

Abstract
The carboxy-terminal domain of the transcription factor Escherichia coli NusA, NusACTD, interacts with the protein N of bacteriophage λ, λN, and the carboxyl terminus of the E. coli RNA polymerase α subunit, αCTD. We solved the solution structure of the unbound NusACTD with high-resolution nuclear magnetic resonance (NMR). Additionally, we investigated the binding sites of λN and αCTD on NusACTD using NMR titrations. The solution structure of NusACTD shows two structurally similar subdomains, NusA(353–416) and NusA(431–490), matching approximately two homologous acidic sequence repeats. Further characterization of NusACTD with 15N NMR relaxation data suggests that the interdomain region is only weakly structured and that the subdomains are not interacting. Both subdomains adopt an (HhH)2 fold. These folds are normally involved in DNA–protein and protein–protein interactions. NMR titration experiments show clear differences of the interactions of these two domains with αCTD and λN, in spite of their structural similarity.
Keywords: NusA, anti-termination, termination, NMR, RNA polymerase, N-protein, phage λ, HhH motif
RNA synthesis in Escherichia coli is catalyzed by RNA polymerase (RNAP), a multiprotein enzyme whose core shows an α2ββ′ ubunit composition (Nudler 1999). After initiation of transcription, the essential transcription factor NusA (N utilization substance A) associates with the RNAP core enzyme, where it modulates transcriptional pausing, termination, and anti-termination (Liu et al. 1996).
The crystal structures of two non-E. coli NusA factors have been solved so far (Thermotoga maritima [Worbs et al. 2001; Shin et al. 2003], Mycobacterium tuberculosis [Gopal et al. 2001]). These structures show a common domain organization, that is, an amino-terminal RNAPbinding domain, followed by one S1 and two KH (K homology) RNA-binding domains. This NusA core organization is conserved in all bacteria for which such sequence information is available. An additional carboxyterminal region, NusACTD, comprising ~160 residues is found in several α-, β-, and γ-proteobacteria like the enterobacterium E. coli, as well as in Chlamydia or Treponema (Mah et al. 2000). Though NusACTD is not as highly conserved as the NusA core, the region is characterized by its acidity and frequently by an internal sequence repeat of ~70 residues.
NusACTD probably serves as a multipurpose protein–protein interaction site as suggested by the existence of different binding partners of NusACTD in distinct elongation complexes (Mogridge et al. 1995, 1998; Mooney et al. 1998; Mah et al. 1999, 2000; Gusarov and Nudler 2001; Bonin et al. 2004). In spite of the fact that NusA harbors three RNA-binding domains, the affinity of isolated NusA to RNA is rather weak (Mogridge et al. 1995). Binding studies with deletion mutants of NusA, however, indicate that RNA binding to E. coli NusA is hindered by the 80 carboxyterminal residues of NusACTD (Mah et al. 2000). Furthermore, αCTD counteracts the inhibitory effect of the carboxy-terminal 80 residues by binding to NusACTD, thus providing a model for the regulation of RNA binding in the NusA:RNAP complex in vivo (Mah et al. 2000).
A similar mechanism to enhance the intrinsic RNAbinding capacity of NusA was suggested for the λN protein (Mah et al. 2000), which binds to NusA during λN-mediated anti-termination (for recent reviews, see Gusarov and Nudler 2001 and Nudler and Gottesman 2002). The minimal fragment of λN required for the λN–NusA interaction consists of residues N34–K47 (Mogridge et al. 1998). Recently, Bonin et al. (2004) determined the X-ray structure of the λN(34–40)•NusA(352–421) complex and presented calorimetric data that indicated an interaction of λN(41–47) with NusACTD outside NusA(352–421). However, in contrast to αCTD, λN(34–47) was not able to stimulate RNA binding to NusA (Mogridge et al. 1998; Bonin et al. 2004). Thus, Bonin et al. (2004) inferred a simple scaffold function for NusACTD in the λN:NusA complex, and suggested different binding sites for αCTD and λN on NusA(431–490). A direct interaction between NusA(431–490) and λN(41–47), however, has not been demonstrated yet, and it is not known whether αCTD and λN bind simultaneously to NusACTD in the antitermination complex.
The modular architecture of the proteins establishing the termination and anti-termination complexes allows dissection and investigation of isolated domains and complexes thereof. We used this fact to determine the three-dimensional solution structure of NusA(339–495) to high resolution, its dynamic properties, and its interaction sites with λN(1–53) and αCTD(233–329) using NMR spectroscopy.
Results and Discussion
Structure calculation and validation
Initial analysis of 15N relaxation data (Fig. 1 ▶) revealed significant variations of the {1H}15N heteronuclear NOE (HetNOE) and the relaxation rates along the backbone. Values of the HetNOE <0.65 at 600 MHz are indicative of a considerable flexibility on a picosecond timescale (Tjandra et al. 1995) and could be observed for the amino- and carboxy-terminal regions of NusACTD as well as residues I417–P430. Higher average values of the HetNOE ~0.7 were found for NusA(353–416) and NusA(431–490), suggesting that these parts of the peptide chain are structured.
Figure 1.
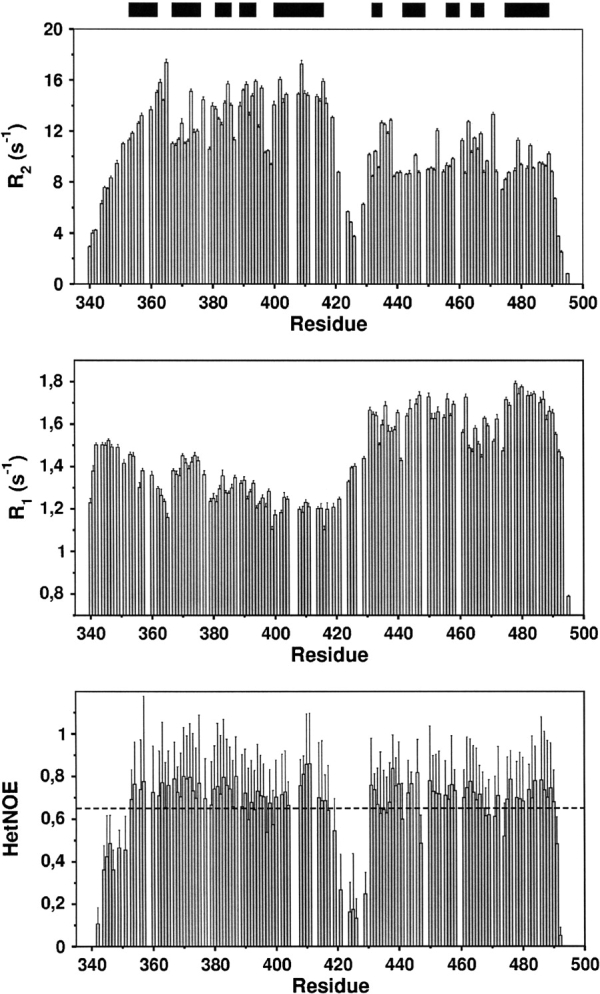
15N transverse (R2, top) and longitudinal (R1, middle) relaxation rates, and {1H}15N heteronuclear NOE (bottom) at 14.1 T. Negative values of the HetNOE for amino- and carboxy-terminal residues (T340, V341, D493, A495) have been omitted for clarity. The black bars represent helices. The evident difference of average R2 values for residues A350–A420 and residues P430–W490, together with the relatively low values (<0.65; see dotted line) of the HetNOE in between residues A420 and P430 corraborates the existence of two subdomains separated by a mobile linker or hinge region. Quantitative analysis of the relaxation data indicates that the two subdomains reorient partially independent in solution and are noninteracting.
BothNusA(353–416) andNusA(431–490) exhibit similar average values of the longitudinal R1 rates, 1.3 ± 0.1 sec−1 and 1.6 ± 0.9 sec−1, but show different average transversal R2 rates, 13.6 ± 1.9 sec−1 and 9.9 ± 1.5 sec−1, respectively. As the R2/R1 ratio for rigid residues is related to the rotational correlation time τc (Kay et al. 1989), the different R2/R1 ratios of NusA(353–416) and NusA(431–490), together with the low HetNOE values for NusA(417–430), suggest that NusACTD consists of two domains that are separated by a flexible linker region.
The hypothesis of NusACTD exhibiting a two-domain structure is supported by the fact that different ranges of residual dipolar couplings (RDCs) were observed for NusA(353–416), −12.4–19.0 Hz, and for NusA(431–490), −8.3–11.0 Hz, indicating different alignment tensors for these domains (Bax et al. 2001). In fact, in the course of molecular dynamics simulations, the magnitude of the alignment tensor Da could be determined as 10.5 ± 0.25 Hz [NusA(353–416)] and 6.5 ± 0.25 Hz [NusA(431–490)], while the rhombicities R were 0.2 ± 0.05 for NusA(353–416) and 0.3 ± 0.05 forNusA(431–490). The average alignment of both domains cannot be described by a single alignment tensor; therefore, the significantly different tensor components (Table 1) imply considerable relative motion of NusA(353–416) and NusA(431–490) (Braddock et al. 2001, 2002).
Table 1.
Alignment data for NusA(353–416) and NusA(431–490)
| Da (Hz) | R | Azz (Hz) | Ayy (Hz) | Axx (Hz) | |
| NusA(353–416) | 10.25 ± 0.25 | 0.20 ± 0.05 | 20.50 ± 0.50 | −13.65 ± 1.11 | −7.35 ± 0.96 |
| NusA(431–490) | 6.25 ± 0.25 | 0.30 ± 0.05 | 12.50 ± 0.50 | −9.43 ± 0.85 | −3.85 ± 0.63 |
Shown are the magnitude of the alignment tensor Da and the rhombicity R, as well as the eigenvalues of the alignment tensor Azz, Ayy, and Axx. Different alignment tensors for individual domains indicate the presence of medium- to large-scale interdomain motions (Braddock et al. 2001).
All NOESY cross-peaks could be interpreted as short distances between protons within the domains NusA(353– 416) and NusA(431–490), respectively, and no NOE between the two domains could be observed. Therefore, though NMR experiments were performed using the complete NusACTD, structures were calculated separately for NusA(351–426) and NusA(426–495). Residues 339–350 were not included in the structure calculation, as this region lacked nontrivial interresidual NOEs and showed low HetNOE values, indicating high flexibility of the amino terminus.
The structure calculation based on NMR-derived restraints resulted in ensembleswithhighprecision ofatomic coordinates for both domains (Fig. 2A ▶). No violations of distance restraints>0.16 Å, dihedral angle restraints >3°, or RDCs >0.72 Hz could be observed (Table 2). Further validation of the structures with PROCHECK 3.5.4 (Morris et al. 1992; Laskowski et al. 1993) showed that 92.1% [NusA(351–426)] and 95.3% [NusA(426–495)] of the nonglycine and nonproline residues adopted a conformation within the most favored regions of the Ramachandran plot.
Figure 2.

Structure of the carboxy-terminal domain of NusA. (A) Backbone overlay of the 19 accepted structures for the two subdomains of the carboxy-terminal domain of NusA, NusA(351–426), and NusA(426–495), respectively. The linker residues are shown in red; flexible residues at the amino and carboxyl terminus are depicted in yellow. (B) Lowest energy structures of the relatively rigid parts of NusACTD, NusA(353–416), and NusA(431–490) in ribbon representation. Each subdomain contains two HhH motifs that are linked together by a so-called connector helix, thus adopting a compact (HhH)2 fold. Helices h1/h1′ and h2/h2′ constitute the first and h4/h4′ and h5/h5′ the second HhH motif, whereas h3/h3′ represent the connector helices. The linker between the two subdomains seems to be structured (see A, red part of overlay); however, 15N relaxation data indicate that the interdomain region comprising about 14 amino acids is flexible. The picture was drawn with MOLMOL (Koradi et al. 1996).
Table 2.
Summary of the structure calculation of NusA(351–426) and NusA(426–495)
| Experimental restraints used for the structure calculation | |||
| NusA(351–426) | NusA(426–495) | ||
| Distance restraints | Total | 883 | 699 |
| Sequential | 328 | 285 | |
| Medium-range | 278 | 191 | |
| Long-range | 268 | 199 | |
| Ambiguous/intramol. | 9 | 24 | |
| Dihedral angles | 39 | 28 | |
| Dipolar couplings | 37 | 32 | |
| Hydrogen bonds (2 restraints each) | 19 | 20 | |
| Molecular dynamics statistics | |||
| Energies (kcal/mol) | |||
| Epot | 23.1324 ± 1.7314 | 17.4150 ± 1.1643 | |
| Ebond | 1.0164 ± 0.0879 | 0.8981 ± 0.0962 | |
| Eangle | 8.7168 ± 0.8408 | 6.5624 ± 0.6648 | |
| Eimpr | 2.6457 ± 0.2801 | 1.6269 ± 0.2001 | |
| EvdW | 5.2270 ± 1.0387 | 2.8016 ± 0.6186 | |
| ENOE | 4.0465 ± 0.5038 | 3.4700 ± 0.5815 | |
| Eedih | 0.0322 ± 0.0495 | 0.0752 ± 0.0846 | |
| Esani | 1.4478 ± 0.4740 | 1.9808 ± 0.4104 | |
| RMSDs from ideal distances (Å) | Bond lengths | 0.0007 ± 0.0001 | 0.0009 ± 0.0001 |
| Distance restraints | 0.0054 ± 0.0004 | 0.0097 ± 0.0008 | |
| RMSDs from ideal angles (deg) | Bond angles | 0.1169 ± 0.0065 | 0.1520 ± 0.0075 |
| Dihedral angle restraints | 0.0655 ± 0.0303 | 0.3179 ± 0.2822 | |
| RMSDs from dipolar couplings (Hz) | 0.1978 ± 0.0327 | 0.2488 ± 0.0249 | |
| Atomic RMSDs of structural ensemble (Å) | |||
| Backbone heavy atomsa | 0.34 (H353–T416)b | 0.36 (A431–W490)b | |
| Heavy atomsa | 0.70 (H353–T416)b | 0.72 (A431–W490)b | |
| Ramachandran Plot statistics | |||
| Residues in | |||
| Most favored regions | 92.1% | 95.3% | |
| Allowed regions | 7.0% | 4.4% | |
Except for the experimental restraints and the atomic RMSD data, all values are average values over the 19 accepted structures in the format average value ± standard deviation.
a Structural precision was calculated with reference to the structure with lowest value of the target function.
b All residues were included for calculation of the structural superposition.
The solution structures of NusA(351–426) and NusA(426–495) were deposited in the Protein Data Bank (PDB) (Berman et al. 2000), accession codes 1WCL and 1WCN.
Structure of NusACTD
NusACTD consists of two subdomains, NusA(353–416) and NusA(431–490), that are connected by a linker region (Fig. 2B ▶). Either subdomain contains two helix-hairpin-helix (HhH) motifs, each formed by two anti-parallel α-helices connected by a short hairpin (Doherty et al. 1996).
The amino-terminal HhH motif of NusA(353–416), HhH1, is composed of helices h1 (H353–Y362) and h2 (E367–E376) and interacts with the carboxy-terminal HhH motif of the first domain, HhH2, constituted by helices h4 (M389–E394) and h5 (E400–T416). Analogously, the amino-terminal HhH motif of NusA(431–490), HhH1′, which comprises helices h1′ (D432–L435) and h2′ (R442–A449), packs on the carboxy-terminal HhH motif, HhH2′, which includes helices h4′ (I464– A468) and h5′ (D475–C489).
The interhelical anglesof theHhHmotifs range from119° to 129° (HhH1, 119°; HhH2, 124°; HhH1′, 129°; HhH2′, 122°), slightly less than the interhelical angles of 130°–155° described for HhHmotifs earlier (Doherty et al. 1996; Shao and Grishin 2000). These deviations from canonical HhH motifs can be interpreted in the context of the observed flexibility and the small database used for the determination of the interhelical angles in Doherty et al. (1996).
The tight packing of the two HhH motifs results in the formation of a compact hydrophobic core, which is completed by the helix connecting the two HhH motifs in each subdomain, helix h3 (L381–Y386) and helix h3′ (L456–A460). This arrangement of two HhH motifs linked by a connector helix has been classified as a separate fold termed (HhH)2 (Shao and Grishin 2000).
(HhH)2 folds are mainly implicated in DNA binding (Shao and Grishin 2000), as confirmed by a search of the PDB (Berman et al. 2000) using the average structures of NusA(353–416) and NusA(431–490) with the DALI program (Holm and Sander 1996). However, both domains show similarity to the SAM (sterile α motif) domain of the ephrin 2 receptor (PDB 1b4f; Thanos et al. 1999). This is a typical representative of proteins whose (HhH)2 fold mediates protein–protein interactions.
Comparison of the two subdomains of NusACTD
Overlay of the average structures of NusA(356–415) and NusA(431–490) results in a backbone RMSDof 1.9Å, and thus confirms that the sequence homology of the acidic repeats is reflected in the three-dimensional structure. The superposition of the two domains (Fig. 3 ▶) shows similar positions and orientations for helices h2–h5 and h2′–h5′, but not for helices h1 and h1′, though all helices tend to be shorter in the second domain.
Figure 3.

The sequence homology of the two subdomains in the carboxy-terminal part of NusA is mirrored in the structural similarity of NusA(353–416) and NusA(431–490). (A) Overlay of NusA(353–416) and NusA(431–490). The arrangement of helices [NusA(353–416), h1–h5; NusA(431–490), h1′–h5′], which are represented as red and blue cylinders for NusA(353–416) and NusA(431–490), respectively, reveals a common overall fold. In contrast to helices h1 and h1′, helices h2–h5 share similar positions and orientations with h2′–h5′, though helices in the second subdomain tend to be shorter. The superposition was calculated for the backbone atoms of I356–A415 and A431–W490 and drawn using MOLMOL (Koradi et al. 1996). (B) Alignment of NusA(353–416) and NusA(431–490) with T-Coffee (Notredame et al. 2000) shows a sequence identity and homology of 31.3% and 60.9%, respectively. The secondary structure (H, helix), the helix-hairpin-helix (HhH) motifs as well as the connector helix (C) are reported beneath the sequence with primes for the second subdomain. For comparison, the 16-residue sequence characteristic of HhH motifs (Doherty et al. 1996) is shown above the sequence and the Gly—hydrophobic amino acid—Gly (GhG) pattern in the hairpin is highlighted in blue. Note that in all HhH motifs occuring in NusACTD, the second Gly that serves as a helix cap (Doherty et al. 1996) is replaced by more typical helix-capping residues like Asp or Thr (Kumar and Bansal 1998).
Both domains are considerably acidic with net charges of −11e− and −9e− and theoretical pIs of about 4. The two domains show clear differences in the distribution of polar and hydrophobic as well as charged residues (Fig. 4 ▶), which may explain their differential recognition of binding partners.
Figure 4.

Surface representations of the lowest energy structures of NusA(353–416) and NusA(431–490) showing the distribution of charged (A) and polar residues (B). As a reference, the corresponding orientations of the subdomains are drawn in ribbon style (C). Both charged (negative, red; positive, blue; neutral, gray) and polar (polar, blue; hydrophobic, green) residues are differently distributed on the surface. The preparation of the figure was done using MOLMOL (Koradi et al. 1996).
Oligomeric state of NusACTD
(HhH)2 motifs have been implicated in oligomerization (Kim and Bowie 2003), which would have to be considered in the analysis of relaxation data.
To examine the possible formation of oligomers, we performed translational diffusion measurements (Wilkins et al. 1999) at concentrations of 0.175, 0.35, and 0.70 mM NusACTD. The resulting average hydrodynamic radius of rH = 25.5 ± 0.3 Å was found to be independent of the protein concentration, which clearly indicates the virtual absence of a fast monomer–oligomer equilibrium in the protein solution. In addition, the value of rH cannot be explained in terms of dimer or higher oligomer formation of NusACTD. Although the hydrodynamic radius of a hypothetical spherical NusACTD can be estimated to be 20.7 Å for a monomer and 25.3 Å for a dimer (Wilkins et al. 1999), a more realistic calculation with an elongated NusA(353–490) structure using a shell model (de la Torre and Carrasco 2000) leads to a hydrodynamic radius of 22.7 Å. The difference between this value and the experimental value of 25.5 Å may easily be explained by the fact that the flexible amino- and carboxy-terminal residues could not be taken into account in this model. Thus, the experimental value for the hydrodynamic radius is virtually identical to the one calculated from a monomer model.
Rotational diffusion of NusACTD
15N relaxation data of anisotropically tumbling proteins contain information about the orientation of each N-HN vector relative to the overall diffusion frame of the molecule. Therefore, this data is frequently used to determine the relative orientation of individual domains in multidomain proteins (for review, see Fushman et al. 2004). This approach, however, assumes domains with fixed relative orientations. In all other cases, the diffusion of the domains will not only be governed by the diffusion of the particle as a whole, but will be influenced to a certain extent by the diffusional properties of the single domains. Thus, by calculating the diffusion tensor separately for each domain within a multidomain protein, it is possible to determine whether the domains are mutually independent.
This is clearly the case for NusA(353–416) and NusA(431–490). Quantitative analysis of the 15N data reveals significantly different diffusion tensors for these domains (Table 3), leading to the conclusion that their movements are highly uncorrelated.
Table 3.
Diffusion tensor analysis from R2 and R1 data at 600 MHz of NusA(353–416) and NusA(431–490)
| Diffusion tensor statisticsa | ||||||||
| Isotropic | Axially symmetric (prolate) | Axially symmetric (oblate) | Anisotropic | |||||
| χ2(5%) | χ2exp | χ2(5%) | χ2exp | χ2(5%) | χ2exp | χ2(5%) | χ2exp | |
| NusA(353–416) | 44.2 | 207.6 | 40.0 | 48.5 | 39.7 | 48.5 | 37.0 | 19.0 |
| NusA(431–490) | 31.4 | 55.98 | 26.6 | 24.3 | 28.0 | 39.7 | 23.4 | 22.9 |
| Diffusion tensor statisticsb | ||||||
| Dzz(107s−1) | Dyy(107s−1) | Dxx(107s−1) | ζ | η | τc,eff[ns] | |
| a χ2(5%) corresponds to the α= 0.05 confidence limit for the fit derived from 500 Monte Carlo simulations, χ2exp refers to the value of the target function used for the fit. Values of χ2(5%) and χ2exp for accepted models are shown in bold italic. An F-statistic was used to differentiate between the prolate (four parameters) and the fully asymmetric model (six parameters) for NusA(431–490). The critical values for the F-statistic with α= 0.1 amount to Fexp = 4.6 and F(10%) = 22.3, indicating that the improvement of the fit upon using the six-parameter model is not statistically relevant (Mandel et al. 1995; Dosset et al. 2000). NusA(431–490) is thus best described by a prolate axially symmetric diffusion model, NusA(353–416) by an anisotropic diffusion tensor. | ||||||
| b Diffusion tensor components from R2 and R1 data at 600 MHz [NusA(353–416): 33 N-HN vectors; NusA(431–490): 21 N-HN vectors] for accepted models of NusA(353–416) and NusA(431–490). For a prolate diffusion tensor (Dzz − Dyy ≥ Dyy − Dxx) the anisotropy and rhombicity are defined as ζ = 2Dzz/(Dxx+Dyy) and η = 3/2 · (Dyy − Dxx)/Dzz · ζ/(ζ − 1), respectively, and characterize the deviations from a spherical top (Fushman et al. 2004). The effective rotational correlation time is related to the diffusion tensor components via τc,eff = 1/(2Dxx+2Dyy+2Dzz). Parameter uncertainties of the diffusion parameters were taken from 500 Monte Carlo simulations. | ||||||
| NusA(353–416) | 2.31 ± 0.05 | 1.73 ± 0.06 | 1.22 ± 0.05 | 1.6 ± 0.1 | 0.91 ± 0.19 | 9.5 ± 0.3 |
| NusA(431–490) | 3.11 ± 0.12 | 2.01 ± 0.07 | 2.01 ± 0.07 | 1.6 ± 0.1 | 0 | 7.0 ± 0.2 |
The rotational correlation times were calculated from the 15N relaxation rates R1 and R2 that were determined for residues inside NusA(353–416) and NusA(431–490) in the intact NusACTD. The resulting correlation times were 9.5 ± 0.3 nsec and 7.0 ± 0.2 nsec, respectively, both values being higher than the roughly 4.5 nsec expected for globular, compact proteins of 64 and 60 residues (Maciejewski et al. 2000). This difference, however, may easily be explained by the fact that unstructured regions precede and follow the two domains of NusACTD, and that these domains are connected by a linker. The linker and the terminal extensions affect the rotational correlation times of the isolated domains in different ways. First, the linker restricts the relative movements of the two domains, which leads to an increase in the tumbling times of NusA(353–416) and NusA(431–490). Second, flexible termini can enhance the rotational correlation times significantly (Tjandra et al. 1995), which is reflected in the tumbling time of NusA(353–416) compared with NusA(431–495).
Taken together, the alignment and relaxation data strongly suggest that NusA(353–416) and NusA(431– 490) are not interacting or are preferentially orientated relative to each other. This justifies our initial approach to treat the domains separately.
Comparison of free and complexed NusACTD
Recently, the structure of the complex of λN(34–47) with two fragments of NusA, NusA(352–419), and NusA(352– 421), respectively, has been determined by X-ray crystallography (Bonin et al. 2004). The NusA molecules are crystallographically independent, but structurally similar with an RMSD of the backbone heavy atoms of 0.67 Å for H353–T416. Both structures deviate only slightly from the uncomplexed solution structure of NusA(353–416), with an RMSD of the backbone heavy atoms of 1.20 Å and 1.10 Å (Fig. 5 ▶). Only minor differences between the free and the complexed structures could be observed, and most of these differences are located in the amino-terminal helix and the hairpin of the second HhH motif, involving M389–D399. These residues feature decreased HetNOE values in the free NusACTD, reflecting increased flexibility on a pico- to nanosecond time scale. λN(34– 47) contacts this region via L40 by side-chain–side-chain interactions with L398, supporting the idea that the flexibility of the helix and the adjacent hairpin might play an important role in recognition (Atkinson and Kieffer 2004). Thus, in general, conformational rearrangements upon complex formation are minor, in line with the suggested function of NusA(353–416) as a scaffold conferring additional stability to the complex (Bonin et al. 2004).
Figure 5.

Overlay of the uncomplexed average structure of NusA(353–416) solved by NMR (this work, red) with the crystal structure 1U9L (Bonin et al. 2004), in which NusA(352–419) (chain A in 1U9L, blue) and NusA(352–421) (chain B in 1U9L, green) form a complex with λN(34–47), respectively. Chains A and B are crystallographically independent, but almost identical to each other with an RMSD of the backbone heavy atoms of 0.67 over residues H353–T416. Both structures deviate only slightly from the uncomplexed NusA(353–416), with a backbone RMSD of 1.20 and 1.10, for chains A and B, respectively. The overlay was calculated and drawn with MOLMOL.
Interactions of NusACTD with αCTD and λN
In order to elucidate the sites of NusACTD that bind λN and αCTD, 15N-labeled NusACTD was titrated with unlabeled αCTD or unlabeled λN(1–53). HSQC spectra of the sample were taken after each titration step (Fig. 6 ▶).
Figure 6.

Titration of NusACTD with αCTD and λN(1–53). (A) Interaction of NusACTD with αCTD. 15N-labeled GP-NusA(339–495) was titrated with unlabeled αCTD until no further changes were observed in the spectra, resulting in a NusACTD:α CTD ratio of 1:2.1. Shown are the overlays of the 1H15N-HSQC spectra at 700 MHz of the end points of the titrations, corresponding to the free (blue) and complexed (red) NusACTD. (B) Interaction of NusACTD with λN(1–53). 15N-labeled GP-NusA(339–495) was titrated with λN(1–53) until no further changes were observed in the spectra, resulting in a NusACTD: λN(1–53) ratio of 1:3.4. Shown are the overlays of the 1H15N-HSQC spectra at 700 MHz of the end points of the titrations, corresponding to the free (blue) and complexed (red) NusACTD.
Line broadening or chemical-shift changes could be observed for approximately two-thirds of the resonances of NusACTD throughout the titration with λN(1–53), beginning with a NusACTD: λN(1–53) ratio of 0.4 and ending with a ratio of 3.4. Most resonances were broadened beyond detection starting from a NusACTD: λN(1–53) ratio of 1.3 and thus could not be tracked during the titration. From a ratio of 2.6, resonances appeared at new positions in the spectra, probably from the NusACTD: λN(1–53) complex. This broadening and reappearance of resonances is typical for intermediate exchange processes (Zuiderweg 2002) and prevented detection of resonances for a wide range of concentration ratios.
During the titration, 98% and 70%, respectively, of the assigned backbone amide cross-peaks in the first and second domain showed significant changes in resonance intensity or position (≥0.05 ppm in 1H or ≥0.4 ppm in 15N dimension). As most residues were affected by the presence of λN(1–53), it was impossible to delineate a contact interface of λN(1–53) on NusACTD. Widespread chemical-shift perturbations may indicate allosteric processes (Stevens et al. 2001). Comparison of the free and complexed NusA(353–416), however, clearly shows that the first domain of NusACTD does not undergo major rearrangements upon complex formation. Although no complex structure is available for the second domain of NusACTD, λN is believed to bind in a similar fashion to both domains (Bonin et al. 2004), rendering large-scale structural changes unlikely. Instead, the scattered chemical-shift perturbations might be explained by subtle changes in the tertiary structure. Nevertheless, given that λN(1–53) and NusACTD are strongly oppositely charged, unspecific interactions cannot be ruled out. In summary, both domains of NusACTD are affected by the presence of λN(1–53), providing direct evidence for an interaction of λN(1–53) with NusA(353–416) and NusA(431–490).
Addition of αCTD to 15N-labeled NusACTD resulted in changes in HSQC resonance intensity or position for approximately one-third of the total NusA(339–495) backbone amide resonances from the first titration step (NusACTD:α CTD ratio of 0.5). Most of the perturbed resonances exhibited severe line broadening above a NusACTD:α CTD ratio of 1.1, and thus, could not be followed during the titration. Some of the affected resonances remained visible at all examined NusACTD: αCTD ratios up to the end point of the titration (NusACTD:α CTD ratio of 2.1), again indicating intermediate exchange rather than protein aggregation.
In total, chemical-shift changes could be observed for 98% of the resolved backbone amide resonances of the second domain, respectively, whereas the resonances in the first domain remained unaffected by the presence of αCTD. Though no defined contact interface of αCTD on NusACTD could be derived by the titration experiments, the data clearly argue for αCTD binding solely to NusA(431–490). Together with structural data and the fact that αCTD is able to relieve the inhibitory effect of NusACTD on RNA binding (Mah et al. 2000), the titration data strongly suggest that NusA(431–490) functions as a domain regulating RNA binding.
Implications for the role of NusACTD in termination and anti-termination
In summary, the structural data provided here suggests how NusACTD functions as a versatile protein–protein interaction site in different transcription complexes. The domain structure offers an explanation of how NusA(431–490) regulates RNA binding to NusA. The flexible linker allows rearrangement of NusA(431–490) relative to the rest of the protein to facilitate access to the RNA-binding sites of NusA on complexation with αCTD. Spatial adaptability might also play a role in switching from the termination-competent NusA:RNAP complex to the λN:NusA:RNAP complex, which is capable of anti-termination.
In addition to its regulatory function, the NusA(431–490):α CTD interaction possibly enhances the stability of the NusA:RNAP complex (Liu et al. 1996). This could explain why a mutant NusA with residues L344–A495 missing prevents bacterial growth at elevated temperatures (Tsugawa et al. 1988).
We demonstrated in NMR-titration experiments that λN(1–53) binds to both domains of NusACTD, NusA(353–416), and NusA(431–490). The region responsible for the NusACTD interaction on λN is presumably located within residues N34–K47 (Mogridge et al. 1998). Despite its probable binding to NusA(431–490), λN(34–47) cannot stimulate NusA’s intrinsic RNA-binding capacity (Bonin et al. 2004). NusA, however, does bind sequence specifically to nut RNA in λN:NusA:nut complexes (Mogridge et al. 1995; Mah et al. 2000). The KD of full-length λNand NusA or a λN:nutBoxB:NusA complex is of the order of 50 nM (van Gilst and von Hippel 1997; Xia et al. 2003), suggesting that λN or the λN:nutBoxB complex might simply displace NusA(431–490) in the competition for the RNA-binding site. This is in accordance with the complex-stabilizing function of the NusA(353–416)– λN(34–40) interaction.
To get further insight into the role of NusA(353–416) and NusA(431–490) in termination and anti-termination, our future research will focus on the elucidation of the complexes of NusACTD with λN and αCTD, as well as investigation of the intramolecular interaction of NusA(431–490) and the RNA-binding sites of NusA.
Materials and methods
Protein production
λN(1–53) was expressed in E. coli BL21(DE3) using the pTKK19 expression vector (Kohno et al. 1998). The resulting fusion protein consisted of an amino-terminal deca-histidinetag, followed by ubiquitin and λN(1–53), and was purified by immobilized nickel-affinity chromatography under nonnative conditions. Subsequent cleavage by yeast ubiquitin hydrolase and a further high-performance liquid chromatography step yielded pure λN(1–53). E. coli αCTD(233–329) was expressed as a deca-histidine-tagged protein in BL21(DE3) from pET-19b (Novagen) and purified by immobilized nickel-affinity chromatography under native conditions. The histidine-tag was cleaved off with rhinovirus protease 3C (Cordingley et al. 1990), resulting in two additional amino-terminal residues (Gly-Pro). NusACTD was expressed and purified as GP-NusA(339–495) (Eisenmann et al. 2004). NMR samples contained 0.5–1.5 mM GP-NusA(339–495) in 10 mM potassium phosphate (pH 6.8), 50 mM sodium chloride, 0.02% sodium azide, and 10% D2O.
NMR spectroscopy
All NMR experiments were performed at 298 K on Bruker Avance400, DRX600, Avance700, and Avance800 spectrometers equipped with inverse 1H/13C/15N triple-resonance probes with pulsed-field gradient capabilities. In addition to the spectra needed for backbone and side-chain resonance assignments (Eisenmann et al. 2004), 13C-NOESY-HSQC, 15N-NOESY-HSQC, CNHNOESY, CCH-NOESY, NNH-NOESY, and 4D-C,C-NOESY experiments with mixing times of 120 msec were acquired on uniformly 15N and 15N, 13C-labeled GP-NusA(339–495) at a concentration of 1.5 mM to obtain distance restraints (Sattler et al. 1999). For angle restraints, 3J(HNHA) scalar coupling constants were determined from intensity ratios of diagonal and cross-peaks in the HNHA spectrum (Vuister and Bax 1993). Slowly exchanging amide protons were identified in a series of 15N-HSQC spectra acquired after dissolving freeze-dried 15Nlabeled GP-NusA(339–495) in D2O, and those amide protons still visible after 15min were assumed to be involved in hydrogen bonds. 1D(NHN) RDCs were obtained from J-modulated HSQC experiments at 800 MHz (Tjandra et al. 1996) with a 0.5-mM sample of GP-NusA(339–495) in the presence of 10 mg/mL Pf1 phage (Hansen et al. 1998) by subtracting the 1J(NHN) scalar coupling constants measured in reference spectra without Pf1 phage. A sample of 0.5 mM 15N-labeled GP-NusA(339–495) was used for measuring 15N R1 and R2 rates, as well as the HetNOE, with published pulse sequences (Dayie and Wagner 1994). For the determination of R1 and R2 at a proton frequency of 600 MHz, spectra were recorded with delays of 6.88 (3×), 752.33 (3×), 1020.48 (3×), and 1288.63 (3×) msec, and 16.96 (3×), 84.4 (3×), 101.76 (3×), and 118.72 (3×) msec, respectively. TheHetNOEs were averaged over two independent data sets. The NMR data were processed using NMRPipe (Delaglio et al. 1995) and in-house written software, and analyzed with the program package NMRview5.0.4 (Johnson and Blevins 1994). Diffusion experiments were performed at a proton resonance frequency of 400 MHz on samples of 175, 350, and 700 μM GP-NusA(339– 495) in D2O containing 0.5% dioxan, 10 mM potassium phosphate (pH 6.8), 50mMsodium chloride, and 0.02% sodium azide. For each experiment, five independent series of 1D spectra were acquired using the PG-SLED pulse sequence (Jones et al. 1997) with a gradient time of 7 msec, an echo time of 70 msec, and gradient strengths of 5%–50% in 5% increments, and 50%–100% in 6.25% increments. A single Gaussian was fitted to the decay of the protein signal, and the decay of the dioxane signal was described by two Gaussians to account for the overlap of the dioxane signal with the protein signal. Titrations were performed by adding aliquots of a concentrated stock solution of unlabeled λN(1–53) or αCTD(233–329) to a sample of ~0.5 mM 15N-labeled GP-NusA(339–495) and acquiring 1H15N HSQC spectra at 700 MHz after each titration step. In both titrations, ligands were added to GP-NusA(339–495) until no further changes could be observed in the spectra.
Structure calculation
Distance restraints for structure calculation were derived from NNH-NOESY, 13C- and 15N-NOESY-HSQC spectra, and 4D-C,C-NOESY spectra. NOESY cross-peaks were classified according to their relative intensities and converted to distance restraints with upper limits of 2.7 Å (strong), 3.5 Å (medium), and 5.0 Å (weak). For ambiguous distance restraints, the r−6 summation over all assigned possibilities defined the upper limit (Nilges 1995).
The raw scalar coupling constants were multiplied with a correction factor of 1.1 to take into account the different relaxation rates of in-phase and anti-phase components (Vuister and Bax 1993). Residues with scalar coupling constants below 6 Hz were restrained to dihedral angles between −80° and −40°, residues showing coupling constants above 8 Hz were restricted to dihedral angles of −160° to −80° (Schweimer et al. 2002). Glycines were omitted, since they were not stereospecifically assigned, and since the coupling constants are likely affected by cross-relaxation (Vuister and Bax 1993).
Hydrogen bonds were included in the final structure calculation if the acceptor of a slowly exchanging amide proton could be identified from the results of preceding structure calculations. Thus, a hydrogen bond was assumed if the distance of the carboxyl oxygen and the amide proton was below 2.6 Å, and the angle of the amide proton, the amide nitrogen, and the carboxyl oxygen was <60° in all accepted structures. For each hydrogen bond, the distance between the amide proton and the acceptor was restrained to <2.3 Å, and the distance between the amide nitrogen and the acceptor was restrained to <3.3 Å (Schweimer et al. 2002).
All prolines were considered to adopt the trans conformation as strong HA(i)-HD(i+1) and HN(i) and HD(i+1) NOEs could be observed (Wüthrich 1986).
The structures for NusA(351–426) and NusA(426–495) were calculated separately, since no NOEs could be observed between the two regions, and relaxation data suggested an at least partly independent reorientation of the two regions. GPNusA( 339–350) was omitted in the structure calculation, due to the lack of inter-residual NOEs and low values of the HetNOEs, both indicating a flexible amino terminus.
The structure calculations were performed with the program XPLOR-NIH 1.2.1 (Schwieters et al. 2003) using a three-step simulated annealing protocol (Nilges et al. 1988a,b,c) with floating assignment of prochiral groups (Folmer et al. 1997).
First conformational space, sampling was carried out for 120 psec with a time step of 3 fsec at a temperature of 2000 K, followed by a cooling period of 120 psec down to 1000 K, and 60 psec cooling to 100 K, both with a time step of 2 fsec. A modified conformational database potential for backbone and side-chain dihedral angles was applied (Kuszewski and Clore 2000; Neudecker et al. 2001). The proline angles were modified according to Neudecker et al. (2004). After simulated annealing, the structures were subjected to 1000 steps of Powell minimization (Powell 1977), and the final 500 steps were minimized without conformational database potential.
In a first step, 90 structures were calculated (Table 1) using 883/699 [NusA(351–426)/NusA(426–495)] distance and 39/28 dihedral angle restraints; in a second step, 38/40 additional restraints from hydrogen exchange experiments were included. The 32 structures with the lowest total energy were then refined using 37/32 1D(NHN) RDCs with a harmonic potential (Tjandra et al. 1997). Flexible residues (for definition, see “NMR data relaxation analysis,” below) were excluded from the calculations. The tensor components of the alignment were optimized separately for NusA(351–426) and NusA(426–495) with a grid search by varying the axial component Da and the rhombicity R in steps of 0.5 and 0.1, respectively. The initial values of Da and R were estimated from the distribution of the 1D(NHN) (Clore et al. 1998) and a molecular dynamics run was performed for each pair of Da and R, yielding axial components of 10.5 ± 0.25 Hz [NusA(351–426)] and 6.5 ± 0.25 Hz [NusA(426–495)], and rhombicities of 0.2 ± 0.05 [NusA(351–426)] and 0.3 ± 0.05 [NusA(426–495)] for the energetically most favorable combination of Da and R.
The 19 structures showing the lowest values of the target function excluding the database potential were further analyzed with XPLOR-NIH 1.2.1 (Schwieters et al. 2003) and PROCHECK 3.5.4 (Morris et al. 1992; Laskowski et al. 1993).
Helix angles in the average structure were estimated by interpolating axes through all heavy atoms in each of the helices according to the MOLMOL least-squares algorithm.
NMR relaxation data analysis
Relaxation rates were calculated by least-squares fitting of mono-exponential decays to the peak-heights with the program Curvefit (A.G. Palmer III, Columbia University, NY, unpubl.). Errors were estimated to be 5% for relaxation rates and 10% for the HetNOEs. The coarse filter of Pawley et al. (2001) served to identify flexible residues [six in NusA(353–416), 12 in NusA(431–490)], that is, residues with significant motions on a pico- to nanosecond timescale, and residues probably involved in exchange processes. Additionally, residues outside of regular secondary structure and H353, Y362, L373, and Y386 were not taken into account for the calculation of the diffusion tensors with Tensor2 (Dosset et al. 2000) using standard settings. Hydrodynamic parameters of an elongated NusA(353–490) molecule were computed with HYDRONMR version 5a using a shell model (de la Torre and Carrasco 2000).
Acknowledgments
We gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (Ro617/12-1).
Abbreviations
αCTD, carboxy-terminal domain of the a subunit of the RNAP
E. coli, Escherichia coli
HetNOE, {1H}15N heteronuclear NOE
HhH, helix hairpin helix
HSQC, heteronuclear single quantum coherence
KH, K homology
λN, protein N of phage λ
NOESY, nuclear Overhauser spectroscopy
NusA, N utilization substance A
NusACTD, carboxy-terminal domain of NusA
NMR, nuclear magnetic resonance, PG-SLED, pulse gradient-stimulated echo longitudinal encode-decode
RDC, residual dipolar coupling
RMSD, root meansquare deviation
RNAP, RNA polymerase
SAM, sterile α motif
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051372205.
References
- Atkinson, A.R. and Kieffer, B. 2004. The role of protein motions in molecular recognition: Insights from heteronuclear NMR relaxation measurements. Prog. Nucl. Magn. Reson. Spectrosc. 44 141–187. [Google Scholar]
- Bax, A., Kontaxis, G., and Tjandra, N. 2001. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 339 127–174. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N.,Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin, I., Muhlberger, R., Bourenkov, G.P., Huber, R., Bacher, A., Richter, G., and Wahl, M.C. 2004. Structural basis for the interaction of Escherichia coli NusA with protein N of phage λ. Proc. Natl. Acad. Sci. 101 13762–13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braddock, D.T., Cai, M., Baber, J.L., Huang, Y., and Clore, G.M. 2001. Rapid identification of medium- to large-scale interdomain motion in modular proteins using dipolar couplings. J. Am. Chem. Soc. 123 8634–8635. [DOI] [PubMed] [Google Scholar]
- Braddock, D.T., Louis, J.M., Baber, J.L., Levens, D., and Clore, G.M. 2002. Structure and dynamics of KH domains from FBP bound to single-stranded DNA. Nature 415 1051–1056. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., and Bax, A. 1998. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J. Magn. Reson. 133 216–221. [DOI] [PubMed] [Google Scholar]
- Cordingley, M.G., Callahan, P.L., Sardana, V.V., Garsky, V.M., and Colonno, R.J. 1990. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J. Biol. Chem. 265 9062–9065. [PubMed] [Google Scholar]
- Dayie, K.T. and Wagner, G. 1994. Relaxation-rate measurements for 15N-1H groups with pulsed-field gradients and preservation of coherence pathways. J. Magn. Reson. 111A 121–126. [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- de la Torre, J. and Carrasco, H.B. 2000. HYDRONMR: Prediction of NMR relaxation of globular proteins from atomic level structures and hydrodynamic calculations. J. Magn. Reson. 147B 138–146. [DOI] [PubMed] [Google Scholar]
- Doherty, A.J., Serpell, L.C., and Ponting, C.P. 1996. The helix-hairpinhelix DNA-binding motif: A structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 24 2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosset, P., Hus, J.C., Blackledge, M., and Marion, D. 2000. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR 16 23–28. [DOI] [PubMed] [Google Scholar]
- Eisenmann, A., Schwarz, S., Rösch, P., and Schweimer, K. 2004. Sequencespecific 1H, 13C, 15N resonance assignments and secondary structure of the carboxy-terminal domain of the E. coli transcription factor NusA. J. Biomol. NMR 28 193–194. [DOI] [PubMed] [Google Scholar]
- Folmer, R.H., Hilbers, C.W., Konings, R.N., and Nilges, M. 1997. Floating stereospecific assignment revisited: Application to an 18 kDa protein and comparison with J-coupling data. J. Biomol. NMR 9 245–258. [DOI] [PubMed] [Google Scholar]
- Fushman, D., Varadan, R., Assfalg, M., and Walker, O. 2004. Determining domain orientation in macromolecules by using spin-relaxation and residual dipolar coupling measurements. Prog. Nucl. Magn. Reson. Spectrosc. 44 189–214. [Google Scholar]
- Gopal, B., Haire, L.F., Gamblin, S.J., Dodson, E.J., Lane, A.N., Papavinasasundaram, K.G., Colston, M.J., and Dodson, G. 2001. Crystal structure of the transcription elongation/anti-termination factor NusA from Mycobacterium tuberculosis at 1.7 Å resolution. J. Mol. Biol. 314 1087–1095. [DOI] [PubMed] [Google Scholar]
- Gusarov, I. and Nudler, E. 2001. Control of intrinsic transcription termination by N and NusA: The basic mechanisms. Cell 107 437–449. [DOI] [PubMed] [Google Scholar]
- Hansen, M.R., Mueller, L., and Pardi, A. 1998. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 5 1065–1074. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. Mapping the protein universe. Science 273 595–602. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRview: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Jones, J.A., Wilkins, D.K., Smith, L.J., and Dobson, C.M. 1997. Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR 10 199–203. [Google Scholar]
- Kay, L.E., Torchia, D.A., and Bax, A. 1989. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry 28 8972–8979. [DOI] [PubMed] [Google Scholar]
- Kim, C.A. and Bowie, J.U. 2003. SAM domains: Uniform structure, diversity of function. Trends Biochem. Sci. 28 625–628. [DOI] [PubMed] [Google Scholar]
- Kohno, T., Kusunoki, H., Sato, K., and Wakamatsu, K. 1998. A new general method for the biosynthesis of stable isotope-enriched peptides using a decahistidine-tagged ubiquitin fusion system: An application to the production of mastoparan-X uniformly enriched with 15N and 15N/13C. J. Biomol. NMR 12 109–121. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structure. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Kumar, S. and Bansal, M. 1998. Dissecting α-helices: Position specific analysis of α-helices in globular proteins. Proteins 31 460–476. [DOI] [PubMed] [Google Scholar]
- Kuszewski, J. and Clore, G.M. 2000. Soures of and solution to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J. Magn. Reson. 146 249–254. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Liu, K., Zhang, Y., Severinov, K., Das, A., and Hanna, M.M. 1996. Role of Escherichia coli RNA polymerase α subunit in modulation of pausing, termination, and antitermination by the transcription elongation factor NusA. EMBO J. 15 150–161. [PMC free article] [PubMed] [Google Scholar]
- Maciejewski, M.W., Liu, D., Prasad, R., Wilson, S.H., and Mullen, G.P. 2000. Backbone dynamics and refined solution structure of the N-terminal domain of DNA polymerase β. Correlation with DNA binding and dRP lyase activity. J. Mol. Biol. 296 229–253. [DOI] [PubMed] [Google Scholar]
- Mah, T.F., Li, J., Davidson, A.R., and Greenblatt, J. 1999. Functional importance of regions in Escherichia coli elongation factor NusA that interact with RNA polymerase, the bacteriophage λ N protein and RNA. Mol. Microbiol. 34 523–537. [DOI] [PubMed] [Google Scholar]
- Mah, T.F., Kuznedelov, K., Mushegian, A., Severinov, K., and Greenblatt, J. 2000. The α subunit of E. coli RNA polymerase activates RNA binding by NusA. Genes & Dev. 14 2664–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel, A.M., Akke, M., and Palmer, A.G. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246 144–163. [DOI] [PubMed] [Google Scholar]
- Mogridge, J., Mah, T.F., and Greenblatt, J. 1995. A protein-RNA interaction network facilitates the template-independent cooperative assembly on RNA polymerase of a stable antitermination complex containing the λ N protein. Genes & Dev. 9 2831–2845. [DOI] [PubMed] [Google Scholar]
- Mogridge, J., Legault, P., Li, J., Van Oene, M.D., Kay, L.E., and Greenblatt, J. 1998. Independent ligand-induced folding of the RNA-binding domain and two functionally distinct antitermination regions in the phage λ N protein. Mol. Cell 1 265–275. [DOI] [PubMed] [Google Scholar]
- Mooney, R.A., Artsimovitch, I., and Landick, R. 1998. Information processing by RNA polymerase: Recognition of regulatory signals during RNA chain elongation. J. Bacteriol. 180 3265–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, A.L., MacArthur, M.W., Hutchinson, E.G., and Thornton, J.M. 1992. Stereochemical quality of protein structure coordinates. Proteins 12 345–364. [DOI] [PubMed] [Google Scholar]
- Neudecker, P., Sticht, H., and Rösch, P. 2001. Improving the efficiency of the Gaussian conformational database potential for the refinement of protein and nucleic acid structures. J. Biomol. NMR 21 373–375. [DOI] [PubMed] [Google Scholar]
- Neudecker, P., Nerkamp, J., Eisenmann, A., Nourse, A., Lauber, T., Schweimer, K., Lehmann, K., Schwarzinger, S., Ferreira, F., and Rösch, P. 2004. Solution structure, dynamics, and hydrodynamics of the calcium-bound cross-reactive birch pollen allergen Bet v 4 reveal a canonical monomeric two EF-hand assembly with a regulatory function. J. Mol. Biol. 336 1141–1157. [DOI] [PubMed] [Google Scholar]
- Nilges, M. 1995. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J. Mol. Biol. 245 645–660. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Clore, G.M., and Gronenborn, A.M. 1988a. Determination of three-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamical simulated annealing calculations. FEBS Lett. 229 317–324. [DOI] [PubMed] [Google Scholar]
- ———. 1988b. Determination of three-dimensional structures of proteins from interproton distance data by dynamical simulated annealing from a random array of atoms. Circumventing problems associated with folding. FEBS Lett. 239 129–136. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Gronenborn, A.M., Brunger, A.T., and Clore, G.M. 1988c. Determination of three-dimensional structures of proteins by simulated annealing with interproton distance restraints. Application to crambin, potato carboxypeptidase inhibitor, and barley serine proteinase inhibitor 2. Protein Eng. 2 27–38. [DOI] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D., and Heringa, J. 2000. T-Coffee: A novel method for multiple sequence alignments. J. Mol. Biol. 302 205–217. [DOI] [PubMed] [Google Scholar]
- Nudler, E. 1999. Transcription elongation: Structural basis and mechanisms. J. Mol. Biol. 288 1–12. [DOI] [PubMed] [Google Scholar]
- Nudler, E. and Gottesman, M.E. 2002. Transcription termination and antitermination in E. coli. Genes Cells 7 755–768. [DOI] [PubMed] [Google Scholar]
- Pawley, N.H., Wang, C., Koide, S., and Nicholson, L.K. 2001. An improved method for distinguishing between anisotropic tumbling and chemical exchange in analysis of 15N relaxation parameters. J. Biomol. NMR. 20 149–165. [DOI] [PubMed] [Google Scholar]
- Powell, M.J.D. 1977. Restart procedures for the conjugate gradient method. Math. Progr. 12 241–254. [Google Scholar]
- Sattler, M., Schleucher, J., and Griesinger, C. 1999. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 34 39–158. [Google Scholar]
- Schweimer, K., Hoffmann, S., Bauer, F., Friedrich, U., Kardinal, C., Feller, S.M., Biesinger, B., and Sticht, H. 2002. Structural investigation of the binding of a herpesviral protein to the SH3 domain of tyrosine kinase Lck. Biochemistry 41 5120–5130. [DOI] [PubMed] [Google Scholar]
- Schwieters, C.D., Kuszewski, J.J., Tjandra, N., and Clore, G.M. 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160 66–74. [DOI] [PubMed] [Google Scholar]
- Shao, X. and Grishin, N.V. 2000. Common fold in helix-hairpin-helix proteins. Nucleic Acids Res. 28 2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, D.H., Nguyen, H.H., Jancarik, J., Yokota, H., Kim, R., and Kim, S.H. 2003. Crystal structure ofNusAfromThermotogamaritimaand functional implication of the N-terminal domain. Biochemistry 42 13429–13437. [DOI] [PubMed] [Google Scholar]
- Stevens, S.Y., Sanker, S., Kent, C., and Zuiderweg, E.R. 2001. Delineation of the allosteric mechanism of a cytidylyltransferase exhibiting negative cooperativity. Nat. Struct. Biol. 8 947–952. [DOI] [PubMed] [Google Scholar]
- Thanos, C.D., Faham, S., Goodwill, K.E., Cascio, D., Phillips, M., and Bowie, J.U. 1999. Monomeric structure of the human EphB2 sterile α motif domain. J. Biol. Chem. 274 37301–37306. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Feller, S.E., Pastor, R.W., and Bax, A. 1995. Rotational diffusion anisotropy of human ubiquitin from 15N relaxation. J. Am. Chem. Soc. 117 12562–12566. [Google Scholar]
- Tjandra, N., Grzesiek, S., and Bax, A. 1996. Magnetic field dependence of nitrogen-proton J splittings in 15N-enriched human ubiquitin resulting from relaxation interference and residual dipolar coupling. J. Am. Chem. Soc. 118 6264–6272. [Google Scholar]
- Tjandra, N., Omichinski, J.G., Gronenborn, A.M., Clore, G.M., and Bax, A. 1997. Use of dipolar 1H-15N and 1H-13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Biol. 4 732–738. [DOI] [PubMed] [Google Scholar]
- Tsugawa, A., Saito, M., Court, D.L., and Nakamura, Y. 1988. NusA amber mutation that causes temperature-sensitive growth of Escherichia coli. J. Bacteriol. 170 908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gilst, M.R. and von Hippel, P.H. 1997. Assembly of the N-dependent antitermination complex of phage λ. NusA and RNA bind independently to different unfolded domains of the N protein. J. Mol. Biol. 274 160–173. [DOI] [PubMed] [Google Scholar]
- Vuister, W.G. and Bax, A. 1993. Quantitative J correlation: A new approach for measuring homonuclear three-bond J(HNHA) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 115 7772–7777. [Google Scholar]
- Wilkins, D.K., Grimshaw, S.B., Receveur, V., Dobson, C.M., Jones, J.A., and Smith, L.J. 1999. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 38 16424–16431. [DOI] [PubMed] [Google Scholar]
- Worbs, M., Bourenkov, G.P, Bartunik, H.D., Huber, R., and Wahl, M.C. 2001. An extended RNA binding surface through arrayed S1 and KH domains in transcription factor NusA. Mol. Cell. 7 1177–11789. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. Wiley, New York.
- Xia, T., Frankel, A., Takahashi, T.T., Ren, J., and Roberts, R.W. 2003. Context and conformation dictate function of a transcription antitermination switch. Nat. Struct. Biol. 10 812–819. [DOI] [PubMed] [Google Scholar]
- Zuiderweg, E.R. 2002. Mapping protein-protein interactions in solution by NMR spectroscopy. Biochemistry 41 1–7. [DOI] [PubMed] [Google Scholar]