

Abstract
Lithium has been shown to be neuroprotective against various insults including ethanol exposure. We previously reported that ethanol-induced apoptotic neurodegeneration in the postnatal day 7 (P7) mice is associated with decreases in phosphorylation levels of Akt, glycogen synthase kinase-3β (GSK-3β), and AMP-activated protein kinase (AMPK), and alteration in lipid profiles in the brain. Here, P7 mice were injected with ethanol and lithium, and the effects of lithium on ethanol-induced alterations in phosphorylation levels of protein kinases and lipid profiles in the brain were examined. Immunoblot and immunohistochemical analyses showed that lithium significantly blocked ethanol-induced caspase-3 activation and reduction in phosphorylation levels of Akt, GSK-3β and AMPK. Further, lithium inhibited accumulation of cholesterol ester (ChE) and N-acylphosphatidylethanolamine (NAPE) triggered by ethanol in the brain. These results suggest that Akt, GSK-3β, and AMPK are involved in ethanol-induced neurodegeneration and the neuroprotective effects of lithium by modulating both apoptotic and survival pathways.
Keywords: lithium, ethanol, brain, apoptosis, Caspase-3, Akt, glycogen synthase kinase-3β, AMP-activated protein kinase
Introduction
Ethanol triggers apoptotic neurodegeneration in the P7 rodent brain during the peak period of synaptogenesis that corresponds to the last trimester of pregnancy in humans [1, 2]. The ethanol-induced neuronal loss may partially explain neuropathological conditions in rodents similar to those found in human fetal alcohol spectrum disorders (FASD). This rodent model of FASD has been widely used for elucidating mechanisms of ethanol-induced toxicity in the developing brain [3-5]. It has been shown that ethanol induces Bax-mediated disruption of mitochondrial membranes, cytochrome c release, and caspase-3 activation, leading to neurodegeneration in P7 mouse brains [6]. However, targets of ethanol, and the subsequent signal transduction pathways, which lead to the mitochondrial apoptotic pathways, are largely unknown. Previous studies using cultured neurons indicate that ethanol perturbs functions of neurotrophic factors (such as brain-derived neurotrophic factor and insulin-like growth factor-I) probably through blockade of NMDA receptors [7-9] leading to the inhibition of PI3K [8, 9]. The suppression of Akt [4] and activation of GSK-3 [10], downstream effects of PI3K inhibition, have also been reported in the developing brain exposed to ethanol. Our recent study showed that apoptotic neurodegeneration in P7 mice induced by acute ethanol exposure was accompanied by inactivation of Akt and activation of GSK-3β, which was indicated by decreases in phosphorylation levels of both protein kinases [11].
It has been demonstrated that lithium has neuroprotective effects against a variety of insults in cultured neurons [12-15], in animal models of neurodegenerative diseases [16], and in human studies [16, 17]. Because lithium inhibits GSK-3β activity [18], it has been postulated that part of the neuroprotective effects of lithium is due to its ability to inhibit GSK-3β, a pro-apoptotic enzyme [19]. However, lithium seems to have many other functions including enhanced phosphorylation of Akt by inhibiting protein phosphatase 2A (PP2A) [20, 21]. A recent study showing the effects of lithium on histone methylation and chromatin structure in C. elegans [22] suggests a wide range of effects of lithium. Lithium also protects ethanol-induced apoptosis in P7 mice and cultured neurons [23]. Our previous studies showed that ethanol-induced apoptosis in the brains of P7 mice was associated with alteration in lipid profiles and decreases in phosphorylation levels of AMPK [24]. The alterations in the lipid profiles include increases in triglyceride (TG), ceramide, ChE, and NAPE. Here, we showed that lithium significantly inhibited ethanol-induced caspase-3 activation, reduction in phosphorylation levels of AMPK, Akt, and GSK-3β, and increases in ChE and NAPE in the P7 mouse brains.
Materials and Methods
Animals and treatment
C57BL/6By mice were maintained at the Animal Facility of Nathan S. Kline Institute for Psychiatric Research. All procedures followed guidelines consistent with those developed by the National Institute of Health and the Institutional Animal Care and Use Committee of Nathan S. Kline Institute. An ethanol treatment paradigm, which had been shown to induce robust neurodegeneration in P7 C57BL/6 mice [1], was followed using P7 C57BL/6By mice as described [25]. Each mouse in a litter was assigned to saline, lithium, ethanol, or “ethanol + lithium” group. The mice were injected subcutaneously with saline or ethanol (2.5 g/kg, 20% solution in sterile saline) twice at 0 h and 2 h. Lithium chloride (LiCl, 0.6M, 10 μl/g) or saline was injected intraperitoneally 15 min after the first ethanol injection as described previously [23]. The mice were separated from their dams after the first saline/ethanol injection, and kept in a plastic box floating in a water bath with a temperature of 35 °C. Two to eight hours after the first ethanol injection, the brains were removed and processed for immunoblotting or immmunohistochemical staining. For lipid analyses, the pups were returned to the dams after injections, and the brains were removed 24 h after the first ethanol injection.
Western blot analysis
Western blot analyses were performed as described [24] using forebrain samples taken 2 to 8 h after the first ethanol injection. For cleaved caspase-3 immunoblotting, the 8 h samples were used because ethanol-induced caspase-3 activation peaks around 8 h after the first ethanol injection [1, 25]. Briefly, forebrain samples were homogenized (20% w/v) in ice cold Tris-HCl (20 mM, pH 7.4) buffer containing 1% Triton X-100, 1mM EGTA, 1 mM EDTA, 20 mM NaF, 1 mM Na-orthovanadate, 20 mM β-glycerophosphate, 5 μM microcystin, and 10μl/ml protease inhibitor cocktail (all from Sigma, St. Louis, MO), and centrifuged at 52K× g for 30 min. Samples (50 μg protein) were boiled in SDS-sample buffer, separated on SDS-PAGE, and blotted onto PVDF membranes. The membranes were then blocked with 5% BSA in blocking buffer (0.1% Tween 20 in Tris buffered saline, TBS) and probed with various dilutions of rabbit polyclonal anti-cleaved caspase-3 (Asp175) antibody, rabbit monoclonal anti-phospho (Thr308)-Akt (western blot specific) antibody, rabbit monoclonal anti-Akt antibody, rabbit monoclonal anti-phospho-AMPKα (Thr172) antibody, rabbit monoclonal anti-AMPKα antibody, rabbit polyclonal anti-phospho (Ser9)-GSK-3β antibody, rabbit monoclonal anti-GSK-3β antibody (all from Cell Signaling Technology, Danvers, MA), or mouse monoclonal anti-PHF-1 antibody (a gift from Dr. Peter Davies at Albert Einstein College of Medicine, New York, NY) in blocking buffer. Mouse monoclonal anti-β-actin antibody (loading control, Abcam Inc. Cambridge, MA) was always included in the primary antibody incubation. Antigens were detected by the Odyssey infrared imaging system (LI-COR Inc. Nebraska, NE) using fluorescence-labeled secondary antibodies, goat anti-rabbit IgG-680 (Invitrogen, Carlsbad, CA) and goat anti-mouse IgG-800 (Rockland Immunochemicals, Gilbertsville, PA) in blocking buffer. Intensities of desired bands were quantified using Fuji film MultiGauge V3.2 software. The intensities of phospho-Akt, Akt, phospho-GSK-3β, GSK-3β, phospho-AMPK and AMPK were normalized by intensities of corresponding actin, whereas cleaved caspase-3 intensities were normalized by corresponding uncleaved caspase-3 band intensity. Finally, the ratios of treatment to saline were calculated. The amount of protein was measured by a BCA method (Pierce, Rockford, IL).
Immunohistochemistry
Eight hours after the first ethanol injection, mice were perfused as described previously [25]. Briefly, mice were perfused with a solution containing 4% paraformaldehyde and 4% sucrose in cacodylate buffer (pH 7.2), and the heads were further fixed in the perfusion solution overnight. Then the brains were removed, transferred to phosphate buffered saline solution, and kept at 4°C for 2−5 days until sectioned with a vibratome into 50 μm thick sections. The free-floating sections were rinsed in TBS, quenched for 10 min in methanol containing 3% hydrogen peroxide, and incubated for 1h in a blocking solution (TBS containing 2% BSA, 0.2% milk, and 0.1% Triton X-100). This was followed by incubation overnight with primary antibodies: monoclonal anti-phospho (Thr308)-Akt (immunohistochemistry specific) antibody and other primary antibodies used for immunoblotting experiments described above. Antibodies were diluted 1:500 (except for anti-cleaved caspase-3 antibody which was diluted 1:1500) in a blocking solution. After rinsing with TBS, the sections were incubated for 1 h with biotinylated goat anti-rabbit IgG diluted at 1: 200 in a blocking solution, rinsed with TBS, and stained with ABC reagents (Vectastain ABC Elite Kit, Vector Labs, Burlingame, CA) and a peroxidase substrate (DAB) kit (Vector Labs) following the manufacturer's instructions. No immunostaining was observed when the primary antibodies were omitted from the procedure. All photomicrographs were taken through a 40× objective with a Nikon Eclipse TE2000 inverted microscope attached to a digital camera DXM1200F.
Lipid Analysis
Triglyceride (TG), ceramide, ChE, and NAPE were analyzed using high performance thin layer chromatography as described [24].
Statistical Analysis
All data were expressed as mean ± SEM (n = 3 to 5). Comparisons among groups were performed by student t-tests and one-way ANOVA with Bonferroni post hoc tests. P< 0.05 was considered significant.
Results
Western blot analyses showed that ethanol-induced caspase-3 activation in the forebrain was strongly inhibited by lithium treatment (Fig. 1), as reported earlier [23]. Although the upstream events triggered by ethanol, which leads to caspase-3 activation in the brain, have not been fully elucidated, involvement of the PI3K/Akt pathway has been implicated [4, 8-10]. We found that ethanol reduced phospho-Akt and phospho-GSK-3β levels without changing the levels of Akt and GSK-3β at 4 h after the first ethanol injection (Fig. 1), and the reduction of phosphorylation of Akt and GSK-3β was significantly inhibited by lithium treatment (Fig. 1). No changes were observed by lithium treatment alone when compared with control (data not shown). In order to examine the inhibitory effects of lithium on GSK-3β activity, we measured the phosphorylation levels of tau protein, a known GSK-3β substrate [26, 27], using anti-PHF-1 antibody, which recognizes highly phosphorylated tau proteins found in the embryonic day18 to P11 rat brains [28]. Our results indicated that lithium partially inhibited tau phosphorylation induced by ethanol treatment (data not shown). Recently, we have reported that ethanol decreases phosphorylation of AMPK-α int the P7 mouse brain in a time dependent manner [24]. In the present experiments, we observed that lithium treatment inhibited ethanol-induced reduction of phospho-AMPK, leaving AMPK unaffected (Fig. 1). The effects of ethanol and/or lithium on phosphorylation status of Akt, GSK-3β, and AMPK shown in Fig. 1 were the results of 4 h after ethanol treatment, although we found the similar but less robust effects at 2 h and 8 h after ethanol and “ethanol + lithium” treatments. The effects of lithium were also examined using immunohistochemistry (Fig. 2). P7 mouse brain sections obtained from saline, ethanol, lithium, and “ethanol + lithium” groups 8 h after the first ethanol injection were stained with antibodies as described in Materials and Methods. Areas around the cingulate cortex, which are strongly affected by ethanol exposure [1, 25], are shown here. Ethanol-induced caspase-3 activation and reduction in phosphorylation levels of Akt, GSK-3β were blocked by lithium treatment, confirming the results of immunoblot analyses. Although lithium inhibited ethanol-induced reduction in phospho-AMPK, the subcellular localization of phospho-AMPK in the ethanol and “ethanol + lithium” groups appeared to be different from that of the saline and lithium groups (Fig.2). We have shown previously that ethanol increased the content of TG, ceramide, ChE, and NAPE in the P7 mouse brains. Lithium significantly inhibited ethanol-induced increases in ChE and NAPE (Fig. 3). The effects of lithium on ethanol-induced increases in TG and ceramide levels were not significant (data not shown).
Fig. 1.
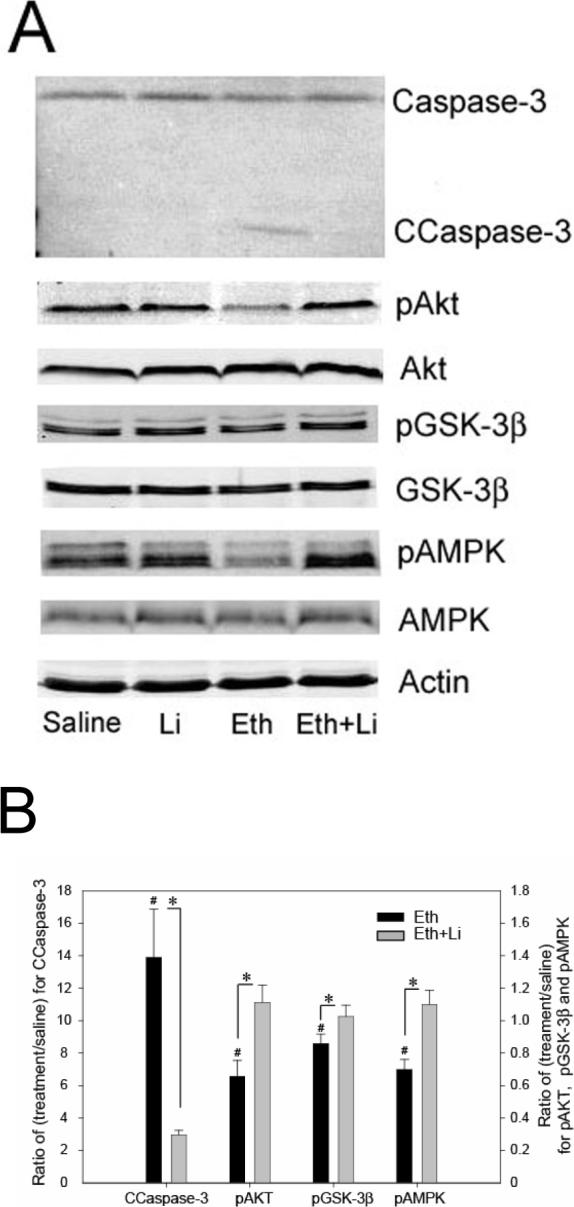
Effects of ethanol and lithium on caspase-3 activation and phosphorylation levels of Akt, GSK-3β, and AMPK. Forebrain samples were taken from P7 mice 8 h and 4 h after the first ethanol/saline injection for immunoblot analyses of cleaved caspase-3 and protein kinases, respectively. Immunoblot analyses were carried out using antibodies specific for phospho (p)- and total proteins for Akt, GSK-3β and AMPK as well as antibody specific for cleaved caspase-3 (CCaspase-3). Anti-actin antibody was always included. Fig. 1A shows representative immunoblots of saline, lithium (Li), ethanol (Eth), and “ethanol + lithium” (Eth+Li) groups. Fig. 1B shows quantitative analysis of immunoblots. Data are expressed as a ratio of treatment to control (saline) after normalized by uncleaved caspase-3 (for CCaspase-3) and with actin (for all others). *Significantly (P< 0.05) different between Eth and “Eth+Li” groups, # significantly (P < 0.05) different between saline and Eth groups by student t-test.
Fig. 2.
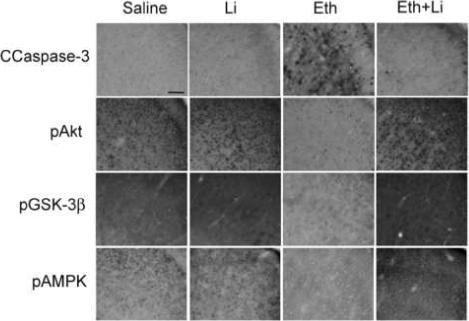
Immunostaining pictures of brain sections from mice treated with saline, lithium (Li), ethanol (Eth), and “ethanol + lithium (Eth+Li)” groups. The mice were treated as described in Materials and Methods, and the brain sections were stained with anti-CCaspase-3, anti-pAkt, anti-pGSK-3β, and anti-pAMPKα antibodies. The pictures show the area of cingulate cortices. The bar indicates 50 μm.
Fig. 3.
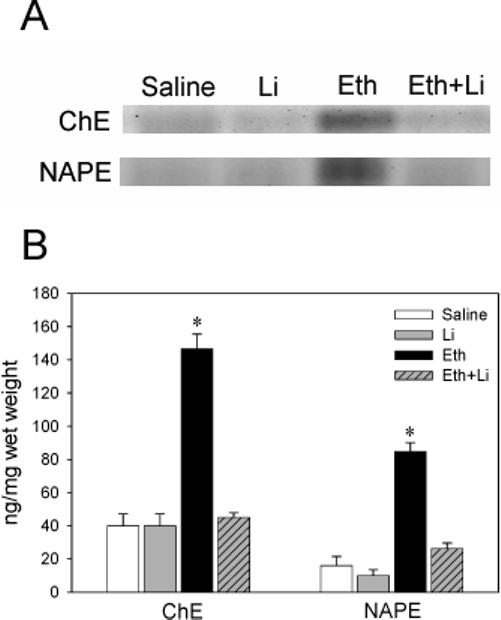
Lithium reduces ethanol-induced accumulation of ChE and NAPE. Lipid extracts from the mice treated with saline, lithium (Li), ethanol (Eth), and “ethanol + lithium (Eth+Li)” groups were separated on HPTLC. Lipid extracts derived from the brains (1 mg and 10 mg wet weight for ChE and NAPE, respectively) were loaded on each lane in the representative HPTLC picture (A). ChE and NAPE in the ethanol group were *significantly (P< 0.05) different from other groups by one-way ANOVA with Bonferroni post hoc tests (B).
Discussion
Although a previous study [23] has demonstrated that ethanol-induced apoptosis in the P7 mouse brain is strongly blocked by lithium, the mechanisms of such neuroprotective action of lithium are unexplained. Here we found that the neuroprotective effects of lithium were accompanied by inhibition of ethanol-induced decreases in phosphorylation levels of Akt, GSK-3β, and AMPK. Such effects of both ethanol and lithium on Akt, GSK-3β, and AMPK have not been reported earlier. Lithium-induced increases in phosphorylation levels of GSK-3β and reduction in phospho-tau recognized by anti-PHF-1 antibody indicated that lithium inhibited GSK-3β activation triggered by ethanol. Because GSK-3β activation is involved in several apoptotic processes including ethanol-induced apoptosis in cultured cortical neurons [29], the neuroprotective effects of lithium on ethanol-induced apoptosis in the P7 brain may be due to GSK-3β inhibition by lithium. However, inhibition of GSK-3β may not be the sole mechanism underlying the lithium-induced neuroprotection. Phosphorylation of Akt, which indicates Akt activation, may partially explain the neuroprotective effects of lithium because activation of Akt is implicated in cell survival [30]. Lithium also inhibited ethanol-induced reduction in phospho-AMPK levels. As far as we know, this is the first observation that lithium affects phosphorylation levels of AMPK. It has been reported that lithium inhibits PP2A activity [20, 21], which has been shown to regulate phosphorylation of AMPK [31]. The observed effects of lithium on AMPK may also be related to the neuroprotective effects of lithium because AMPK activation is connected to cell survival in several systems [32-34]. Immunohistochemical data suggested that the subcellular localization of phospho-AMPK in “ethanol + lithium” group is different from that in control and lithium groups. Functions of AMPK may be influenced by the subcellular localization, as reported previously in muscle cells [35]. Lithium inhibited ethanol-induced accumulation of ChE and NAPE, implicating that the actions of lithium are upstream events of ChE and NAPE accumulation. Lithium did not significantly inhibit ethanol-induced accumulation of TG and ceramide. While activation of AMPK inhibits lipogenesis in peripheral tissues [36], activation of Akt enhances lipogenesis [37]. Further studies on the activities and subcellular localization of AMPK and Akt may give insight into the roles of these protein kinases particularly during neurodegeneration of the developing brain by ethanol and the neuroprotective action of lithium.
Thus, our studies suggest that the strong neuroprotective effects of lithium against ethanol-induced apoptosis in P7 mouse brains may be due to concerted actions of lithium on Akt, GSK-3β, and AMPK activities.
Acknowledgements
This work was supported by NIH/NIAAA grant R01 AA015355 (to M.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002;133:115–126. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- 2.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 3.Carloni S, Mazzoni E, Balduini W. Caspase-3 and calpain activities after acute and repeated ethanol administration during the rat brain growth spurt. J Neurochem. 2004;89:197–203. doi: 10.1111/j.1471-4159.2004.02341.x. [DOI] [PubMed] [Google Scholar]
- 4.Han JY, Jeong JY, Lee YK, Roh GS, Kim HJ, Kang SS, Cho GJ, Choi WS. Suppression of survival kinases and activation of JNK mediate ethanol-induced cell death in the developing rat brain. Neurosci Lett. 2006;398:113–117. doi: 10.1016/j.neulet.2005.12.065. [DOI] [PubMed] [Google Scholar]
- 5.Young C, Roth KA, Klocke BJ, West T, Holtzman DM, Labruyere J, Qin YQ, Dikranian K, Olney JW. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis. 2005;20:608–614. doi: 10.1016/j.nbd.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, Holtzman DM, Roth KA, Olney JW. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003;10:1148–1155. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]
- 7.Bhave SV, Hoffman PL. Ethanol promotes apoptosis in cerebellar granule cells by inhibiting the trophic effect of NMDA. J Neurochem. 1997;68:578–586. doi: 10.1046/j.1471-4159.1997.68020578.x. [DOI] [PubMed] [Google Scholar]
- 8.Bhave SV, Ghoda L, Hoffman PL. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang FX, Rubin R, Rooney TA. N-Methyl-D-aspartate inhibits apoptosis through activation of phosphatidylinositol 3-kinase in cerebellar granule neurons. A role for insulin receptor substrate-1 in the neurotrophic action of n-methyl-D-aspartate and its inhibition by ethanol. J Biol Chem. 1998;273:26596–26602. doi: 10.1074/jbc.273.41.26596. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, de la Monte S. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem. 2003;278:26929–26937. doi: 10.1074/jbc.M300401200. [DOI] [PubMed] [Google Scholar]
- 11.Chakraborty G, Saito M, Mao RF, Wang R, Vadasz C, Saito M. Involvement of sphingomyelin and the Akt-pathway in ethanol-induced neurodegeneration in the neonatal mouse brain. Alcoholism: Clinical and Experimental Research. 2007;31:40A. doi: 10.1111/j.1530-0277.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- 12.Bijur GN, De Sarno P, Jope RS. Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J Biol Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- 13.Centeno F, Mora A, Fuentes JM, Soler G, Claro E. Partial lithium-associated protection against apoptosis induced by C2-ceramide in cerebellar granule neurons. Neuroreport. 1998;9:4199–4203. doi: 10.1097/00001756-199812210-00036. [DOI] [PubMed] [Google Scholar]
- 14.Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci U S A. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marks N, Saito M, Green M, Reilly MA, Yang AJ, Ditaranto K, Berg MJ. Opposite effects of lithium on proximal and distal caspases of immature and mature primary neurons correlate with earlier paradoxical actions on viability. Neurochem Res. 2001;26:1311–1320. doi: 10.1023/a:1014249517926. [DOI] [PubMed] [Google Scholar]
- 16.Chuang DM, Manji HK. In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol Psychiatry. 2007;62:4–6. doi: 10.1016/j.biopsych.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yucel K, Taylor VH, McKinnon MC, Macdonald K, Alda M, Young LT, Macqueen GM. Bilateral Hippocampal Volume Increase in Patients with Bipolar Disorder and Short-term Lithium Treatment. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301405. [DOI] [PubMed] [Google Scholar]
- 18.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Bijur GN, Jope RS. Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord. 2002;4:137–144. doi: 10.1034/j.1399-5618.2002.40201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CL, Lin CF, Chiang CW, Jan MS, Lin YS. Lithium inhibits ceramide- and etoposide-induced protein phosphatase 2A methylation, Bcl-2 dephosphorylation, caspase-2 activation, and apoptosis. Mol Pharmacol. 2006;70:510–517. doi: 10.1124/mol.106.024059. [DOI] [PubMed] [Google Scholar]
- 21.Mora A, Sabio G, Risco AM, Cuenda A, Alonso JC, Soler G, Centeno F. Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cell Signal. 2002;14:557–562. doi: 10.1016/s0898-6568(01)00282-0. [DOI] [PubMed] [Google Scholar]
- 22.McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem. 2007 doi: 10.1074/jbc.M705028200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong J, Yang X, Yao W, Lee W. Lithium protects ethanol-induced neuronal apoptosis. Biochem Biophys Res Commun. 2006;350:905–910. doi: 10.1016/j.bbrc.2006.09.138. [DOI] [PubMed] [Google Scholar]
- 24.Saito M, Chakraborty G, Mao RF, Wang R, Cooper TB, Vadasz C, Saito M. Ethanol alters lipid profiles and phosphorylation status of AMP-activated protein kinase in the neonatal mouse brain. J Neurochem. 2007;103:1208–1218. doi: 10.1111/j.1471-4159.2007.04836.x. [DOI] [PubMed] [Google Scholar]
- 25.Saito M, Mao RF, Wang R, Vadasz C, Saito M. Effects of gangliosides on ethanol-induced neurodegeneration in the developing mouse brain. Alcohol Clin Exp Res. 2007;31:665–674. doi: 10.1111/j.1530-0277.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- 26.Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- 27.Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- 28.Mawal-Dewan M, Henley J, Van de Voorde A, Trojanowski JQ, Lee VM. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem. 1994;269:30981–30987. [PubMed] [Google Scholar]
- 29.Takadera T, Ohyashiki T. Glycogen synthase kinase-3 inhibitors prevent caspase-dependent apoptosis induced by ethanol in cultured rat cortical neurons. Eur J Pharmacol. 2004;499:239–245. doi: 10.1016/j.ejphar.2004.07.115. [DOI] [PubMed] [Google Scholar]
- 30.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–9788. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 32.Blazquez C, Geelen MJ, Velasco G, Guzman M. The AMP-activated protein kinase prevents ceramide synthesis de novo and apoptosis in astrocytes. FEBS Lett. 2001;489:149–153. doi: 10.1016/s0014-5793(01)02089-0. [DOI] [PubMed] [Google Scholar]
- 33.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 34.Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–4327. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase--development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–6481. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]