

Abstract
Measles virus is a negative-sense, single-stranded RNA virus within theMononegavirales order,which includes several human pathogens, including rabies, Ebola, Nipah, and Hendra viruses. Themeasles virus nucleoprotein consists of a structured N-terminal domain, and of an intrinsically disordered C-terminal domain, NTAIL (aa 401–525), which undergoes induced folding in the presence of the C-terminal domain (XD, aa 459–507) of the viral phosphoprotein. With in NTAIL, an α-helical molecular recognition element (α-MoRE, aa 488–499) involved in binding to P and in induced folding was identified and then observed in the crystal structure of XD. Using small-angle X-ray scattering, we have derived a low-resolution structural model of the complex between XD and NTAIL, which shows that most of NTAIL remains disordered in the complex despite P-induced folding within the α-MoRE. The model consists of an extended shape accommodating the multiple conformations adopted by the disordered N-terminal region of NTAIL, and of a bulky globular region, corresponding to XD and to the C terminus of NTAIL (aa 486–525). Using surface plasmon resonance, circular dichroism, fluorescence spectroscopy, and heteronuclear magnetic resonance, we show that NTAIL has an additional site (aa 517–525) involved in binding to XD but not in the unstructured-to-structured transition. This work provides evidence that intrinsically disordered domains can establish complex interactions with their partners, and can contact them through multiple sites that do not all necessarily gain regular secondary structure.
Keywords: measles virus, nucleoprotein, phosphoprotein, intrinsic disorder, induced folding, NMR, CD, SAXS
Measles virus (MV) is an enveloped RNA virus within the Morbillivirus genus of the Paramyxoviridae family. Its nonsegmented, negative-sense, single-stranded RNA genome is encapsidated by the viral nucleoprotein (N) within a helical nucleocapsid. This N–RNA complex is used as a template for both transcription and replication. These latter activities are carried out by the viral polymerase complex, which consists of two components, the large protein (L) and the phosphoprotein (P) (for review, see Lamb and Kolakofsky 2001).
Nucleoproteins of Paramyxoviridae are divided into two regions: a structured N-terminal moiety, NCORE (aa 1–400 in MV), which contains all the regions necessary for self-assembly and RNA-binding (Buchholz et al. 1993; Curran et al. 1993; Bankamp et al. 1996; Liston et al. 1997; Myers et al. 1997b, 1999; Karlin et al. 2002a; Kingston et al. 2004b), and a C-terminal domain, NTAIL (aa 401–525 in MV). NTAIL is intrinsically unstructured (i.e., it lacks any stable secondary and tertiary structure in physiological conditions) (Longhi et al. 2003) and is exposed at the surface of the viral nucleocapsid (Heggeness et al. 1980, 1981). The presence of a flexible region protruding from the viral nucleocapsid allows the establishment of interactions with numerous different viral partners and with several cellular proteins (Moyer et al. 1990; De and Banerjee 1999; tenOever et al. 2002; Zhang et al. 2002; Laine et al. 2003). In Morbilliviruses and Respiroviruses, NTAIL is responsible for binding to P (Curran et al. 1993; Harty and Palese 1995; Bankamp et al. 1996; Liston et al. 1997; Longhi et al. 2003; Kingston et al. 2004b), to the polymerase complex P–L, and to the matrix protein (Coronel et al. 2001). Beyond viral partners, NTAIL also interacts with several cellular proteins, including the interferon regulatory factor 3 (tenOever et al. 2002) and the heat-shock protein Hsp72 (Zhang et al. 2002). Moreover, NTAIL within viral nucleocapsids released from infected cells also binds to a yet unidentified protein receptor expressed at the surface of human thymic epithelial cells (Laine et al. 2003, 2005).
The P protein of Paramyxovirinae plays multiple roles in both transcription and replication: It is an essential subunit of the viral polymerase complex and acts as a bridge between the nucleocapsid template (NNUC) and the polymerase complex L–P. P is a modular protein, consisting of an intrinsically unstructured N-terminal moiety (PNT) (Karlin et al. 2002b, 2003), and of a well conserved C-terminal moiety (PCT) which contains all the regions required for transcription (Curran 1996). Paramyxovirinae PCT have a modular organization, consisting of alternating disordered and structured regions (Karlin et al. 2003). In particular, they possess a coiled-coil domain (referred to as PMD, for P multimerization domain) responsible for both oligomerization and binding to L (Smallwood et al. 1994; Liston et al. 1995), and a C-terminal globular region (referred to as XD), that is involved in binding to both monomeric and assembled forms of N (Curran et al. 1994, 1995a,Curran et al. b; Harty and Palese 1995; Tuckis et al. 2002; Johansson et al. 2003; Kingston et al. 2004b). Figure 1 ▶ shows a schematic representation of the MV NNUC–P complex, which highlights the role of NTAIL in the recruitment of P via the binding to XD.
Figure 1.

Schematic representation of the modular organization of P (A) and of the NNUC–P complex (B) of measles virus. Disordered regions are represented by thin bars (A) or by lines (B). The encapsidated RNA is shown as a dotted line. PMD is represented with a dumbbell shape according to Tarbouriech et al. (2000). The tetrameric P (Rahaman et al. 2004) is shown bound to NNUC through three of its four C-terminal XD “arms,” as in the model of Curran and Kolakofsky (1999). The L protein is shown as a rectangle contacting P through PMD by analogy with SeV (Smallwood et al. 1994).
We have previously reported the crystal structure of the C-terminal globular domain of MV P (XD, aa 459–507): It consists of a monomeric protein composed of three α-helices, forming an anti-parallel three-helix bundle (Johansson et al. 2003). We have also shown that NTAIL undergoes an induced folding in the presence of XD and that this unstructured-to-structured transition implies a gain of α-helicity (Johansson et al. 2003). Using a combination of computational and biochemical approaches, we have identified within NTAIL an α-helical molecular recognition element (α-MoRE, aa 488–499) involved in binding to P and in the induced folding of NTAIL (Bourhis et al. 2004). The α-MoRE has been modeled in the crystal structure of XD, in the long hydrophobic cleft delimited by helices α2 and α3 (Johansson et al. 2003). In this model, the contact of NTAIL with a hydrophobic patch at the surface of XD would be the driving force in the induced folding of the α-MoRE of NTAIL by burying apolar residues at the protein–protein interface. Very recently, Kingston et al. (2004a) have reported the crystal structure of a chimeric protein composed of MV XD and a peptide corresponding to residues 486–505 of MV N. The structure of the complex is a four-helix bundle in which the α-helix of N is bound in the reverse orientation with respect to the model proposed by Johansson et al. (2003). Using NMR and crystallographic studies, Kingston et al. (2004a) deduced that the induced folding of NTAIL is restricted to only 18 residues (out of 125), although the analysis was restricted to the N region encompassing residues 477–505.
In this paper we examine this localized induced folding event in the context of the entire NTAIL domain. A low-resolution structural model of the complex between XD and NTAIL shows the presence of a bulky globular region and of an extended and elongated shape. The model shows that the N-terminal region of NTAIL (residues 401–488) remains predominantly unfolded, and the 489–525 region is packed against XD, thus suggesting that beyond the α-MoRE the C terminus may play a role in the interaction with XD. Indeed, we present several lines of experimental evidence confirming that beyond the region encompassing the α-MoRE, an additional NTAIL region encompassing residues 517–525 also contributes to binding to XD, although without undergoing any gain of regular secondary structure.
Results
Small angle X-ray scattering (SAXS) studies of the NTAIL–XD complex
Small angle X-ray scattering (SAXS) is a valuable technique for the study of flexible, low compactness macromolecules in solution, which has already been successfully used to characterize NTAIL (Longhi et al. 2003). Therefore, we used SAXS to study the NTAIL–XD complex. To this endeavor, we cloned, expressed, and purified from the soluble fraction of Escherichia coli an N-terminally histidine-tagged form of NTAIL (i.e., NTAILHN) (data not shown). The identity of the recombinant products was confirmed by immunoprecipitation (IP) studies using anti-N Cl25 and anti-hexahistindine tag monoclonal antibodies (mAbs) (data not shown).
Beforehand, we checked whether XD possesses the same structure in solution as in the crystal. SAXS experiments performed on XD showed that it has a globular shape with a radius of gyration (Rg), extrapolated at a nil concentration, of 12.1 ± 0.8 Åand a maximum diameter Dmax of 41 ± 1 Å(data not shown). Comparison with the crystal structure using the program CRYSOL (Svergun et al. 1995) indicates that the scattering profile calculated for the crystal structure (PDB code 1OKS) is identical to the experimental one, fitting the data with a χ2 of 1.4 and giving a theoretical Rg of 12.3 Å(data not shown). These results suggest that the overall conformation of XD in solution is similar to that observed in the crystal.
The scattering profile of the NTAILHN–XD complex was obtained as described in Materials and Methods. Analysis of the curve in the low q region with the Guinier approximation gave an Rg of 32.7 ± 0.7 Å. The molecular mass (MM) calculated from the forward scattering intensity I(0) is 23.5 ± 2 kDa, in agreement with the value expected for a 1:1 stoichiometric complex (21.3 kDa), thus suggesting that the complex did form in solution. The high value of Rg indicated that the overall structure of the NTAILHN–XD complex is not compact. The distances distribution function inferred from the scattering curve of the NTAILHN–XD complex exhibits a maximum at 20 Å, with a shoulder at about 30 Å and a long tail up to 146 Å, typical of an elongated object (see Fig. 2A ▶). The bump most probably corresponds to the intramolecular distances within the globular portion of the complex (see below), while the tail indicates that NTAIL possesses regions with an extended conformation.
Figure 2.
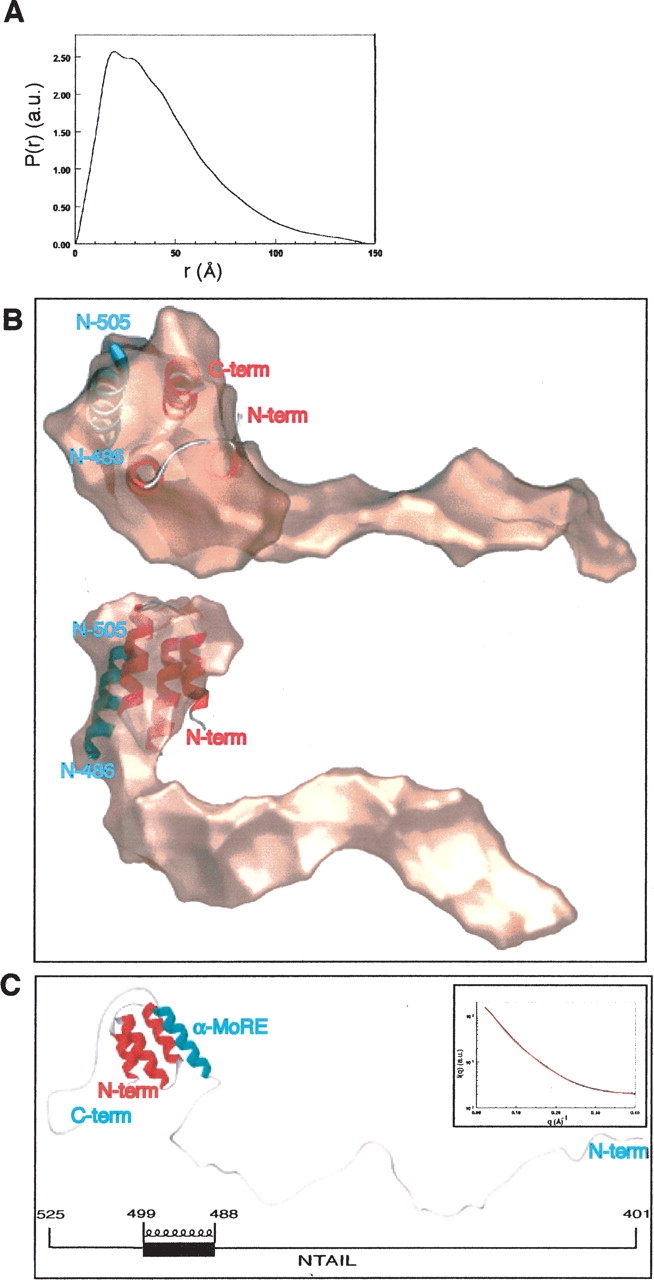
Low-resolution structure of the NTAIL–XD complex derived by SAXS. (A) Distance distribution function of the NTAIL–XD complex. (B) Overall shape of the NTAIL–XD complex, as obtained by GASBOR, shown in two orientations rotated by 90°, with the crystal structure of XD–NTAIL486–505 inserted in the bulky cluster. The XD molecule is shown in red, while the 486–505 region of NTAIL is shown in blue. (C) Low-resolution model of the NTAIL–XD complex provided by CREDO. Colors are as in B. The schematic organization of NTAIL, showing the location of the α-MoRE, is given below. The inset shows the experimental scattering curve (black circles) and fit (red line) obtained by CREDO with the low-resolution model.
The overall envelope of the complex was restored ab initio from its scattering profile using the program GASBOR (Svergun et al. 2001). Several independent runs yielded different shapes with recurrent features: they were all elongated, with a globular cluster of always the same size at one extremity and an elongated protuberance with varying bends and cross-sections. The quality of the fit to the experimental data was similar in all cases, with a χ2 of 1.3 to 1.6. The globular part most probably corresponds to XD packed against the folded region of NTAIL, while the outgrowth corresponds to unfolded regions of the latter. The crystal structure of the XD–NTAIL486–505 complex (PDB code 1T6O), which encompasses the α-MoRE (residues 488– 499), was inserted in the shape using the program SUPCOMB (Kozin and Svergun 2001) (Fig. 2B ▶). Interestingly, only one protuberance was observed, corresponding to the N-terminal area of NTAIL, and no other protuberance was restored from the shape determination in the opposite region of the globular cluster, thus suggesting that the C-terminal region of NTAIL (aa 506–525), although not visible per se, is packed inside the bulky portion of the complex (Fig. 2B ▶). In order to get further insights into the conformation that the regions of NTAIL encompassing residues 401–485 and 506–525 adopt in the complex with XD, we used the program package CREDO, which is an extension of GASBOR and calculates the position of missing loops in crystal structures. The results obtained with several independent runs using the crystal structure of XD–NTAIL486–505 as the template, revealed at one side a peptide chain in an extended conformation and at the other side a chain packed against the bulky portion of the XD–NTAIL486–505 complex. The extended conformation, which protrudes from the globular cluster of the complex and points to the solvent, corresponds to the 92-residue-long N-terminal region of the NTAILHN construct. In contrast, the more compact peptide chain, corresponding to the 20-residue-long C-terminal region of NTAIL (aa 506–525), always packs against the XD– NTAIL486–505 complex but at varying distances and positions. An example of one of the calculated conformations is shown in Figure 2C ▶. The experimental data were fitted by these models with similar χ2 of about 0.8–1.0 (see Fig. 2C ▶, inset). The various possible conformations provided by the program CREDO together with the extended structural properties of the complex (as indicated by the distance distribution function, the Rg and the restored shape, with respect to the number of amino acids of the complex) are typical of disordered polypeptide chains lacking a stable regular structure. Accordingly, the structure shown in Figure 2C ▶ represents only one possible conformation found by CREDO among many others. Indeed, CREDO restores only one conformation per run, and therefore cannot account for multiple conformations due to disorder when fitting the data. Therefore, although a unique conformation of the NTAIL region encompassing residues 506–525 cannot be derived, all the solutions provided by CREDO consistently revealed a packing of the C-terminal region of NTAIL against XD. The recurrence of these packed conformations suggests that the C-terminal region of NTAIL interacts with XD.
Moreover, we checked the influence of the Box3 conformation on the scattering curve by calculating the scattering profile of pseudomodels of the complex. In these pseudomodels, the N-terminal region of NTAIL is in the same conformation as in the credo model shown in Figure 2C ▶. The only difference concerns the orientation of Box3, which points out of the complex, in different solvent-exposed conformations. Noteworthy, the theoretical scattering curves calculated from these pseudomodels using CRYSOL (Svergun et al. 1995) poorly fit with the experimental curve, with an average χ2 of ~7.0 (data not shown). This confirms that the C terminus of NTAIL does contribute to the observed scattering profile, thus attesting the reliability of the model provided by CREDO.
Cloning, expression, and purification of NTAIL deletion constructs
In order to further explore the possible contribution of NTAIL regions other than the α-MoRE to XD binding, we have looked at the regions of homology conserved amongst members of the Morbillivirus genus (Diallo et al. 1994). These regions of homology are herein referred to as Box1, Box2, and Box3 and span N residues 401–420, 489–506, and 517–525, respectively (see Fig. 3A ▶), with the α-MoRE being located within Box2. We have then designed three deletion constructs bearing different combinations of these homology boxes (Fig. 3A ▶).
Figure 3.

(A) Schematic representation of NTAIL deletion proteins. (Top) Domain organization of N showing that it is composed of two regions, NCORE (aa 1–399) and NTAIL (aa 401–525). The epitope recognized by the anti-N mAbs, Cl 25 mAb (aa 457–476) is shaded. NTAILΔ3, NTAILΔ2,3, and NTAILΔ1 are devoid of Box3, Box2 plus Box3 and of Box1, respectively. The three NTAIL deletion proteins contain an N-terminal hexahistidine tag and a C-terminal Flag. The predicted α-helix (residues 489–504), as well as the α-MoRE (aa 488–499), i.e., the region shown to be involved in induced folding of NTAIL through binding to P (see Bourhis et al. 2004), are indicated. The position (aa 518) targeted for the Tyr → Trp substitution is highlighted by a black diamond. (B) Purification of NTAIL deletion proteins from E. coli. Coomassie blue staining of a 12% SDS-PAGE. (TF) Bacterial lysate (total fraction); (SN) clarified supernatant (soluble fraction); (IMAC) eluent from immobilized metal affinity chromatography; (GF) eluent from gel filtration.
The gene fragments encoding the different NTAIL deletion proteins were cloned into the pDest14 vector (Invitrogen) to yield N-terminally histidine-tagged and C-terminally flag-tagged recombinant products, the expression of which is under the control of the T7 promoter. In all cases, most recombinant protein was recovered from the soluble fraction of bacterial lysates (Fig. 3B ▶, lanes SN). The NTAIL deletion proteins were purified to homogeneity (>95%) in two steps: Immobilized Metal Affinity Chromatography (IMAC), and gel filtration (Fig. 3B ▶). The identity of the recombinant products was confirmed by IP studies using anti-N Cl25, anti-flag, and antihexahistindine tag mAbs (data not shown). As shown in Figure 3B ▶, the three NTAIL deletion proteins migrate in SDS-PAGE with an apparent MM of either 20 kDa (NTAILΔ3 and NTAILΔ1) or 18 kDa (NTAILΔ2,3) (expected MMs are 14.5, 13.4, and 11.5 kDa, respectively). This abnormal migratory behavior has already been documented for NTAIL, where mass spectrometry analysis and N-terminal sequencing gave the expected results (Longhi et al. 2003). The anomalous electrophoretic mobility is therefore due to a rather high content of acidic residues, as frequently observed in intrinsically disordered proteins (Tompa 2002). Likewise, the behavior of the truncated NTAIL proteins can probably be accounted for by this sequence bias composition.
The number of different conformations of the NTAIL deletion proteins is limited, as indicated by the sharpness of the peaks observed in gel filtration (data not shown). As expected for intrinsically disordered protein subdomains, the Stokes radius (RS) values, as inferred by gel filtration (27 ± 3 Å, 22 ± 3 Å, and 27 ± 3 Å for NTAILΔ3, NTAILΔ2,3, and NTAILΔ1, respectively), are consistent with extended conformations (see Materials and Methods). Thus, these deletion proteins share similar hydrodynamic properties with full-length NTAIL, all possessing an elongated shape.
Surface plasmon resonance studies
In order to directly measure the contribution of the different NTAIL boxes to binding, we have studied binding reactions between XD and NTAIL deletion proteins. Changes in surface plasmon resonance were monitored in real time as the NTAIL proteins passed over sensor chips to which XD was covalently coupled. This analytical approach is ideally suited to study reversible, low-affinity protein–protein interactions that typify interactions involving intrinsically disordered proteins (Wright and Dyson 1999; Dunker and Obradovic 2001; Dunker et al. 2001; Uversky 2002). Moreover, reaction rate and equilibrium constants were calculated for reactions with different protein substrates, allowing differences in XD binding affinities to be readily quantified.
Binding affinities between XD and NTAIL constructs were established using 180–225 RU of immobilized XD and NTAIL concentrations ranging from 0.1 to 10 μM (see Materials and Methods). Dosage-dependent binding was observed in this range. Reactions conformed to a 1:1 ligand-substrate (Langmuir) binding model, exhibiting an excellent fit (i.e., a χ2 value <1 and residuals within the range of ±2) following global analysis of sensorgrams. Binding reactions between XD and NTAILHNFC exhibit an equilibrium dissociation constant of 80 nM (see Table 1). The XD binding affinity for NTAILΔ1 (50 nM) is similar to that of NTAILHNFC indicating that Box1 does not participate in binding. In contrast, removal of either Box3 alone or Box2 plus Box3 results in a strong decrease (three orders of magnitude) in the equilibrium dissociation constant, where NTAILΔ3 and NTAILΔ2,3 display similar binding affinities (see Table 1). The strong decrease in the affinity resulting from removal of Box3 clearly indicates that Box2 is not the sole region involved in binding to XD, and attests that Box3 also plays a role in the interaction with XD, as already suggested by SAXS studies.
Table 1.
Calculated equilibrium dissociation constants (KD) between XD and NTAIL proteinsss
| Quality of fit | |||
| Analyte | Residuals | χ2 | KD (M) |
| NTAILHNFC | −1.5–1.5 | 0.676 | 8.1 × 10−8 |
| NTAILΔ3 | −0.6–0.4 | 0.043 | 1.2 × 10−5 |
| NTAILΔ2,3 | −2.4–2.4 | 0.919 | 4.1 × 10−5 |
| NTAILΔ1 | −1.2–1.2 | 0.239 | 4.9 × 10−8 |
Folding propensities of NTAIL deletion proteins
The far-UV circular dichroism (CD) spectra of NTAIL deletion proteins at neutral pH are typical of unstructured proteins, as seen by their large negative ellipticity at 198 nm and very low ellipticity at 185 nm (Fig. 4A–C ▶). The solvent 2,2,2-trifluoroethanol (TFE) mimics the hydrophobic environment experienced by proteins in protein–protein interactions, and is therefore widely used as a probe to unveil disordered regions having a propensity to undergo an induced folding (Hua et al. 1998). Thus, we have recorded CD spectra of NTAIL deletion proteins in the presence of increasing concentrations of TFE (Fig. 4A–C ▶). All proteins show an increasing gain of α-helicity upon addition of TFE, as indicated by the characteristic maximum at 190 nm and minima at 208 nm and 222 nm (Fig. 4A–C ▶). In the case of NTAILΔ3 and NTAILΔ1, most unstructured-to-structured transitions take place in the presence of 20% TFE, a concentration at which the α-helical content is estimated to be about 13% (using the ellipticity at 222 nm) (Fig. 4D ▶). On the other hand, in the case of NTAILΔ2,3, TFE concentrations as high as 30% are required for most pronounced unstructured-to-structured transitions to take place (see Fig. 4B ▶). Moreover, in the presence of 30% TFE, the α-helical content of NTAILΔ2,3 is not only lower (11%) than that (16%) of NTAILΔ1 and NTAILΔ3, but also lower than the α-helical content observed at 20% TFE for the two other NTAIL deletion proteins (Fig. 4D ▶). Therefore, NTAILΔ2,3 displays the lowest α-helical potential, while NTAILΔ1 and NTAILΔ3 exhibit a folding propensity similar to that observed for NTAIL (Longhi et al. 2003; Bourhis et al. 2004). These results, beyond confirming that the region spanning residues 489–516 affects the folding potential of NTAIL (Bourhis et al. 2004), suggest that neither Box1 nor Box3 contribute to the α-helical propensity of NTAIL, in agreement with the secondary structure prediction provided by PSIPRED (McGuffin et al. 2000) and PHD (Rost 1996), which both predict an α-helix (residues 489–504, see Fig. 3A ▶) as the sole secondary structure element within NTAIL.
Figure 4.

Far-UV CD spectra and analysis of the α-helical propensities of NTAIL deletion proteins. Far-UV CD spectra of NTAILΔ3 (A) NTAILΔ2,3 (B) and NTAILΔ1 (C) at 0.1 mg/mL in 10 mM sodium phosphate at pH 7 in the presence of increasing concentrations of TFE (0%, 10%, 20%, and 30%) recorded at 20°C. Each spectrum is the mean of three independent acquisitions. (D) α-Helical content of NTAIL deletion proteins in the presence of increasing TFE concentrations. The α-helical content was derived from the ellipticity value at 222 nm as described in Myers et al. (1997a).
Induced folding of NTAIL deletion proteins in the presence of XD
In order to investigate whether the NTAIL deletion proteins retained the ability to undergo induced folding in the presence of XD, we have used far-UV CD spectroscopy. The NTAILHNFC protein, bearing an N-terminal hexahistidine tag and a C-terminal flag was purified from the soluble fraction of E. coli (data not shown) and used as the reference in these experiments to allow direct comparison with the NTAIL truncated proteins. Noticeably, the CD spectrum of NTAILHNFC is fully superimposable on that of NTAILHN (data not shown), thus ruling out the possibility that the flag sequence might affect the folding properties of the protein.
The far-UV CD spectrum of XD (Fig. 5A–D ▶, gray line) is typical of a structured protein with a predominant α-helical content, as indicated by the positive ellipticity between 185 nm and 200 nm, and by the two minima at 208 nm and 222 nm. After mixing NTAILHNFC with different molar excesses of XD, the observed CD spectra differed from the corresponding theoretical average curves calculated from the individual spectra. Since the theoretical average curves correspond to the spectra that would be expected if no structural variations occur, deviations from these curves indicate structural transitions. The results obtained in the presence of a threefold molar excess of XD indicate a random coil to α-helix transition, as judged by the much more pronounced minima at 208 nm and 222 nm, and by the higher ellipticity at 190 nm of the experimentally observed spectrum compared to the corresponding theoretical average curve (see Fig. 5A ▶). Although even more dramatic structural transitions of full-length NTAIL have been previously observed with a twofold molar excess of XD (Johansson et al. 2003), the results obtained with a threefold molar excess have been selected for presentation in order to allow direct comparison with the NTAIL deletion proteins (see below).
Figure 5.

Induced folding on NTAIL deletion proteins in the presence of XD. Far-UV CD spectra of NTAILHNFC (A), NTAILΔ3 (B), NTAILΔ2,3 (C), and NTAILΔ1 (D) either alone (black line) or in the presence of a threefold molar excess of XD (full circles). The CD spectrum of XD alone (gray line), as well as the theoretical average curves calculated by assuming that no structural variations occur (see Materials and Methods) are also shown (open circles).
Figure 5 ▶, panels B–D, show the results obtained for the three NTAIL deletion proteins in the presence of a threefold molar excess of XD, a condition leading to the most dramatic structural transitions. Removal of Box3 significantly reduces, but does not abrogate, the folding ability of NTAIL. Indeed, the experimental CD spectrum does not significantly deviate from the average curve in the 200–260 nm region, but it considerably deviates from the average curve in the 185–195 region (58% mean increase of ellipticity) (Fig. 5B ▶), thus supporting partial folding ability of NTAILΔ3 in the presence of XD.
Further removal of Box2 results in a truncated NTAIL form, which has completely lost its ability to fold in the presence of XD, as indicated by the good superimposition between the experimental and the average spectra (Fig. 5C ▶). Conversely, removal of Box1 does not affect the folding ability of NTAIL, as the deviations from the average spectrum are similar to those observed with the full-length form (Fig. 5 ▶, cf. D and A). The mean increase in ellipticity in the 185–195 nm region (77%) and the decrease in the ellipticity value at 220 nm (46%) observed with NTAILΔ1, are comparable to the corresponding values observed with NTAILHNFC (70% and 46%, respectively).
As a control, we recorded CD spectra of NTAILHNFC in the presence of lysozyme (data not shown). The absence of significant structural variations even with molar excesses as high as 5, confirms the specificity of the deviations observed upon addition of XD to the NTAIL proteins.
A two- and threefold molar excess of XD is required to induce the most pronounced structural transitions of full-length and truncated forms of NTAIL, respectively. In contrast, an equimolar amount of PCT is sufficient to produce the same effect (Longhi et al. 2003; Bourhis et al. 2004). This difference can be accounted for by a lower affinity of NTAIL towards XD, compared to PCT, rather than to the formation of a 1:2 or 1:3 stoichiometric complex. Indeed, formation of a 1:1 stoichiometric complex between XD and a peptide corresponding to residues 477–505 of N, has been documented using isothermal titration calorimetry (Kingston et al. 2004b). The higher affinity of NTAIL for PCT compared to XD could be ascribed either to cooperativity phenomena among the different XDs within the PCT tetramer (Rahaman et al. 2004) or to the possible contribution of other PCT regions to binding. This latter possibility can be ruled out based on recent data pointing out XD as the sole PCT region contributing to binding (Kingston et al. 2004b; S. Longhi and M.J. Oglesbee, unpubl.).
In conclusion, these results indicate that (1) Box1 is fully dispensable for binding to XD and induced folding, (2) Box2 is strictly required for induced folding to take place, and (3) Box3 contributes to binding to XD, as pointed out by the reduced ability of NTAILΔ3 to undergo induced folding. Moreover, the fact that NTAILΔ3, NTAILΔ1, andNTAIL (Longhi et al. 2003; Bourhis et al. 2004) share similar folding propensities suggests that the contribution of Box3 to the interaction can be accounted for more in terms of binding rather than of induced folding.
Fluorescence spectroscopy studies
In order to further characterize the contribution of Box3 to binding, we have used fluorescence spectroscopy. Accordingly, we have designed an N-terminally hexahistidine- tagged NTAIL variant form bearing a tyrosine to tryptophan substitution at position 518 (see also Fig. 3A ▶). Introduction of a tryptophan residue in Box3 allowed binding events to be followed by fluorescence spectroscopy, while maximizing the conservative nature of the substitution (note that neither NTAIL nor XD contain any tryptophan residue).
Most recombinant product was purified from the soluble fraction of the bacterial lysate (Fig. 6A ▶). The identity of the recombinant product was confirmed by IP studies using anti-N Cl25, and anti-hexahistindine tag mAbs (data not shown). As already observed in the case of full-length NTAIL and NTAIL deletion proteins, NTAILW518 migrates in SDS-PAGE with an apparent MM higher than expected (Fig. 6A ▶). The mutated protein, displays the same gel filtration elution profile as the wt form (data not shown), leading to an estimated RS of 27 ± 3 Å.
Figure 6.

(A) Purification of NTAILW518 from E. coli. Coomassie blue staining of a 12% SDS-PAGE. Purified NTAILW518 protein. (B) Far-UV CD spectra of NTAILHN and NTAILW518. The spectra were recorded on a 0.1 mg/mL protein solution in 10 mM sodium phosphate (pH 7) at 20°C, and represent the mean of three independent acquisitions. (C) Fluorescence spectroscopy studies of NTAILW518. The relative fluorescence increase of NTAILW518 (1 μM in 10 mM sodium phosphate at pH 7) is plotted as a function of the XD concentration. The KDapp value (see text) results from the fitting of the data to a single exponential.
As shown in Figure 6B ▶, the far-UV CD spectrum of NTAILW518 is almost perfectly superimposable on that of NTAILHN, thus indicating that the introduction of the tryptophan residue does not affect the overall secondary structure content of the protein. In order to investigate whether the variant form retained the ability to undergo induced folding in the presence of XD, we have added various molar excesses (ranging from 1 to 4) of XD to NTAILW518, and recorded the corresponding far-UV CD spectra (data not shown). These latter studies indicate that the tyrosine to tryptophan substitution does not affect the ability of the protein to undergo induced folding in the presence of the partner, thus supporting the biochemical relevance of this variant form.
Fluorescence spectroscopy studies showed that NTAILW518 has a maximum of emission at 356 nm, indicating that Trp 518 is fully exposed to the solvent (data not shown). Addition of gradually increasing XD concentrations triggers an increase in the fluorescence intensity in a dose-dependent manner, which indicates a modification in the pattern of interactions with neighboring groups. At the same time, addition of XD causes a progressive shift in the emission maximum from 356 nm to 352 nm (data not shown), thus indicating that Trp 518 becomes only slightly less exposed to the solvent.
No significant variations are observed in the fluorescence spectrum obtained after addition to NTAILW518 of a 2 μM solution of an irrelevant protein (SARS virus, unclassified protein 5) of similar size and devoid of tryptophan residues (data not shown). After plotting the relative fluorescence intensity increase as a function of the XD concentration (Fig. 6C ▶), an apparent constant equilibrium dissociation (KDapp) value of 133 ± 32 μM is derived. This value is in good agreement with the value obtained by surface plasmon resonance studies with wt NTAIL. Although the environment of Trp 518 remains mostly polar upon binding to XD, the observed increase in the fluorescence intensity indicates that the chemical environment of Trp 518 is affected, thus further suggesting that the C terminus of NTAIL interacts with XD.
Two-dimensional Heteronuclear Magnetic Resonance (NMR)
In order to further explore the nature of the interaction established between Box3 and XD, we have used NMR spectroscopy. To this endeavor, we have recorded a HSQC spectrum of 15N uniformly labeled NTAILHN either alone or in the presence of a twofold molar excess of XD, as well as of 15N uniformly labeled NTAILΔ3 in the same conditions.
This analysis allowed a quantitative estimation of the number of residues involved in the interaction with XD by following chemical shift changes in the backbone amide and proton resonances upon addition of unlabeled XD. Upon addition of XD to NTAILHN, 16 correlation peaks are displaced. Among them, 11 undergo an upfield shift (see Fig. 7 ▶, stars), which indicates a random coil to α-helix transition, and two correspond to the side chain of either a Gln or an Asn. Additionally, at least seven additional peaks undergo a less dramatic displacement (see Fig. 7 ▶, diamonds). Because of its small amplitude, this shift most likely does not reflect a conformational change. Nevertheless, it is indicative of a change in the chemical environment of these residues resulting either by direct interaction with the partner or by local magnetic perturbations of residues spatially close to the newly formed α-helix. Based on this experiment, however, the determination of NTAIL residues involved in these two types of shifts is not possible. The precise identification of these residues would require the full assignment of the NTAIL HSQC spectrum, which is complicated by the strong signal overlapping inherent in the predominantly unfolded nature of NTAIL.
Figure 7.

2D-HSQC NMR spectra. HSQC spectrum of a 0.5 mM solution of purified 15N-NTAILHN alone (black) or in the presence of a twofold molar excess of XD (red), of a 0.125-mM solution of purified 15N-NTAILΔ3 alone (green) or in the presence of a twofold molar excess of XD (blue). All proteins were in 10 mM sodium phosphate at pH 7. All spectra are recorded at 283 K. ppm quotes for resonance shifts in parts per million of the spectrophotometer frequency. Some peaks are labeled as follows: stars indicate peaks present in spectra of both complexes only, while diamonds quote for peaks present in the spectrum recorded with the 15N-NTAILHN–XD complex, but not with the 15N-NTAILΔ3–XD complex. The inset shows the purified 15N-NTAILΔ3 and 15N-NTAILHN proteins.
Comparison of present results to those of the NMR spectroscopy studies of Kingston et al. (2004a) suggest that the 11 residues undergoing the random coil to α-helix transition are most likely located within the 486–503 region. In order to assess whether the additional NTAIL residues undergoing a less pronounced displacement upon binding to XD are located within Box3, we recorded a HSQC spectrum of 15N uniformly labeled NTAILΔ3 either alone or with a twofold molar excess of XD. As shown in Figure 7 ▶, the same peaks undergoing the large displacement in the NTAILHN–XD complex are also observed in the NTAILΔ3–XD complex. In particular, among these peaks, the occurrence in both complexes of the 11 peaks that undergo the random coil to α-helix transition (see Fig. 7 ▶, stars), provides further support that the helical folding occurs within Box2. On the other hand, the seven peaks that undergo a less dramatic displacement upon complex formation with XD (see Fig. 7 ▶, diamonds) are not present in the spectrum recorded on the NTAILΔ3–XD complex. This latter observation indicates that these seven peaks correspond to residues that are located within Box3.
In conclusion, these experiments reveal that complex formation between NTAIL and XD implies two types of interaction: one, mediated by residues belonging to Box2, involves a significant gain of α-helicity, while the other, attributable to Box3 residues, is not accompanied by a significant structural transition.
Discussion
In this paper we show that NTAIL remains predominantly unfolded after binding to XD, with two distinct sites being involved in the interaction. Although the XDinduced gain of regular secondary is restricted to Box2 (aa 489–504), the region encompassing residues 489–504 is not the only NTAIL region involved in binding to XD. In particular, we present several lines of evidence indicating that the extreme C terminus of NTAIL (Box3, aa 517–525) also contributes to binding, without however gaining any regular secondary structure.
The C terminus of NTAIL is an additional XD binding site
SAXS studies suggest that the C terminus of NTAIL interacts with XD. In particular, the calculated overall envelope of the complex shows the presence of a globular cluster of invariant size at one extremity and of an elongated protuberance with varying shapes. The elongated protuberance corresponds to the 92-residue-long N-terminal region of NTAIL, while the more compact region accommodates the structure of the XD–NTAIL486–505 complex as well as the 20 C-terminal residues of NTAIL. Data from overall shape calculations thus clearly indicate that the C terminus is not protruding towards the solvent, and remains close to XD. On the other hand, attempts to more precisely model the conformation adopted by the C-terminal region of NTAIL within the complex led to an ensemble of solutions. In all these solutions, the C-terminal region of NTAIL always packs against the XD–NTAIL486–505 complex, rather than being extended and exposed to the solvent. Identification of the possible gain of regular secondary structure within the C-terminal region of NTAIL is beyond the resolution limits of SAXS. However, results provided by CD, fluorescence spectroscopy, and heteronuclear NMR all converge to suggest that the C-terminal region of NTAIL does not gain any regular secondary structure (see below). Besides supporting a role for the C-terminal region of NTAIL in the interaction with XD, these data also indicate that most of NTAIL remains disordered within the complex. The prevalent disorder of NTAIL within the complex is in agreement with other data reported in the literature where an intrinsically disordered protein largely preserves its overall extended conformation even after interaction with a binding partner (see Tompa 2002, and references cited therein; Permyakov et al. 2003). Finally, it is noteworthy that in the SAXS model the N terminus of XD is exposed to the solvent (see Fig. 2B ▶), a position that would accommodate the remaining part of P.
The involvement of additional NTAIL regions other than the α-MoRE in binding to XD is confirmed by surface plasmon resonance studies, where the contribution of Box3 to XD binding can be quantitatively estimated. Removal of either Box3 alone or Box2 plus Box3 leads to a strong decrease (three orders of magnitude) in the affinity as compared to full-length NTAIL, thus indicating that Box3 contribution to binding is similar to that of Box2.
The additional XD binding site does not gain any regular secondary structure
Spectroscopy studies showed that Box3 contributes to binding to XD but does not undergo any gain of regular secondary structure. Although Box3 does not affect the folding potential of NTAIL, as indicated by CD studies in the presence of TFE, the removal of Box3 significantly reduces the ability of NTAIL to undergo induced folding in the presence of XD. This supports a role for Box3 in binding to XD. Further removal of Box2 results in a truncated form that has a significantly decreased folding potential and has lost the ability to undergo induced folding in the presence of XD. These results are consistent with the unique α-helical forming potential of Box2 and its role as primary binding site for XD.
Contribution of Box3 to binding without dramatic structural change is supported by fluorescence spectroscopy data, which show an increase in the fluorescence intensity of NTAILW518 upon addition of XD. Increases in the fluorescence intensity upon binding to a partner/ligand have been already documented, for both folded (Bette et al. 2002) and intrinsically unstructured proteins (Raggett et al. 1998), and indicate that the chemical environment of the Trp, although remaining mostly polar, is changed as a result of the addition of the partner. However, the fluorescence data do not enable us to discriminate between a direct Trp 518–XD interaction and secondary effects resulting from local perturbations triggered by a direct Box2–XD interaction.
That complex formation with XD involves both Box2 and Box3, and that only Box2 undergoes a gain of α-helicity, is confirmed by NMR studies. Heteronuclear NMR experiments show that upon complex formation with XD, 11 NTAIL residues, belonging to Box2, undergo a random coil to α-helix transition, while at least seven additional residues undergo a shift in their chemical environment not accompanied by the gain of regular secondary structure elements. Our data show that these latter peak displacements correspond to residues belonging to Box3, as indicated by the absence of such peaks in both the spectra of NTAILΔ3 alone and in complex with XD. Thus, the NMR studies, beyond confirming the role of Box3 in the interaction with XD, also highlight that binding of NTAIL to XD implies two types of interaction, where gain of regular secondary structure is restricted to only one of the binding determinants, Box2.
Structural and functional insights into the interaction between NTAIL and XD
The accommodation of the 486–505 region of N within XD (Kingston et al. 2004a) triggers some minor rearrangements at the surface of the latter, compared to the crystal structure of the uncomplexed form (Johansson et al. 2003). In particular, the largest movements are observed for the side chains of residues Arg15 and Glu17 of XD (corresponding to residues 472 and 474 of P). The Arg15 NH2 atom moves 9.7 Å away in the chimeric structure compared to the structure of XD alone, while the Glu17 Cδ atom undergoes a shift of 2.5 Å. These two residues are both located within the loop connecting α1 and α2 helices, and occur at least 9 Å away from Box2, thus ruling out the possibility that the observed spatial rearrangements they undergo could be ascribed to Box2 embedding. The significant displacement they undergo in the complex therefore indicates that binding of Box2 induces local conformation changes in XD that could favor interaction with Box3. The surface displaced residues of XD could, in fact, be part of a Box3 binding site.
We tentatively propose that binding to XD might take place through a sequential mechanism involving successive binding of disordered domains, as it has been recently reported for p27 upon binding to the CdK2–cyclin A complex (Lacy et al. 2004).
The KD value between NTAIL and XD, as measured by both fluorescence spectroscopy and surface plasmon resonance, is in the 100 nM range. Surprisingly, this value is considerably lower than that reported by Kingston et al. (2004b) (13 μM) and derived from isothermal titration calorimetry studies. A weak binding affinity, associated with a fast association rate, would ideally fulfill the requirements of a polymerase complex, which has to cartwheel on the nucleocapsid template during both transcription and replication. However, a KD in the μM range would not seem to be physiologically relevant considering the low intracellular concentrations of P in the early phases of infection. Moreover, such a weak affinity is not consistent with the ability to readily purify nucleocapsid–P complexes using rather stringent techniques such as CsCl isopycnic density centrifugation (Robbins and Bussell 1979; Stallcup et al. 1979; Robbins et al. 1980; Oglesbee et al. 1989). Our data support a higher affinity between P and N, resulting in a stable P–NTAIL complex that would be predicted to hinder processive movement of P along the nucleocapsid template. In agreement with this prediction, Box3 has been shown to have an inhibitory role upon transcription and replication, as indicated by previous minireplicon experiments, where deletion of Box3 enhanced basal reporter gene expression (Zhang et al. 2002). We can speculate that the transient nature of the NTAIL–XD interaction might be ensured by the possible intervention of cellular and/or viral cofactors. Indeed, the requirement for cellular or viral cofactors in both transcription and replication has been already documented in the case of measles (Vincent et al. 2002), respiratory syncytial (Fearns and Collins 1999), and Ebola viruses (Hartlieb et al. 2003). These cofactors may serve as processivity or transcription elongation factors and could act by modulating the strength of the interaction between the polymerase complex and the nucleocapsid template. Such a mechanism may explain the stimulatory effect of hsp72 on MV transcription and genome replication, an effect mediated by Box3–hsp72 interaction (Zhang 2002). In this capacity, hsp72 may neutralize the contribution of Box3 to a more stable complex between P and NTAIL, thereby promoting successive cycles of P binding and release that are essential to polymerase processivity.
Conclusion
Using different physico-chemical approaches we have shown that the interaction of NTAIL with XD involves an additional, previously unreported site located at the extreme C terminus of NTAIL. While the primary site (i.e., Box2) gains α-helical structure upon binding to XD, this additional site does not gain any regular secondary structure elements. Nevertheless, it plays a crucial role in stabilizing the complex.
We have previously shown that NTAIL belongs to the premolten globule subfamily, e.g., it possesses a certain extent of residual secondary and/or tertiary structure (Longhi et al. 2003; Bourhis et al. 2004). In agreement with these findings, NMR studies showed that the NTAIL region encompassing residues 486–503 is not a statistical random coil (Kingston et al. 2004a). These results, supporting a restricted conformational freedom of this NTAIL region, are in agreement with the speculation that residual structure within NTAIL may play a role for efficient binding to P (Longhi et al. 2003; Bourhis et al. 2004). That residual structure within intrinsically disordered proteins may favor the folding process triggered by binding to a partner has already been reported (Bienkiewicz et al. 2002; Fuxreiter et al. 2004; Csizmok et al. 2005).
NTAIL provides an interesting model system for the study of the interaction of an intrinsically disordered protein and its partners. Its multiple-site mode of interaction well illustrates the complexity of the contacts established by intrinsically disordered proteins and emphasizes the need for thorough investigations of the molecular mechanisms underlying recognition of the partner. Indeed, binding of disordered regions to their targets involves different types of interactions, such as binding coupled to folding, binding with no gain of regular secondary structure, or coexistence of both interaction mechanisms (for a review, see Dyson and Wright 2005).
Binding coupled to folding has been reported for the phosphorylated kinase-inducible domain (pKID) of CREB, which undergoes a coil to α-helix folding transition upon binding to the KIX domain of the transcription coactivator CBP (CREB Binding Protein) (Radhakrishnan et al. 1997). The amphipathic helix αB of pKID interacts with a hydrophobic groove defined by helices α1 and α3 of the partner. The other pKID helix, αA, contacts a different face of the α3 helix. This mode of interaction is reminiscent of that occurring between NTAIL and XD. Similarities concern the involvement of two distinct sites within the disordered domain (where Box2 and Box3 resemble helices αB and αA, respectively) and embedding of an α-helix within a hydrophobic cleft delimited by α-helices from the structured partner. The difference concerns the mode of interaction of the second site, where Box3 of NTAIL does not gain any regular secondary structure element, contrary to αA of pKID. Nevertheless, complexes with dual interaction have already been described in the literature. Among them, we mention the case of the binding of p27 to the cyclinA–Cdk2 complex (Lacy et al. 2004), and that of the activation domain of CITED2 to the TAZ1 domain of CBP (De Guzman et al. 2004).
On the other hand, binding without any concomitant gain of regular secondary structure, has been also reported. This is the case of the unfolded proteins 4E–BP1 (4E binding protein 1), an inhibitor of translation. Its binding to eIF4E does not involve any transition to stable regular secondary structure (Fletcher et al. 1998). Interestingly, when eIF4E binds to another partner, namely eIF4G, it induces a coil-to-helix transition in the latter (Marcotrigiano et al. 1999), highlighting the diversity of the structural transitions that can be triggered by a structured partner.
Finally, there are a few examples of proteins that undergo different structural transitions as a function of the partner they bind. Notably, HIFα (hypoxia-inducible factor α) possesses an intrinsically disordered domain, which can interact with two different partners. This unstructured domain adopts an α-helical structure when bound to the TAZ1 domain of CBP. However, binding of the same region of HIFα to an asparagine hydroxylase results in a highly extended conformation, which is required for the enzymatic activity of the latter. These findings provide an elegant example of the plasticity that intrinsically disordered proteins display for different partners involved in different functions. We can speculate on a similar behavior in the case of NTAIL. In particular, Box2 and Box3 of NTAIL interact both with at least two distinct partners, P and Hsp72, and competition between XD and Hsp72 for binding to NTAIL has been recently shown (Zhang et al. 2005). It is conceivable that binding of NTAIL to P or to Hsp72 may involve a different structural transition, as in the case of HIFα.
Last, but not least, our findings, beyond contributing to elucidate the dynamics of the interactions established by intrinsically disordered proteins, provide an interesting target site for mutational studies aimed at exploring further the mode of interaction between NTAIL and XD.
Materials and methods
Bacterial strains and media
The E. coli strains DH5α (Stratagene) was used for selection and amplification of DNA constructs. The E. coli strains Rosetta [DE3] pLysS (Novagen) and C41 [DE3] (Avidis) were used for expression of recombinant proteins. E. coli was grown either in Luria-Bertani (LB) medium, or in minimal M9 medium supplemented with 15NH4Cl.
Chemicals and antibodies
Pfu polymerase was from Promega. Primers were purchased from Invitrogen. The anti-hexahistidine tag mAb was purchased from Qiagen. The anti-flag mAb was purchased from Sigma. The anti-N Cl 25 (Giraudon et al. 1988; Buckland et al. 1989) mAb was kindly provided by D. Gerlier.
Construction of protein expression plasmids
The XD gene construct, encoding residues 459–507 of the MV P protein (strain Edmonston B) with an hexahistidine tag fused to its C terminus, has already been described (Johansson et al. 2003).
All NTAIL constructs were obtained by PCR using the MV N gene, strain Edmonston B, as template. The NTAILHN gene construct, encoding residues 401–525 of the MV N protein with a hexahistidine tag fused to its N terminus, was obtained using the plasmid pET21a/N (encoding the MV N protein; Karlin et al. 2002a) as template. Forward primer (5′-gatagaaccatgCATCATCATCATCATCATactactgaggacaagatcagtaga-3′) was designed to introduce a hexahistidine tag encoding sequence (upper case) at the N terminus of NTAIL, while reverse primer (5′-ggggaccactttgtacaagaaagctgggtcttagtctagaagatttctgtcattgta-3′) was designed to introduce an AttB2 site (bold). The PCR amplification product was further used as template in a second PCR step, using forward primer (5′-ggggacaagtttgtacaaaaaagcaggcttcgaaggagatagaaccatgCATCATCATCAT-3′), designed to introduce an AttB1 site (bold), and reverse primer as above. After purification (PCR Purification Kit; Qiagen), the PCR product was cloned into the pDest14 vector (Invitrogen) using the Gateway recombination system (Invitrogen). The final construct is referred to as pDest14/NTAILHN.
The NTAILHNFC gene construct, encoding residues 401–525 of the MV N protein with an N-terminal hexahistidine tag and a C-terminal Flag sequence (dykddddk) (Brizzard et al. 1994), was obtained using the plasmid pDest14/NTAILHN as template. Forward primer (5′-ggggacaagtttgtacaaaaaagcaggcttcgaaggagatagaaccatgCATCATCATCAT-3′) was designed to introduce an AttB1 site (bold), and reverse primer (5′-gtcttaTTTGTCGTCATCGTCTTTATAATCgtctagaagatttctgtcattgta-3′) was designed to introduce a Flag encoding sequence (upper case). The PCR amplification product was further used as template in a second PCR step, using the same forward primer as above, and reverse primer (5′-ggggaccactttgtacaagaaagctgggtcttaTTTGTCGTCATCGTCTTT-3′), which was designed to introduce an AttB2 site (bold). After purification, the PCR product was cloned into the pDest14 vector to yield pDest14/NTAILHNFC.
The NTAILΔ3 gene construct, encoding residues 401–516 of the MV N protein with an N-terminal hexahistidine tag plus a C-terminal Flag sequence, was obtained using the plasmid pet21a/N as template. Forward primer (5′-gatagaaccatgCATCATCATCATCATCATactactgaggacaagatcagtaga-3′) was designed to introduce a hexahistidine tag encoding sequence (upper case) at the N terminus of NTAIL, and reverse primer (5′-gtcttaTTTGTCGTCATCGTCTTTATAATCtataggggtgtccgtgtctgagcc-3′) was designed to introduce a Flag encoding sequence. The PCR amplification product was further used as template in a second PCR step, using forward primer (5′-ggggacaagtttgtacaaaaaagcaggcttcgaaggagatagaaccatgCATCATCATCAT-3′) and reverse primer (5′-ggggaccactttgtacaagaaagctgggtcttaTTTGTCGTCATCGTCTTT-3′), which were designed to introduce an AttB1 and an AttB2 site (bold), respectively. After purification, the PCR product was cloned into the pDest14 vector to yield pDest14/NTAILΔ3.
The NTAILΔ2,3 gene construct, encoding residues 401–488 of the MV N protein with an N-terminal hexahistidine tag plus a C-terminal Flag sequence, previously referred to as NTAIL2 (Bourhis et al. 2004), was obtained using the plasmid pet21a/N as template. Forward primer (5′-gatagaaccatgCATCATCATCATCATCATactactgaggacaagatcagtaga-3′) was designed to introduce a hexahistidine tag encoding sequence (upper case) at the N terminus of NTAIL, and reverse primer (5′-gtcttaTTTGTCGTCATCGTCTTTATAATCactgtcctgcggatcttggctgga-3′) was designed to introduce a Flag encoding sequence (upper case). The PCR amplification product was further used as template in a second PCR step, using the same pair of primers as in the second PCR step, which yielded the NTAILΔ3 amplification product. After purification, the PCR product was cloned into the pDest14 vector to yield pDest14/NTAILΔ2,3.
The NTAILΔ1 gene construct, encoding residues 421–525 of the MV N protein with an N-terminal hexahistidine tag plus a C-terminal Flag sequence, was obtained using the plasmid pet21a/N as template. Forward primer (5′-gatagaaccatgCATCATCATCATCATCATcacggtgatcaaagtgagaatgag-3′) was designed to introduce a hexahistidine tag encoding sequence (upper case) at the N terminus of NTAIL, and reverse primer (5′-gtcttaTTTGTCGTCATCGTCTTTATAATCgtctagaagatttctgtcattgta-3′) was designed to introduce a Flag-encoding sequence (upper case). The PCR amplification product was further used as template in a second PCR step, using the same pair of primers as in the second PCR step, which yielded both NTAILΔ3 and NTAILΔ2,3 amplification products. After purification, the PCR product was cloned into the pDest14 vector to yield pDest14/NTAILΔ1.
The NTAILW518 gene construct, encoding residues 401–525 of the MV N protein with a Tyr → Trp substitution at position 518 and with a hexahistidine tag fused to its N terminus, was obtained by PCR, using the plasmid pET21a/N as template. Two separate PCR steps were carried out in parallel, yielding amplification products A and B. Product A was obtained using forward primer (5′-gatagaaccatgCATCATCATCATCATCATactactgaggacaagatcagtaga-3′), designed to introduce a hexahistidine tag-encoding sequence (upper case) at the N terminus of NTAIL, while reverse primer (5′-aaga tttctgtcattCCAcactatTggggtgtc-3′) was designed to introduce a Trp at position 518 (bold and upper case) and a silent mutation at nucleotide position 1545 (upper case) thus resulting in the introduction of a BstXI site (underlined). Product B was obtained using forward primer (5′-gacaccccAatagtgTGGaatgacagaaatctt-3′) designed to introduce a Trp at position 518 (bold and upper case) and a silent mutation at nucleotide position 1545 (upper case) thus resulting in the introduction of a BstXI site (underlined), and reverse primer (5′-ccgggcatgcatccggatatagttcctcctt-3′) designed to anneal with nucleotide positions 1755–1775 of pet21a/N (where the A of the ATG codon of the N ORF was set as nucleotide position 1). A further PCR step was carried out using amplification products A plus B as template, to yield the NTAIL W518 amplification product. Forward primer (5′-ggggacaagtttgtacaaaaaagcaggcttcgaaggagatagaaccatgCATCATCATCAT-3′) was designed to introduce an AttB1 site (bold), while reverse primer (5′-ggggaccacttttgtacaagaaagctgggtcttagtctagaagatttctgtcattCCA-3′) was designed to introduce an AttB2 site (bold) and a Trp at position 518 (upper case). After purification, the PCR product was cloned into the pDest14 vector using the Gateway recombination system. The final construct is referred to as pDest14/NTAILW518. Candidate clones bearing the desired mutation were selected on the basis of the ability of their recombinant plasmids to be restricted by BstXI, a unique restriction site introduced by PCR together with the Tyr to Trp substitution.
The sequence of the coding region of all expression plasmids was verified by sequencing (MilleGen).
Expression of NTAIL constructs
E. coli strain Rosetta [DE3] (Novagen) was used for the expression of NTAIL constructs. Since the MV N gene contains several rare codons that are used with a very low frequency in E. coli, coexpression ofNTAIL constructs with the plasmid pLysS (Novagen) was carried out. This plasmid, which supplies six rare tRNAs, carries also the lysozyme gene, thus allowing a tight regulation of the expression of the recombinant gene, as well as a facilitated lysis. Cultures were grown overnight to saturation in LB medium containing 100 μg/mL ampicilin and 17 μg/mL chloramphenicol. An aliquot of the overnight culture was diluted 1/25 in LB medium and grown at 37°C. At OD600 of 0.7, isopropyl β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.2 mM, and the cells were grown at 37°C for 3 h. The induced cells were harvested, washed, and collected by centrifugation. The resulting pellets were frozen at −20°C.
Isotopically substituted (15N) NTAILHN and NTAILΔ3 were prepared by growing transformed bacteria in minimal M9 medium supplemented with 15NH4Cl (0.8 g/L). A 50-mL preculture grown overnight to saturation in LB medium containing 100 μg/mL ampicillin and 17 μg/mL chloramphenicol, was harvested, washed in minimal M9 medium, and inoculated into 1 L of minimal M9 medium supplemented with ampicillin and chloramphenicol. The culture was grown at 37°C. At OD600 of 0.5, IPTG was added to a final concentration of 0.2 mM and the cells were grown first at 37°C for 3 h, and then over night at 28°C. The induced cells were harvested, washed, and collected by centrifugation. The resulting pellets were frozen at −20°C.
Expression of tagged XD was carried out as described in Johansson et al. (2003).
Purification of NTAIL proteins
Cellular pellets from bacteria transformed with the different NTAIL expression plasmids were resuspended in 5 volumes (v/w) buffer A (50 mM sodium phosphate at pH 8, 300 mM NaCl, 10 mM Imidazole, 1 mM phenyl-methyl-sulphonyl-fluoride [PMSF]) supplemented with lysozyme 0.1 mg/mL, DNase I 10 μg/mL, protease inhibitor cocktail (Sigma) (50 μL/g cells). After a 20-min incubation with gentle agitation, the cells were disrupted by sonication (using a 750 W sonicator and four cycles of 30 sec each at 60% power output). The lysate was clarified by centrifugation at 30,000g for 30 min. Starting from a 1-L culture, the clarified supernatant was incubated for 1 h with gentle shaking with 4 mL Chelating Sepharose Fast Flow Resin preloaded with Ni2+ ions (Amersham Pharmacia Biotech), previously equilibrated in buffer A. The resin was washed with buffer A, and the NTAIL proteins were eluted in buffer A containing 250 mM imidazole. Eluates were analyzed by SDS-PAGE for the presence of the desired product. The fractions containing the recombinant product were combined, and concentrated using Centricon Plus-20 (molecular cutoff, 5000 Da) (Millipore). The proteins were then loaded onto a Superdex 75 HR 10/30 column (Amersham Pharmacia Biotech) and eluted in either 10 mM sodium phosphate at pH 7 or 10 mM Tris/HCl at pH 8. The proteins were stored at −20°C.
Purification of histidine-tagged XD was carried out as described in Johansson et al. (2003).
All purification steps, except for gel filtrations, were carried out at 4°C.
Apparent molecular mass of proteins eluted from gel filtration columns was deduced from a calibration carried out with LMW and HMW calibration kits (Amersham Pharmacia Biotech). The theoretical Stokes radii (RS) of a native (RSN) and fully unfolded (RSU) protein with a MM (in Daltons) were calculated according to (Uversky 1993): log(RSN)=0.369 • log(MM) − 0.254 and log(RSU)=0.533 • log(MM) − 0.682.
Determination of protein concentration
Protein concentrations were calculated either using the theoretical absorption coefficients ɛ (mg/mL • cm) at 280 nm as obtained using the program ProtParam at the EXPASY server (http://www.expasy.ch/tools), or the Biorad protein assay reagent (Bio-Rad).
Immunoprecipitation studies of NTAIL proteins
IP experiments were carried out using the anti-N Cl 25, the anti-flag, and the anti-hexahistidine tag mAbs, and bacterial lysates expressing NTAIL proteins as described in Longhi et al. (2003).
Small angle X-ray scattering
All protein samples were prepared by dilution of the purified NTAILHN and XD solutions in buffer 10 mM Tris/HCl at pH 8, 40 mM NaCl, with 1 mM DTT as radiation scavenger. The complex NTAILHN–XD was prepared by mixing XD and NTAILHN with a molar ratio of 2:1 in the same buffer and at a final protein concentration of 10 mg/mL. The samples were filtered prior to each measurement (Millex syringe filters 0.22 μm, Millipore) to eliminate possibly existing large aggregates.
SAXS experiments were carried out on beamline ID02 (Narayanan et al. 2001) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. The wavelength was 1.0 Å and the sample-to-detector distance was 3.0 m and 1.0 m, leading to scattering vectors q ranging from 0.02 to 0.20 Å−1 and 0.05 to 0.40 Å−1, respectively. The scattering vector is defined as q=4 π/λ sin θ, where 2θ is the scattering angle. The detector was an X-ray image intensified optically coupled to an ESRF developed FReLoN CCD camera. Forty successive frames of 1.0 sec with a 5-sec pause between each frame were recorded for each sample. The protein solution was circulated through an evacuated quartz capillary between each frame. Thus, no protein solution was irradiated longer than 1.0 sec. Each frame was then carefully inspected to check for possible bubble formation or radiation-induced aggregation. No such effect was observed, and individual frames could then be averaged. Absolute calibration was made with a Lupolen sample. A series of measurements at different protein concentrations ranging from 1.8 to 10 mg/mL were performed for every protein (XD, NTAILHN, and the mixture NTAILHN–XD) to check for interparticle interaction. Background scattering was measured before or after each protein sample using the buffer solution and then subtracted from the protein scattering patterns after proper normalization and correction from detector response. All the experiments were carried out at 20°C. The data acquired at both sample-to-detector distances of 3 m and 1 m were merged and extrapolated to zero concentration for the calculations using the entire scattering spectrum.
The scattering pattern of the NTAILHN–XD complex was obtained following this process: to avoid any possible bias on the absolute intensities due to the concentration, the experimental merged scattering curves obtained as described above were normalized by their theoretical I(0). The scattering pattern of XD was then subtracted twice from the scattering pattern of the mixture NTAILHN–XD (molar ratio 1:2). Given the rather low KD between NTAIL and XD (~100 nM; see Results section), we assumed that the concentration of uncomplexed NTAILHN in solution is negligible at the concentration used in the SAXS experiments (2 to 10 mg/mL). The scattering pattern of the complex thus obtained was then used for further calculations.
The value of the Rg was derived from the Guinier approximation (Guinier and Fournet 1955): I(q)=I(0) exp(−q2Rg2/3), where I(q) is the scattered intensity and I(0) is the forward scattered intensity. The Rg and I(0) are inferred, respectively, from the slope and the intercept of the linear fit of Ln[I(q)] versus q2 at low q values (q Rg<1.0). The distance distribution function P(r) is the histogram of all the interatomic distances within a molecule. This function also provides the maximum dimension Dmax of the molecule, which is defined as the point where P(r) becomes zero. The P(r) function was calculated by the Fourier inversion of the scattering intensity I(q) using GNOM (Svergun 1992) and GIFT (Bergmann et al. 2000) on the entire scattering spectra.
3D modeling
The low-resolution shape of the NTAILHN–XD complex was determined ab initio from the scattering curve using the program GASBOR (Svergun et al. 2001). This program restores lowresolution shapes of protein and calculates a volume filled with densely packed spheres (dummy residues) fitting the experimental scattering curve by a simulated annealing minimization procedure and considering the protein as an assembly of dummy residues centered on the Cα positions (spheres of 3.8 Å diameter) with a nearest-neighbour distribution constraint. Several independent fits were run with no symmetry restriction with an input of 188 dummy residues, corresponding to the 56 residues of XD and 132 residues of NTAILHN. The program package CREDO (CHADD and GLOOPY) was used to restore the low resolution model of NTAILHN in complex with XD, with the crystal structure of the chimeric protein between XD and the region of NTAIL encompassing residues 486–505 (XD–NTAIL486–505) (Kingston et al. 2004a) as template. CREDO is an extension of the original program GASBOR. It calculates the structure of missing domains or loops of crystal structures from the experimental scattering curve of the entire particle and represents them by an ensemble of dummy residues forming a chain-compatible model.
Surface plasmon resonance studies
Binding between purified XD and purified NTAIL proteins was analyzed by using BIAcore 3000 (Amersham Pharmacia Biotech), a system for real-time biomolecular interaction analysis that is based upon surface plasmon resonance technology. Purified XD (1.4 μg/mL in acetate buffer at pH 5.5) was covalently bound to carboxy-methyl groups of CM5 sensor chips using amine-coupling chemistry (Biosensor AB, Amersham Pharmacia Biotech). The levels of immobilized XD were comprised between 180 and 225 RU (1000 RU equal a change in mass of 1 ng/mm2 on the sensor surface) (Zhang et al. 2002). Kinetic and equilibrium constants were calculated from global analysis of reactions with multiple analyte concentrations (0.1, 0.2, 0.5, 1, 2, 5, and 10 μM). Reactions were performed at 25°C. Remaining flow channels on the sensor chip included a control for a nonspecific interaction with the sensor chip (i.e., activated/blocked flow channel) and a control for nonspecific interactions with irrelevant protein targets (i.e., lactoferrin conjugated flow channel). Protein analytes were passed over the sensor chip in HBS-P buffer (0.01MHEPES at pH 7.4, 0.15MNaCl, 0.005% surfactant P-20) containing 2.5 mM magnesium acetate and 2.5 mM ATP. Sensorgrams plotted changes in surface plasmon resonance (measured in RU) as a function of time. Multiple sensorgrams representing various analyte concentrations were analyzed by using BIAevaluation 3.1 software. Background interaction of NTAIL proteins (i.e., analytes) with the sensor surfaces were measured on flow channels that were activated and subsequently blocked under buffer conditions used to immobilize XD. This background was subtracted from all binding curves (i.e., sensorgrams) prior to global analyses. Immobilized lactoferrin was used as a specificity control, and the resultant sensorgrams ruled out high affinity interactions between NTAIL constructs or peptides and this irrelevant protein ligand (data not shown). Global fitting of experimental data to well-characterized binding reactions was used to define reaction rate and equilibrium constants. Signal changes on the activated/blocked control channel were subtracted from the peptide–XD and NTAIL protein–XD interactions using in-line reference and the subtracted sensorgrams were analyzed. Curves generated with serial analyte concentrations were applied globally to the 1:1 Langmuir binding model with or without correction for baseline drifting depending on baseline status. χ2 and residual values were used to evaluate the quality of fit between experimental data and individual binding models. Plots of residuals indicate the difference between the experimental and reference data for each point in the fit. The χ2 value represents the sum of squared differences between the experimental data and reference data at each point. A good fit between experimental and reference data has small residuals in the −2 to +2 range that randomly distribute about the X-axis, and χ2 values are less than 10.
Circular dichroism
The CD spectra were recorded on a Jasco 810 dichrograph using 1-mm thick quartz cells in 10 mM sodium phosphate (pH 7) at 20°C. Structural variations of NTAIL proteins were measured as a function of changes in the initial CD spectrum upon addition of either increasing concentrations of TFE (Fluka), or different amounts of XD or lysozyme (Sigma). CD spectra were measured between 185 and 260 nm, at 0.2 nm/min and were averaged from three independent acquisitions. Mean ellipticity values per residue ([Θ]) were calculated as [Θ]=3300 m ΔA/(l c n), where l (path length)=0.1 cm, n=number of residues, m=molecular mass in daltons, and c=protein concentration expressed in mg/mL. Number of residues (n) are 140 for NTAILHNFC, 131 for NTAILΔ3, 103 for NTAILΔ2,3, 120 for NTAILΔ1, 132 for NTAIL W518, 56 for XD, and 129 for lysozyme, while m values are 15,626 Da for NTAILHNFC, 14,523 Da for NTAILΔ3, 11,539 Da for NTAILΔ2,3, 13,440 Da for N TAILΔ1, 6690 Da for XD, and 14,300 Da for lysozyme. Protein concentrations of 0.1 mg/mL were used when recording spectra of both individual and protein mixtures. In the case of protein mixtures, mean ellipticity values per residue ([Θ]) were calculated as [Θ]=3300 ΔA/{[(c1 n1/m1)+(c2 n2/m2)] l}, where l (path length)=0.1 cm, n1 or n2=number of residues, m1 or m2=molecular mass in daltons, and c1 or c2=protein concentration expressed in mg/mL for each of the two proteins in the mixture. The theoretical average ellipticity values per residue ([Θ]Ave), assuming that neither unstructured-to-structured transitions nor secondary structure rearrangements occur, were calculated as follows: [Θ]Ave=[([Θ]1 n1)+([ Θ]2 n2 R)]/(n1+n2 R), where [Θ]1 and [Θ]2 correspond to the measured mean ellipticity values per residue, n1 and n2 to the number of residues for each of the two proteins, and R to the excess molar ratio of protein 2. The α-helical content was derived from the ellipticity at 220 nm as described in Morris et al. (1999).
Fluorescence spectroscopy
Fluorescence intensity variations of the single tryptophan in NTAILW518 was measured by using a Cary Eclipse (Varian) equipped with a front-face fluorescence accessory at 20°C, by using 2.5 nm excitation and 10 nm emission bandwidths. The excitation wavelength was 290 nm and the emission spectra were recorded between 300 nm and 540 nm. Titrations were performed in a 1-mL quartz fluorescence cuvette containing 1 μM NTAILW518 in 10 mM sodium phosphate buffer at pH 7, and by gradually increasing the XD concentration from 0.05 μM to 3 μM. Experimental fluorescence intensities were corrected by subtracting the spectrum obtained with XD alone (note that XD is devoid of tryptophan residues). Data were analyzed by plotting the relative fluorescence intensities at the maximum of emission at increasing XD concentrations. The dissociation equilibrium constant (KDapp) value was determined from data fitted to a single exponential equation, by using the PRISM 3.02 nonlinear regression tool (GraphPad).
Two-dimensional heteronuclear magnetic resonance
2D-HSQC spectra (Mori et al. 1995) were recorded on a 500-MHz DRX Bruker spectrometer either on 0.5 mM uniformly 15N-labeled NTAILHN or on 0.125 mM uniformly 15N-labeled NTAILΔ;3 in 10 mM sodium phosphate buffer at pH 7.0 containing 10% D2O (v/v) alone or after addition of a twofold molar excess of XD. The temperature was set to 283 K and the spectra were recorded with 2048 complex points in the directly acquired dimension and 256 points in the indirectly detected dimension, for 6 h each. Solvent suppression was achieved by the WATERGATE 3-9-19 pulse (Piotto et al. 1992). The data were processed using the UXNMR software; they were multiplied by a sine-squared bell and zero-filled to 1 K in first dimension prior to Fourier transformation. For the spectra recorded after addition of XD, the number of scans was multiplied by 4.
Sequence analysis and secondary structure predictions
The accession number of MV N is P04851. Secondary structure predictions were carried out using both PSI-PRED (McGuffin et al. 2000) and PHD (Rost 1996).
Acknowledgments
We thank Aurelia Salomoni, Cécile Durousseau, and Fabienne Tocque for technical help in cloning the NTAIL constructs. We are grateful to Valérie Campanacci for critical advice with fluorescence spectroscopy, and to Denis Gerlier for kindly providing us with the anti-N monoclonal antibodies. We also thank Charles Brooks for critical advice with surface plasmon resonance analyses and Javier Perez for fruitful discussion. We wish to thank David Karlin, who is the author of Figure 1B ▶. We also thank Barbara Selisko and Frédéric Carrié re for critical reading of the manuscript. Finally, we thank Richard L. Kingston for kindly providing us with the PDB file of the chimeric construct between XD and the NTAIL peptide (PDB code 1T6O). This study has been carried out with financial support from the Commission of the European Communities, specific RTD program “Quality of Life and Management of Living Resources,” QLK2-CT2001-01225, “Towards the design of new potent antiviral drugs: Structure–function analysis of Paramyxoviridae RNA polymerase.” It does not necessarily reflect its views, and in no way anticipates the Commission’s future policy in this area. This work was also in part supported by the CNRS and by funds from the National Institute of Neurological Disorders and Stroke (R01 NS31693).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051411805.
References
- Bankamp, B., Horikami, S.M., Thompson, P.D., Huber, M., Billeter, M., and Moyer, S.A. 1996. Domains of the measles virus N protein required for binding to P protein and self-assembly. Virology 216 272–277. [DOI] [PubMed] [Google Scholar]
- Bergmann, A., Fritz, G., and Glatter, O. 2000. Solving the generalized indirect Fourier transformation (GIFT) by Boltzmann simplex simulated annealing (BSSA). J. Appl. Crystallogr. 33 1212–1216. [Google Scholar]
- Bette, S., Breer, H., and Krieger, J. 2002. Probing a pheromone binding protein of the silkmoth Antheraea polyphemus by endogenous tryptophan fluorescence. Insect Biochem. Mol. Biol. 32 241–246. [DOI] [PubMed] [Google Scholar]
- Bienkiewicz, E.A., Adkins, J.N., and Lumb, K.J. 2002. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1). Biochemistry 41 752–759. [DOI] [PubMed] [Google Scholar]
- Bourhis, J., Johansson, K., Receveur-Bréchot, V., Oldfield, C.J., Dunker, A.K., Canard, B., and Longhi, S. 2004. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 99 157–167. [DOI] [PubMed] [Google Scholar]
- Brizzard, B.L., Chubet, R.G., and Vizard, D.L. 1994. Immunoaffinity purification of FLAG epitope-tagged bacterial alkaline phosphatase using a novel monoclonal antibody and peptide elution. Biotechniques 16 730–735. [PubMed] [Google Scholar]
- Buchholz, C.J., Spehner, D., Drillien, R., Neubert, W.J., and Homann, H.E. 1993. The conserved N-terminal region of Sendai virus nucleocapsid protein NP is required for nucleocapsid assembly. J. Virol. 67 5803–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckland, R., Giraudon, P., and Wild, F. 1989. Expression of measles virus nucleoprotein in Escherichia coli: Use of deletion mutants to locate the antigenic sites. J. Gen. Virol. 70 435–441. [DOI] [PubMed] [Google Scholar]
- Coronel, E.C., Takimoto, T., Murti, K.G., Varich, N., and Portner, A. 2001. Nucleocapsid incorporation into parainfluenza virus is regulated by specific interaction with matrix protein. J. Virol. 75 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csizmok, V., Bokor, M., Banki, P., Klement, E., Medzihradszky, K.F., Friedrich, P., Tompa, K., and Tompa, P. 2005. Primary contact sites in intrinsically unstructured proteins: The case of calpastatin and microtubule-associated protein 2. Biochemistry 44 3955–3964. [DOI] [PubMed] [Google Scholar]
- Curran, J. 1996. Reexamination of the Sendai virus P protein domains required for RNA synthesis: A possible supplemental role for the P protein. Virology 221 130–140. [DOI] [PubMed] [Google Scholar]
- Curran, J. and Kolakofsky, D. 1999. Replication of paramyxoviruses. Adv. Virus Res. 54 403–422. [DOI] [PubMed] [Google Scholar]
- Curran, J., Homann, H., Buchholz, C., Rochat, S., Neubert, W., and Kolakofsky, D. 1993. The hypervariable C-terminal tail of the Sendai paramyxovirus nucleocapsid protein is required for template function but not for RNA encapsidation. J. Virol. 67 4358–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, J., Pelet, T., and Kolakofsky, D. 1994. An acidic activation-like domain of the Sendai virus P protein is required for RNA synthesis and encapsidation. Virology 202 875–884. [DOI] [PubMed] [Google Scholar]
- Curran, J., Boeck, R., Lin-Marq, N., Lupas, A., and Kolakofsky, D. 1995a. Paramyxovirus phosphoproteins form homotrimers as determined by an epitope dilution assay, via predicted coiled coils. Virology 214 139–149. [DOI] [PubMed] [Google Scholar]
- Curran, J., Marq, J.B., and Kolakofsky, D. 1995b. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J. Virol. 69 849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De, B.P. and Banerjee, A.K. 1999. Involvement of actin microfilaments in the transcription/replication of human parainfluenza virus type 3: Possible role of actin in other viruses. Microsc. Res. Tech. 47 114–123. [DOI] [PubMed] [Google Scholar]
- De Guzman, R.N., Martinez-Yamout, M.A., Dyson, H.J., and Wright, P.E. 2004. Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: Regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J. Biol. Chem. 279 3042–3049. [DOI] [PubMed] [Google Scholar]
- Diallo, A., Barrett, T., Barbron, M., Meyer, G., and Lefevre, P.C. 1994. Cloning of the nucleocapsid protein gene of peste-des-petits-ruminants virus: Relationship to other morbilliviruses. J. Gen. Virol. 75( Pt 1): 233–237. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K. and Obradovic, Z. 2001. The protein trinity—Linking function and disorder. Nat. Biotechnol. 19 805–806. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Lawson, J.D., Brown, C.J., Williams, R.M., Romero, P., Oh, J.S., Oldfield, C.J., Campen, A.M., Ratliff, C.M., Hipps, K.W., et al. 2001. Intrinsically disordered protein. J. Mol. Graph. Model. 19 26–59. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6 197–208. [DOI] [PubMed] [Google Scholar]
- Fearns, R. and Collins, P.L. 1999. Role of the M2–1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J. Virol. 73 5852–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, C.M., McGuire, A.M., Gingras, A.C., Li, H., Matsuo, H., Sonenberg, N., and Wagner, G. 1998. 4E binding proteins inhibit the translation factor eIF4E without folded structure. Biochemistry 37 9–15. [DOI] [PubMed] [Google Scholar]
- Fuxreiter, M., Simon, I., Friedrich, P., and Tompa, P. 2004. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 338 1015–1026. [DOI] [PubMed] [Google Scholar]
- Giraudon, P., Jacquier, M.F., and Wild, T.F. 1988. Antigenic analysis of African measles virus field isolates: Identification and localisation of one conserved and two variable epitope sites on the NP protein. Virus Res. 10 137–152. [DOI] [PubMed] [Google Scholar]
- Guinier, A. and Fournet, F. 1955. Small angle scattering of X-rays. Wiley-Interscience, New York.
- Hartlieb, B., Modrof, J., Muhlberger, E., Klenk, H.D., and Becker, S. 2003. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J. Biol. Chem. 278 41830–41836. [DOI] [PubMed] [Google Scholar]
- Harty, R.N. and Palese, P. 1995. Measles virus phosphoprotein (P) requires the NH2- and COOH-terminal domains for interactions with the nucleoprotein (N) but only the COOH terminus for interactions with itself. J. Gen. Virol. 76 2863–2867. [DOI] [PubMed] [Google Scholar]
- Heggeness, M.H., Scheid, A., and Choppin, P.W. 1980. Conformation of the helical nucleocapsids of paramyxoviruses and vesicular stomatitis virus: Reversible coiling and uncoiling induced by changes in salt concentration. Proc. Natl. Acad. Sci. 77 2631–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1981. The relationship of conformational changes in the Sendai virus nucleocapsid to proteolytic cleavage of the NP polypeptide. Virology 114 555–562. [DOI] [PubMed] [Google Scholar]
- Hua, Q.X., Jia, W.H., Bullock, B.P., Habener, J.F., and Weiss, M.A. 1998. Transcriptional activator-coactivator recognition: Nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry 37 5858–5866. [DOI] [PubMed] [Google Scholar]
- Johansson, K., Bourhis, J.M., Campanacci, V., Cambillau, C., Canard, B., and Longhi, S. 2003. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 278 44567–44573. [DOI] [PubMed] [Google Scholar]
- Karlin, D., Longhi, S., and Canard, B. 2002a. Substitution of two residues in the measles virus nucleoprotein results in an impaired self-association. Virology 302 420–432. [DOI] [PubMed] [Google Scholar]
- Karlin, D., Longhi, S., Receveur, V., and Canard, B. 2002b. The N-terminal domain of the phosphoprotein of morbilliviruses belongs to the natively unfolded class of proteins. Virology 296 251–262. [DOI] [PubMed] [Google Scholar]
- Karlin, D., Ferron, F., Canard, B., and Longhi, S. 2003. Structural disorder and modular organization in Paramyxovirinae N and P. J. Gen. Virol. 84 3239–3252. [DOI] [PubMed] [Google Scholar]
- Kingston, R.L., Hamel, D.J., Gay, L.S., Dahlquist, F.W., and Matthews, B.W. 2004a. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. 101 8301–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston, R.L., Walter, A.B., and Gay, L.S. 2004b. Characterization of nucleocapsid binding by the measles and the mumps virus phosphoprotein. J. Virol. 78 8615–8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin, M.B. and Svergun, D.I. 2001. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34 33–41. [Google Scholar]
- Lacy, E.R., Filippov, I., Lewis, W.S., Otieno, S., Xiao, L., Weiss, S., Hengst, L., and Kriwacki, R.W. 2004. p27 binds cyclin–CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol. 11 358–364. [DOI] [PubMed] [Google Scholar]
- Laine, D., Trescol-Biémont, M., Longhi, S., Libeau, G., Marie, J., Vidalain, P., Azocar, O., Diallo, A., Canard, B., Rabourdin-Combe, C., et al. 2003. Measles virus nucleoprotein binds to a novel cell surface receptor distinct from FcgRII via its C-terminal domain: Role in MV-induced immunosuppression. J. Virol. 77 11332–11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine, D., Bourhis, J., Longhi, S., Flacher, M., Cassard, L., Canard, B., Sautès-Fridman, C., Rabourdin-Combe, C., and Valentin, H. 2005. Measles virus nucleoprotein induces cell proliferation arrest and apoptosis through NTAIL/NR and NCORE/FcgRIIB1 interactions, respectively. J. Gen. Virol. 86 1771–1784. [DOI] [PubMed] [Google Scholar]
- Lamb, R.A. and Kolakofsky, D. 2001. Paramyxoviridae: The viruses and their replication. In Fields virology (eds. B.N. Fields, D.M. Knipe, and P.M. Howley), 4th ed., pp. 1305–1340. Lippincott-Raven, Philadelphia, PA.
- Liston, P., DiFlumeri, C., and Briedis, D.J. 1995. Protein interactions entered into by the measles virus P, V, and C proteins. Virus Res. 38 241–259. [DOI] [PubMed] [Google Scholar]
- Liston, P., Batal, R., DiFlumeri, C., and Briedis, D.J. 1997. Protein interaction domains of the measles virus nucleocapsid protein (NP). Arch. Virol. 142 305–321. [DOI] [PubMed] [Google Scholar]
- Longhi, S., Receveur-Brechot, V., Karlin, D., Johansson, K., Darbon, H., Bhella, D., Yeo, R., Finet, S., and Canard, B. 2003. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 278 18638–18648. [DOI] [PubMed] [Google Scholar]
- Marcotrigiano, J., Gingras, A.C., Sonenberg, N., and Burley, S.K. 1999. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell 3 707–716. [DOI] [PubMed] [Google Scholar]
- McGuffin, L.J., Bryson, K., and Jones, D.T. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16 404–405. [DOI] [PubMed] [Google Scholar]
- Mori, S., Abeygunawardana, C., Johnson, M.O., and van Zijl, P.C. 1995. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108 94–98. [DOI] [PubMed] [Google Scholar]
- Morris, M.C., Mery, J., Heitz, A., Heitz, F., and Divita, G. 1999. Design and synthesis of a peptide derived from positions 195–244 of human cdc25C phosphatase. J. Peptide Sci. 5 263–271. [DOI] [PubMed] [Google Scholar]
- Moyer, S.A., Baker, S.C., and Horikami, S.M. 1990. Host cell proteins required for measles virus reproduction. J. Gen. Virol. 71 775–783. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1997a. Helix propensities are identical in proteins and peptides. Biochemistry 36 10923–10929. [DOI] [PubMed] [Google Scholar]
- Myers, T.M., Pieters, A., and Moyer, S.A. 1997b. A highly conserved region of the Sendai virus nucleocapsid protein contributes to the NP-NP binding domain. Virology 229 322–335. [DOI] [PubMed] [Google Scholar]
- Myers, T.M., Smallwood, S., and Moyer, S.A. 1999. Identification of nucleocapsid protein residues required for Sendai virus nucleocapsid formation and genome replication. J. Gen. Virol. 80 1383–1391. [DOI] [PubMed] [Google Scholar]
- Narayanan, T., Diat, O., and Boesecke, P. 2001. SAXS and USAXS on the high brilliance beamline at the ESRF. Nucl. Instr. Methods Phys. Res. A 467 1005–1009. [Google Scholar]
- Oglesbee, M., Tatalick, L., Rice, J., and Krakowka, S. 1989. Isolation and characterization of canine distemper virus nucleocapsid variants. J. Gen. Virol. 70 2409–2419. [DOI] [PubMed] [Google Scholar]
- Permyakov, S.E., Millett, I.S., Doniach, S., Permyakov, E.A., and Uversky, V.N. 2003. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins 53 855–862. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan, I., Perez-Alvarado, G.C., Parker, D., Dyson, H.J., Montminy, M.R., and Wright, P.E. 1997. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell 91 741–752. [DOI] [PubMed] [Google Scholar]
- Raggett, E.M., Bainbridge, G., Evans, L.J., Cooper, A., and Lakey, J.H. 1998. Discovery of critical Tol A-binding residues in the bactericidal toxin colicin N: A biophysical approach. Mol. Microbiol. 28 1335–1343. [DOI] [PubMed] [Google Scholar]
- Rahaman, A., Srinivasan, N., Shamala, N., and Shaila, M.S. 2004. Phosphoprotein of the rinderpest virus forms a tetramer through a coiled coil region important for biological function. A structural insight. J. Biol. Chem. 279 23606–23614. [DOI] [PubMed] [Google Scholar]
- Robbins, S.J. and Bussell, R.H. 1979. Structural phosphoproteins associated with purified measles virions and cytoplasmic nucleocapsids. Intervirology 12 96–102. [DOI] [PubMed] [Google Scholar]
- Robbins, S.J., Bussell, R.H., and Rapp, F. 1980. Isolation and partial characterization of two forms of cytoplasmic nucleocapsids from measles virus-infected cells. J. Gen. Virol. 47 301–310. [DOI] [PubMed] [Google Scholar]
- Rost, B. 1996. PHD: Predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 266 525–539. [DOI] [PubMed] [Google Scholar]
- Smallwood, S., Ryan, K.W., and Moyer, S.A. 1994. Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology 202 154–163. [DOI] [PubMed] [Google Scholar]
- Stallcup, K.C., Wechsler, S.L., and Fields, B.N. 1979. Purification of measles virus and characterization of subviral components. J. Virol. 30 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun, D. 1992. Determination of the regularization parameters in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25 495–503. [Google Scholar]
- Svergun, D.I., Baraberato, C., and Koch, M.H. 1995. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28 768– 773. [Google Scholar]
- Svergun, D.I., Petoukhov, M.V., and Koch, M.H. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80 2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbouriech, N., Curran, J., Ruigrok, R.W., and Burmeister, W.P. 2000. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat. Struct. Biol. 7 777–781. [DOI] [PubMed] [Google Scholar]
- tenOever, B.R., Servant, M.J., Grandvaux, N., Lin, R., and Hiscott, J. 2002. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J. Virol. 76 3659–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa, P. 2002. Intrinsically unstructured proteins. Trends Biochem. Sci. 27 527–533. [DOI] [PubMed] [Google Scholar]
- Tuckis, J., Smallwood, S., Feller, J.A., and Moyer, S.A. 2002. The C-terminal 88 amino acids of the Sendai virus P protein have multiple functions separable by mutation. J. Virol. 76 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V.N. 1993. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry 32 13288–13298. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 11 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, S., Tigaud, I., Schneider, H., Buchholz, C.J., Yanagi, Y., and Gerlier, D. 2002. Restriction of measles virus RNA synthesis by a mouse host cell line: Trans-complementation by polymerase components or a human cellular factor(s). J. Virol. 76 6121–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, P.E. and Dyson, H.J. 1999. Intrinsically unstructured proteins: Reassessing the protein structure–function paradigm. J. Mol. Biol. 293 321–331. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Glendening, C., Linke, H., Parks, C.L., Brooks, C., Udem, S.A., and Oglesbee, M. 2002. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 76 8737–8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., Bourhis, J.M., Longhi, S., Carsillo, T., Buccellato, M., Morin, B., Canard, B., and Oglesbee, M. 2005. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 337 162–174. [DOI] [PubMed] [Google Scholar]