new spectrum of mutations by intermittent administration of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in combination with a high fat diet. Carcinogenesis. 2002;23:197–200. doi: 10.1093/carcin/23.1.197. [DOI] [PubMed] [Google Scholar]
- 22.Ochiai M, Ushigome M, Fujiwara K, Ubagai T, Kawamori T, Sugimura T, Nagao M, Nakagama H. Characterization of dysplastic aberrant crypt foci in the rat colon induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Am J Pathol. 2003;163:1607–1614. doi: 10.1016/S0002-9440(10)63517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugiyama Y, Farrow B, Murillo C, Li J, Watanabe H, Sugiyama K, Evers BM. Analysis of differential gene expression in colon cancer and cancer stroma using microdissected tissues. Gastroenterology. 2005;128:480–486. doi: 10.1053/j.gastro.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Notterman DA, Alon U, Sierk AJ, Levine AJ. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 2001;61:3124–3130. [PubMed] [Google Scholar]
- 25.Li Q, Dashwood WM, Zhong X, Al-Fageeh M, Dashwood RH. Cloning of the rat β-catenin gene (Ctnnb1) promoter and its functional analysis compared with the Catnb and CTNNB1 promoters. Genomics. 2004;83:231–242. doi: 10.1016/j.ygeno.2003.08.004. [DOI] [PubMed] [Google Scholar]