

Abstract
Human γD crystallin (HγD-Crys) is a two domain, β-sheet eye lens protein that must remain soluble throughout life for lens transparency. Single amino acid substitutions of HγD-Crys are associated with juvenile-onset cataracts. Features of the interface between the two domains conserved among γ-crystallins are a central six-residue hydrophobic cluster, and two pairs of interacting residues flanking the cluster. In HγD-Crys these pairs are Gln54/Gln143 and Arg79/Met147. We previously reported contributions of the hydrophobic cluster residues to protein stability. In this study alanine substitutions of the flanking residue pairs were constructed and analyzed. Equilibrium unfolding/refolding experiments at 37°C revealed a plateau in the unfolding/refolding transitions, suggesting population of a partially folded intermediate with a folded C-terminal domain (C-td) and unfolded N-terminal domain (N-td). The N-td was destabilized by substituting residues from both domains. In contrast, the C-td was not significantly affected by substitutions of either domain. Refolding rates of the N-td were significantly decreased for mutants of either domain. In contrast, refolding rates of the C-td were similar to wild type for mutants of either domain. Therefore, domain interface residues of the folded C-td probably nucleate refolding of the N-td. We suggest that these residues stabilize the native state by shielding the central hydrophobic cluster from solvent. Glutamine and methionine side chains are among the residues covalently damaged in aged and cataractous lenses. Such damage may generate partially unfolded, aggregation- prone conformations of HγD-Crys that could be significant in cataract.
Keywords: human γD crystallin, domain interface, partially folded intermediate, cataract, equilibrium unfolding/refolding transitions
The α-, β- and γ-crystallins are structural proteins of the vertebrate eye lens whose solubility and stability are required to maintain eye lens transparency throughout life. The β- and γ-crystallins act solely as structural proteins in the lens, while the α-crystallins are related to small heat shock proteins and probably have an additional in situ function as passive chaperones (Horwitz 1992; Boyle and Takemoto 1994). The α-crystallin sub-units associate in the lens to form polydisperse, high-molecular-weight complexes (Haley et al. 1998; Aquilina et al. 2005).
Lens transparency is established via short-range ordering of natively folded crystallins in the fibrous cells of the lens (Delaye and Tardieu 1983; Fernald and Wright 1983). The protein concentration in these cells approaches 70% g/g wet weight, with the crystallins accounting for >90% of the total protein (Oyster 1999). The major mass of crystallin proteins in the lens are expressed in utero, do not turnover during adulthood, and thus must remain soluble and stable in the continued presence of environmental stresses for a lifetime. The crystallins in aged and cataractous lenses are covalently damaged, presumably as a result of age and exposure to radiative and oxidative stress (Hoenders and Bloemendal 1983). Covalent damage found in the crystallins of aged and cataractous lenses include nonnative disulfide bonding, backbone cleavage, methionine oxidation and glutamine and asparagine deamidation (Hanson et al. 1998, 2000; Lampi et al. 1998, 2001, 2002; Ma et al. 1998; Harms et al. 2004). The eye disease mature-onset cataract is associated with the aggregation and condensation of such damaged crystallin proteins (Hoenders, and Bloemendal 1983).
The βγ-crystallin superfamily comprises >50% of the crystallins in the vertebrate lens (Wistow et al. 1983; Slingsby et al. 1997). Members of this superfamily exhibit a conserved two-domain, primarily β-sheet fold that likely arose from a gene duplication event (Lubsen et al. 1988; Wistow and Piatigorsky 1988; Norledge et al. 1996). The highly homologous N- (N-td) and C-(C-td) terminal domains are composed of two Greek-key motifs each and exhibit pseudo-twofold pairing. The domains are covalently linked by a six- to eight-amino-acid linker and interact noncovalently through interdomain side-chain contacts. The γ-crystallins are monomeric, with the N-tds and C-tds pairing intramolecularly, while the β-crystallins associate into multimers (Wistow et al. 1983; Bax et al. 1990; Slingsby and Bateman 1990). For instance, bovine βB2 crystallin forms dimers by domain swapping where the N-tds and C-tds pair in a similar manner as in the γ-crystallins, except that pairing occurs between domains from different subunits (Bax et al. 1990). Despite differences in oligomeric state, side chains that make contact across the domain interfaces of the β- and γ-crystallins play a critical role in intra- or inter-molecular domain interactions.
Human γD crystallin (HγD-Crys) is a monomeric, 173-amino-acid protein and a member of the βγ-crystallin superfamily. HγD-Crys is present at highest concentrations in the lens nucleus, the region formed earliest in development. Mutations resulting in single amino acid substitutions of HγD-Crys are associated in juvenile-onset cataract (Stephan et al. 1999; Pande et al. 2001; Santhiya et al. 2002). The crystal structure of HγD-Crys has been solved to 1.25 Å resolution (Basak et al. 2003) and displays the βγ-crystallin superfamily’s characteristic two-domain fold, with the domains interacting intramolecularly (Fig. 1 ▶). The two domains of HγD-Crys are covalently joined by an extended six-residue linker and the side chains of 10 amino acids interact noncovalently across the domain interface. Buried in the center of the interface is a hydrophobic cluster of Met43, Phe56, and Ile81 from the N-td, and Val132, Leu145, and Val170 from the C-td. Additionally, there are pair-wise interactions on the periphery of the hydrophobic cluster between Gln54 (N-td) and Gln143 (C-td) at the top of the interface, and Arg79 (N-td) and Met147 (C-td) nearer to the linker peptide (Fig. 1 ▶). These peripheral interactions flank the central hydrophobic cluster and appear to shield it from solvent. Chemical properties of the amino acids in these interface positions are highly conserved among γ-crystallins from diverse organisms, suggesting that they are critical for folding, stability, or solubility. An exception to this is Met147 in the human sequence, as most γ-crystallins have an arginine in this position. The amino acids Gln54, Gln143, and Met147 are all potential sites of covalent damage in aged lenses.
Figure 1.

The crystal structure of wild-type HγD-Crys (Basak et al. 2003) shown in ribbon representation with the peripheral interface residues in spacefill (PDB code 1HK0). www.proteinscience.org 2031 Domain interactions of human γD crystallin
Folding and stability of the β- and γ-crystallins have been extensively studied and, in general, the γ-crystallins display higher intrinsic stability than the β-crystallins (for review, see Bloemendal et al. 2004). During refolding, rat βB2 crystallin, bovine γB crystallin (BγB-Crys), and HγD-Crys display competing aggregation reactions (Rudolph et al. 1990; Jaenicke 1999; Kosinski-Collins and King 2003). Aggregation may have also been observed but not reported for other β- and γ-crystallins. Domain stablities of several β- and γ-crystallins have also been studied both in the context of the full-length proteins as well as in isolation (Rudolph et al. 1990; Sharma et al. 1990; Mayr et al. 1997; Wieligmann et al. 1999). The domains of these proteins are often less stable in isolation than when paired with their partner domain in the full-length proteins.
We previously investigated wild-type HγD-Crys unfolding/refolding in near-physiological conditions (37°C, and phosphate buffer [pH 7.0]) to best relate in vitro and in vivo properties (Kosinski-Collins and King 2003; Kosinski-Collins et al. 2004; Flaugh et al. 2005). Refolding to concentrations of GuHCl below 1.0 M revealed an aggregation pathway that competed with productive refolding, which may provide an in vitro model for the involvement of HγD-Crys in cataract (Kosinski-Collins and King 2003). In productive kinetic refolding to 1.0 M GuHCl, HγD-Crys exhibited a sequential domain refolding pathway (Fig. 2 ▶) where the C-td refolded first followed by the N-td (Kosinski-Collins et al. 2004). Subsequently, a partially folded intermediate was detected in equilibrium unfolding/ refolding experiments that likely had a folded C-td and unfolded N-td (Flaugh et al. 2005). Since partially folded species are often involved in off-pathway aggregation, the single folded domain species is an attractive candidate for the species that aggregates during refolding (Fig. 2 ▶).
Figure 2.

A schematic illustration of the in vitro kinetic refolding pathway of HγD-Crys. The C-td refolds first, followed by the N-td (Kosinski-Collins et al. 2004). The conformation of the aggregation-prone intermediate is currently unknown but may be related to the single folded domain conformer.
Given that the two domains of HγD-Crys refold sequentially and display differential stability, association of the residues that make contact across the domain interface may be critical in folding and stability. This has been confirmed by a mutational study of hydrophobic domain interface residues of HγD-Crys (Flaugh et al. 2005). Several site-directed alanine mutants of the hydrophobic domain interface residues displayed significantly reduced refolding rates for the N-td but not the C-td. Additionally, these mutants also displayed reduced midpoints for unfolding/refolding of the N-td (Flaugh et al. 2005). Similarly, mutating domain interface residues of BγB-Crys and human βB1 crystallin (HβB1-Crys) destabilized the proteins (Palme et al. 1997; Kim et al. 2002).
In this study we use site-specific mutagenesis to study the two pairs of peripheral interface residues in HγD-Crys that flank the hydrophobic cluster, Gln54/Gln143 and Arg79/Met147. Similar to results of the hydrophobic interface residues, substituting these residues with alanine caused selective destabilization of the N-td and a decrease in the rate of refolding for the N-td but not the C-td.
Results
Protein expression and purification
All of the proteins used in this study had an exogenous N-terminal peptide with the sequence MKHHHHHHQ to aid in purification. Previous studies confirmed that the addition of the His-tag did not perceptibly affect the structure of the native protein or its thermodynamic or kinetic refolding properties (Kosinski-Collins and King 2003; Kosinski-Collins et al. 2004).
The mutant proteins expressed at levels comparable to that of wild type and behaved similarly to wild type during purification. The proteins were found primarily in the soluble fraction after cell lysis (>90%) and were purified by Ni-NTA affinity chromatography to >98% homogeneity (data not shown). As described further below, the proteins were in soluble native-like conformations. Thus, these residues were not required for the in vivo folding of HγD-Crys in Escherichia coli.
Circular dichroism and fluorescence spectroscopy
The structures of single and double alanine mutants of HγD-Crys were probed by circular dichroism (CD) and fluorescence spectroscopy. In accord with previous results, the far-UV CD spectrum of wild-type HγD-Crys at 37°C was reminiscent of a primarily β-sheet protein with a prominent minimum at 218 nm (Andley et al. 1996; Pande et al. 2000). The far UV-CD spectra of the single and double mutants, Q54A and R79A/ M147A, closely resembled that of wild-type HγD-Crys (Fig. 3A ▶). In contrast, the ellipticity intensities of Q143A, Q54A/Q143A, R79A, and M147A differed slightly from wild-type HγD-Crys and the other mutants. Despite the difference in intensity, the minima of all proteins were indistinguishable. The discrepancy in intensity may reflect disturbances of the domain interface or other structural rearrangements that do not grossly disrupt overall β-sheet content.
Figure 3.

(A) Far-UV CD spectra of wild-type (⋄), R79A (○), M147A (▪), R79A/M147A (▸), Q54A (▵), Q143A (◂), and Q54A/Q143A (▾) HγD-Crys. Samples contained 100 μg/mL protein in 10 mM sodium phosphate, 5 mM DTT, and 1 mM EDTA (pH 7.0) at 37°C. A 0.25-cm pathlength cuvette was used for all measurements. (B) Fluorescence spectroscopy of wild-type and mutant HγD-Crys. Symbols are the same as in A. Samples contained 10 μg/mL protein in 10 mM sodium phosphate, 5 mM DTT, and 1 mM EDTA (pH 7.0) at 37°C.
Fluorescence spectra of the wild-type and mutant proteins were measured by using an excitation wave-length of 295 nm with emission monitored from 310 to 420 nm. Consistent with previous results, the spectrum of wild-type HγD-Crys had an emission maximum of 325 nm (Kosinski-Collins and King 2003). The fluorescence spectra of the mutant proteins had emission maxima and similar intensities as those of wild-type HγD-Crys (Fig. 3B ▶). These results suggest that the tryptophan side chains are buried in the hydrophobic cores of the mutant proteins and are located in environments similar to that of the wild-type protein. Unfolding the proteins in 5.5 M GuHCl shifted the emission maxima to 350 nm and increased the emission intensities (data not shown). This native state quenching has been previously described for HγD-Crys and other β- and γ-crystallins (Kim et al. 2002; Bateman et al. 2003; Kosinski-Collins et al. 2004).
Equilibrium unfolding/refolding of wild-type HγD-Crys
Previous equilibrium unfolding/refolding experiments of wild-type HγD-Crys used fluorescence spectroscopy to monitor structural changes in GuHCl (Kosinski-Collins and King 2003; Evans et al. 2004; Kosinski-Collins et al. 2004; Flaugh et al. 2005). The fluorescent residues of HγD-Crys include four buried tryptophans and 14 primarily surface-exposed tyrosines. An excitation wavelength of 295 nm was used to selectively monitor exposure of the tryptophan side chains to solvent. Structural transitions observable by this approach include complete unfolding of themolecule or structural transitions of a single domain.
Previous equilibrium unfolding/refolding experiments of HγD-Crys were performed at 37°C by using an equilibration time of 24 h, and the data were analyzed by plotting the concentration of GuHCl versus a ratio of fluorescence intensities at 360 and 320 nm (Flaugh et al. 2005). Using these parameters, a slight inflection in the transitions was observed at ~2.3 M GuHCl, suggesting population of a partially folded intermediate (Fig. 4 ▶). The transitions were best fit by a three-state model where population of a partially folded intermediate was assumed to occur in equilibrium with the native and unfolded states. When fit to a three-state model, the native to intermediate transition had a midpoint of 2.2 M GuHCl, and the intermediate to unfolded transition had a midpoint of 2.8 MGuHCl (Table 1). The first transition likely corresponded to unfolding/refolding of the N-td and the second transition to unfolding/refolding of the C-td (Flaugh et al. 2005).
Figure 4.

Equilibrium unfolding (♦) and refolding (⋄) of wild-type HγD-Crys in GuHCl probed by fluorescence emission. Fluorescence spectra were collected by using an excitation wavelength of 295 nm, and fluorescence intensity at 360/320 nm was used for data analysis. Protein was present at 10 μg/mL in 10 mM sodium phosphate, 5 mM DTT, 1 mM EDTA (pH 7.0), and GuHCl from 0–5.5 M at 37°C, and samples were incubated for 24 h prior to measurement. The solid line is a three-state fit of the unfolding data. Residuals of the three-state fit are shown.
Table 1.
Equilibrium unfolding/refolding parameters for wild-type and mutant HγD-Crys
| Transition 1 | Transition 2 | |||||||
| Protein | [GuHCl]1/2a | Apparent m valueb | Apparent ΔG°c | Δ ΔG°d | Δ Δ Inte | [GuHCl]1/2a | Apparent m valueb | Apparent ΔG°c |
| Wild typef | 2.2 ± 0.1 | 3.6 ± 0.1 | 7.7 ± 0.2 | – | 2.8 ± 0.1 | 3.1 ± 0.4 | 8.9 ± 1.3 | |
| R79A | 1.8 ± 0.1 | 3.4 ± 0.4 | 6.7 ± 1.6 | −1.0 | 2.9 ± 0.1 | 2.7 ± 0.2 | 7.6 ± 0.6 | |
| M147A | 1.8 ± 0.1 | 3.7 ± 0.4 | 6.6 ± 1.3 | −1.1 | 2.8 ± 0.1 | 3.1 ± 0.3 | 8.9 ± 1.0 | |
| Q54A | 2.0 ± 0.1 | 3.8 ± 0.2 | 7.5 ± 0.2 | −0.2 | 2.9 ± 0.1 | 3.4 ± 0.3 | 9.6 ± 1.1 | |
| Q143A | 1.9 ± 0.1 | 3.8 ± 0.2 | 7.2 ± 0.3 | −0.5 | 3.0 ± 0.1 | 3.0 ± 0.2 | 9.0 ± 0.7 | |
| R79A/M147A | 1.7 ± 0.1 | 3.2 ± 0.4 | 5.6 ± 0.5 | −2.1 | 0.0 | 2.8 ± 0.1 | 3.2 ± 0.3 | 8.9 ± 0.8 |
| Q54A/Q143A | 1.8 ± 0.1 | 3.5 ± 0.1 | 6.3 ± 0.1 | −1.4 | −0.7 | 3.1 ± 0.1 | 3.1 ± 0.2 | 9.6 ± 0.7 |
a Transition midpoints in units of M.
b Apparent m values in units of kcal/mol/M.
c Free energy of unfolding in the absence of GuHCl in units of kcal/mol.
d ΔΔG° = ΔG°Mutant − ΔG°Wild-type in units of kcal/mol.
e Free energy of interaction for peripheral amino acids calculated using the equation: ΔΔGInt = ΔG°Wild-type+ΔG°XY − ΔG°X −ΔG°Y in units of kcal/mol, where X, Y, and XY are the single and double mutants of the putatively interacting side-chains. A negative value represents a favorable interaction energy in the native state.
fFrom Flaugh et al. (2005).
Equilibrium unfolding/refolding of Arg79/Met147 mutants
In order to assess the contributions of peripheral domain interface amino acids to the stability of HγD-Crys, equilibrium unfolding/refolding experiments were performed. These experiments were performed on all alanine substitution mutants by using analogous methodologies as those described for wild-type HγD-Crys.
The equilibrium unfolding/refolding transitions of R79A deviated slightly from that observed for wild type (Fig. 5 ▶). Similar to wild type, a plateau was evident in the unfolding/refolding curves at ~2.3 M GuHCl. However, the range of GuHCl concentrations over which the intermediate was populated was increased for R79A. The unfolding/refolding transitions were best fit to a three-state model similar to wild type, with a native to intermediate transition midpoint of 1.8 M GuHCl and an intermediate to unfolded transition mid-point of 2.9 MGuHCl (Table 1). Alanine substitution of Arg79 decreased the free energy of unfolding (ΔG°) of the native to intermediate transition by ~1.0 kcal/mol compared with wild-type HγD-Crys (Table 1).
Figure 5.
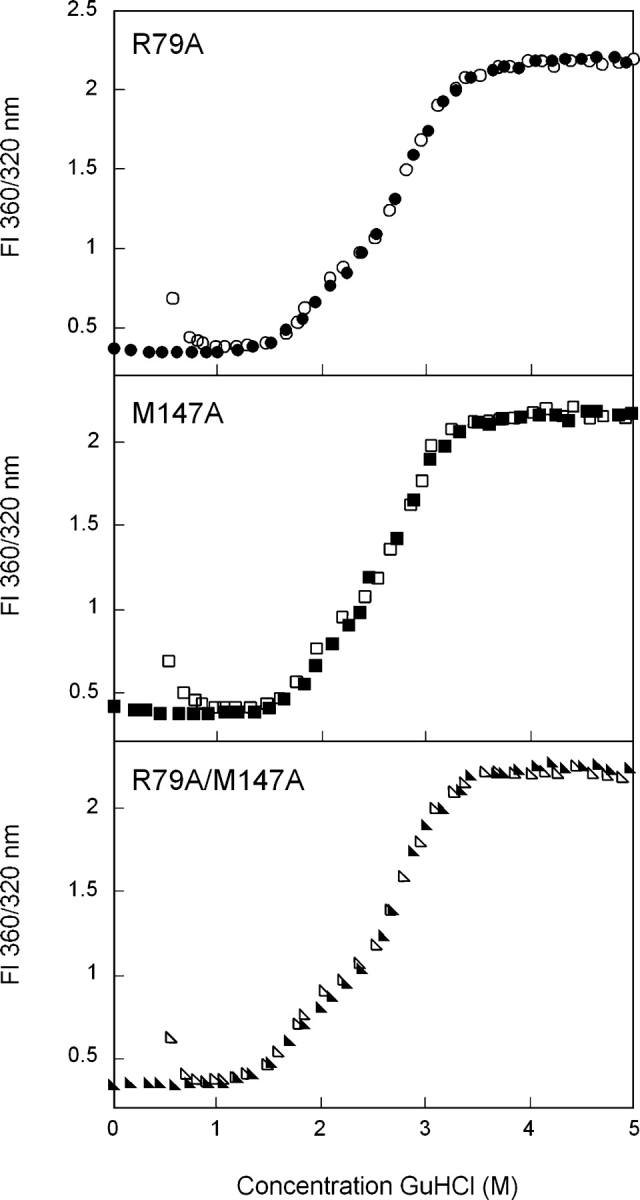
Equilibrium unfolding/refolding of R79A (•), M147A (▪), and R79A/M147A (▸) HγD-Crys in GuHCl probed by fluorescence emission. Data was analyzed by fluorescence intensity at 360/320 nm using an excitation wavelength of 295 nm. Protein was present at 10 μg/mL in 10 mM sodium phosphate, 5 mM DTT, 1 mM EDTA (pH 7.0), and GuHCl from 0–5.5 M at 37°C. Samples were equilibrated for 24 h. The unfolding transitions are shown as closed symbols; the refolding transitions as open symbols.
The equilibrium unfolding/refolding transitions of M147A also displayed a significant plateau, suggesting population of a partially folded intermediate (Fig. 5 ▶). Similar to R79A, the intermediate was populated over a larger range of GuHCl concentrations than wild-type HγD-Crys. When fit to a three-state model, the native to intermediate transition had a midpoint of 1.8 M GuHCl, and the intermediate to unfolded transition had a midpoint of 2.8 M GuHCl (Table 1). The ΔG° of the native to intermediate transition was decreased by ~1.1 kcal/mol compared with wild type (Table 1).
The double alanine mutant R79A/M147A also populated a partially folded intermediate during equilibrium unfolding/refolding (Fig. 5 ▶). The native to intermediate transition was calculated to have a midpoint of 1.7 M GuHCl and the transition from intermediate to unfolded had a transition of 2.8 M GuHCl (Table 1). The ΔG° of the native to intermediate transition was decreased by ~2.1 kcal/mol compared with wild-type HγD-Crys (Table 1).
Equilibrium unfolding/refolding of Gln54/Gln143 mutants
Equilibrium unfolding/refolding of Q54A HγD-Crys also occurred by an apparent three-state mechanism with population of an intermediate at ~2.3 M GuHCl (Fig. 6 ▶). The transition from native to intermediate had a midpoint of 2.0 M GuHCl while the intermediate to unfolded transition had amidpoint of 2.9 M GuHCl (Table 1).This corresponded to a decrease in the native to intermediate ΔG° of ~0.2 kcal/ mol relative to wild type. In the crystal structure of HγD-Crys (Basak et al. 2003), the side chain of Gln54 participates in a hydrogen bond with the main-chain nitrogen of Leu145.
Figure 6.
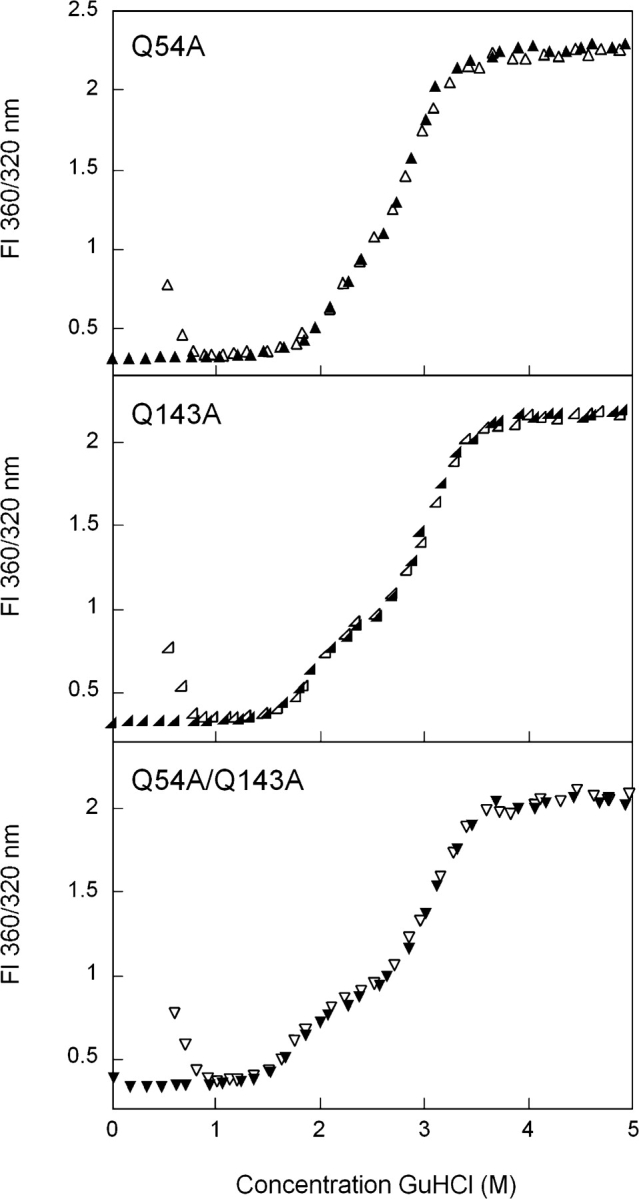
Equilibrium unfolding/refolding of Q54A (▴), Q143A (◂), and Q54A/Q143A (▾) HγD-Crys in GuHCl probed by fluorescence emission. The unfolding transitions are shown as closed symbols; the refolding transitions as open symbols. Data was analyzed by fluorescence intensity at 360/320 nm using an excitation wavelength of 295 nm. Protein was present at 10 μg/mL in 10 mM sodium phosphate, 5 mM DTT, 1 mM EDTA (pH 7.0), and GuHCl from 0–5.5 M at 37°C, with a 24-h equilibration time.
The side chain of Gln143 lies in very close proximity to that of Gln54. However, Gln143 does not participate in hydrogen bonding to any main-chain or side-chain atoms as observed in the crystal structure. There was a plateau in equilibrium unfolding/refolding curves of Q143A at 2.3 M GuHCl (Fig. 6 ▶). When fit to a three-state model, the native to unfolded transition had a midpoint of 1.9 M GuHCl and the intermediate to unfolded transition had a midpoint of 3.0 M GuHCl (Table 1). The native to intermediate transition had a ΔG° ~0.5 kcal/mol less than that for wild type.
The double mutant protein, Q54A/Q143A, also displayed a plateau in the equilibrium unfolding/refolding transitions (Fig. 6 ▶). The presence of a partially unfolded intermediate is more obvious for Q54A/Q143A than for any of the other mutants, as it appears to be populated over a range of 2.2–2.6 M GuHCl. The two transitions were calculated to have midpoints of 1.8 and 3.1 M GuHCl (Table 1). The ΔG° of the native to intermediate transition was decreased ~1.4 kcal/mol relative to wild type.
In vitro aggregation
HγD-Crys did not significantly aggregate or self-associate in the transition region during unfolding and refolding. However, consistent with previous results, when refolded from 5.5 M GuHCl to <1.0 M GuHCl, wild-type Hγ-DCrys aggregated into a high-molecular-weight species that significantly scattered light (Kosinski-Collins and King 2003). This causes a sharp increase in the FI 360/320 nm values of samples refolded to <1.0 M GuHCl due to right-angle light scattering by the aggregates (Fig. 4 ▶). All of the peripheral domain interface mutants displayed similar aggregation behavior (Figs. 5 ▶, 6 ▶).
Refolding to the native state below 1.0 M GuHCl is masked by the light scattering from the aggregated chains. The aggregation samples of all the mutant proteins were tested for the presence of native-like protein by fluorescence spectroscopy after centrifugation at 12,000 rpm. The fluorescence spectra of the soluble protein present in the supernatant after centrifugation were consistent with the presence of native protein (data not shown). Thus, the productive refolding and aggregation pathways compete under these conditions. All spectra displayed an emission maximum of ~325 nm and decreased fluorescence intensity, reflecting the loss of protein molecules into the aggregate.
Productive refolding kinetics of wild-type HγD-Crys
To analyze the role of peripheral domain interface residues in kinetic refolding of HγD-Crys, productive kinetic refolding experiments were performed analogous to previous experiments with wild type (Kosinski-Collins et al. 2004). Proteins were fully unfolded in 5.5 M GuHCl and subsequently diluted into refolding buffer. Burial of the tryptophan residues was monitored by observing a decrease in fluorescence intensity at 350 nm over time. Rapid dilution was performed with a syringe-injection port instead of a stopped-flow apparatus because the major transitions in HγD-Crys refolding have been shown to occur on a second, and not a millisecond, timescale (Kosinski-Collins et al. 2004). Potential refolding intermediates populated on a millisecond timescale were not addressed in these experiments.
Previous experiments determined that kinetic refolding of wild-type HγD-Crys was best fit with two exponentials, suggesting population of a partially folded intermediate (Kosinski-Collins et al. 2004). Upon dilution into refolding buffer, the fluorescence intensity at 350 nm rapidly decreased with a t1/2 of 15 sec. This first phase presumably corresponded to refolding into an intermediate conformation. The partially folded intermediate was more fluorescent than the native state and less fluorescent than the unfolded state at 350 nm. Following the fast phase, a second phase was observed where the fluorescence intensity decreased slowly with a t1/2 of 190 sec. This transition presumably corresponded to refolding into the native conformation.
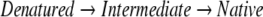 |
(1) |
Productive refolding kinetics of Arg79/Met147 mutants
Kinetic refolding of R79A is shown in Figure 7 ▶. Consistent with previous results of wild-type HγD-Crys, the curve was best fit with a three-state model. An initial rapid decrease in fluorescence intensity at 350 nm occurred with a t1/2 of 19 sec. A second slower phase occurred with a t1/2 of 890 sec (Fig. 7 ▶; Table 2). Similar to wild-type HγD-Crys, the intermediate had a fluorescence emission signal at 350 nm unique from both the native and unfolded conformations (Fig. 7 ▶). The t1/2 for transition from the denatured to intermediate state was approximately equal to that of wild-type HγD-Crys, while the t1/2 for the intermediate to native transition was increased more than fourfold.
Figure 7.

Productive kinetic refolding of wild-type (♦), R79A (•), M147A (▪), and R79A/M147A (▸) HγD-Crys. Proteins were initially unfolded at a concentration of 100 μg/mL in 5.5 M GuHCl at 37°C. Refolding was initiated by dilution of unfolded proteins into refolding buffer to give a final concentration of 10 μg/mL. Refolding buffer contained 10 mM sodium phosphate, 5 mM DTT, 1 mM EDTA (pH 7.0), and 1.0 M GuHCl at 37°C. Refolding was monitored by changes in fluorescence emission at 350 nm.
Table 2.
Kinetic refolding parameters for wild-type and mutant HγD-Crys
| Protein | k1 (sec−1) | t1/2 (sec) | k2 (sec−1) | t1/2 (sec) |
| Wild typea | 0.048 ± 0.001 | 15 ± 1 | 0.0037 ± 0.0001 | 190 ± 10 |
| R79A | 0.037 ± 0.006 | 19 ± 3 | 0.0008 ± 0.0001 | 890 ± 50 |
| M147A | 0.032 ± 0.002 | 21 ± 1 | 0.0010 ± 0.0001 | 680 ± 20 |
| Q54A | 0.040 ± 0.001 | 17 ± 1 | 0.0023 ± 0.0002 | 310 ± 30 |
| Q143A | 0.041 ± 0.003 | 17 ± 1 | 0.0016 ± 0.0001 | 430 ± 10 |
| R79A/M147A | 0.030 ± 0.004 | 23 ± 3 | 0.0004 ± 0.0001 | 1700 ± 20 |
| Q54A/Q143A | 0.033 ± 0.004 | 20 ± 2 | 0.0012 ± 0.0001 | 600 ± 40 |
aFrom Flaugh et al. (2005).
Refolding of M147A was also best fit with a three-state model (Fig. 7 ▶). A t1/2 of 21 sec was calculated for the initial rapid decrease in fluorescence corresponding to a transition from the denatured to intermediate state. The second transition from partially folded intermediate to native was significantly slower with a t1/2 of 680 sec (Table 2). Similar to the results for R79A, the t1/2 for the unfolded to intermediate transition of M147A was approximately equal to that of wild-type HγD-Crys and the t1/2 for the intermediate to native transition was notably increased.
The double mutant, R79A/M147A, displayed the most significantly altered refolding kinetics (Fig. 7 ▶). The denatured to partially folded intermediate transition was similar to that of wild-type HγD-Crys with a t1/2 of 23 sec. Conversely, the partially folded intermediate to native transition was markedly slower, occurring with a t1/2 of 1700 sec (Table 2).
Productive refolding kinetics of Gln54/Gln143 mutants
Productive kinetic refolding of the mutant proteins Q54A, Q143A, and Q54A/Q143A are shown in Figure 8 ▶. The data were best fit with a three-state model similar to wild type. Changes in fluorescence intensity at 350 nm upon refolding were similar to those described above for wild type. The first phase was characterized by a rapid decrease in fluorescence intensity and was followed by a second phase with a slower decrease in intensity. The transition from denatured to partially folded intermediate occurred with t1/2 values of 17 sec for both Q54A and Q143A and 20 sec for Q54A/Q143A (Table 2). These values are not significantly different from that measured for wild-type HγD-Crys. Conversely, the t1/2 values for the transition from partially folded intermediate to native were slightly increased compared with wild-type HγD-Crys. The t1/2 values were 310 sec for Q54A, 430 sec for Q143A, and 600 sec for Q54A/Q143A (Table 2).
Figure 8.

Productive kinetic refolding of wild-type (♦), Q54A (▴), Q143A (◂), and Q54A/Q143A (▾) HγD-Crys. Unfolded stock solutions of proteins were initially prepared at a concentration of 100 μg/mL in 5.5 M GuHCl at 37°C. Refolding was initiated by diluting the unfolded stock solutions into refolding buffer to give a final protein concentration of 10 μg/mL. Refolding buffer contained 10 mM sodium phosphate, 5 mM DTT, 1mMEDTA (pH 7.0), and 1.0MGuHCl at 37°C. Refolding was monitored by changes in fluorescence emission at 350 nm.
Discussion
Based on crystal structure and sequence alignment data, most domain interfaces of the β- and γ-crystallins are composed of a central hydrophobic cluster surrounded by peripheral paired interactions. The paired residues of HγD-Crys, identified by proximity in the crystal structure, are Gln54/Gln143 and Arg79/Met147 (Fig. 1 ▶). The side chains of these amino acids are in close proximity to both the exterior of the protein and the interface hydrophobic cluster. The side chains of Arg79 and Met147 are located toward the bottom of the domain interface and interact by packing the hydrophobic side chain of Met147 against the β, γ, and δ methylene groups of Arg79 (Fig. 1 ▶). The hydrophobic portions of the side chains of Arg79 and Met147 are also in close contact with the central hydrophobic cluster of the domain interface. Gln54 and Gln143 are located at the top of the domain interface and are in proximity of the hydrophobic cluster and the exterior of the protein (Fig. 1 ▶).
Destabilizing effects of alanine substitutions
The far-UV CD minima of all single and double alanine substitution mutants of peripheral interface residues were analogous to that of wild type. These data suggest that the mutants folded into a native-like structure with similar β-sheet content as that of wild-type HγD-Crys (Fig. 3A ▶). However, the far-UV CD spectra of the mutants R79A, M147A, Q143A, and Q54A/Q143A deviated moderately in overall intensity from that of wild type and the other mutants. Mutations of domain interface residues may disrupt or alter domain pairing, which could cause these differing CD signals. An alternative interpretation is that the differences in the spectra are due to discrepancies in individual solution conditions and not actual changes in the native state structures.
The fluorescence emission spectra of the mutant proteins indicated that they adopted native-like conformations similar to that of wild-type HγD-Crys. All single and double alanine mutants displayed a fluorescence emission maximum of ~325 nm and native-state quenching similar to the wild-type protein. These results suggest that the tryptophan side chains are buried in the hydrophobic cores of all mutants. However, native state fluorescence intensities of the mutants differed slightly from that of wild type (Fig. 3B ▶). This may be due to a structural rearrangement or relaxation around the tryptophans responsible for the quenching (Kosinski-Collins et al. 2004). Conclusive evidence for structural modifications will require obtaining high-resolution structures of the mutant proteins. The overall behavior of these mutant proteins suggests that, individually, these side chains are not critical determinants of the native state fold.
Stabilities of the mutant proteins were determined by equilibrium unfolding/refolding in GuHCl. An inflection in the unfolding/refolding curves was evident at 2.3 M GuHCl for the wild-type and mutant proteins, suggesting population of a partially folded intermediate in equilibrium with the native and unfolded states. Previous investigations of triple tryptophan mutants of HγD-Crys indicated that the C-td is more stable than the N-td (Kosinski-Collins et al. 2004). Preliminary studies of the isolated N-td and C-td of HγD-Crys have confirmed this observation (I.A. Mills, S.L. Flaugh, and J. King, unpubl.). Thus, we postulate that the intermediate populated during equilibrium unfolding/refolding of wild-type HγD-Crys is a single folded domain conformer with a folded C-td and unfolded N-td (Flaugh et al. 2005). According to this hypothesis, the native to intermediate transition corresponded to unfolding/refolding of N-td and the intermediate to unfolded transition corresponded to unfolding/refolding of the C-td. From here on we discuss the results according to these terms.
All single and double alanine substitution mutants displayed unfolding/refolding transitions that differed from that of wild type. The midpoint of N-td unfolding/ refolding was consistently decreased for all mutants, thus causing the partially folded intermediate to be populated over a larger range of GuHCl concentrations (Figs. 5 ▶, 6 ▶; Table 1). Similar effects were observed with alanine substitution mutants of hydrophobic domain interface residues (Flaugh et al. 2005). Therefore, stability of the N-td was dependent on correct association of both peripheral and hydrophobic domain interface residues.
In contrast to the consistent destabilization of the N-td, midpoints of C-td unfolding/refolding remained the same or increased slightly with the mutations. Thus, the C-td of full-length wild-type HγD-Crys was not stabilized by domain interface contacts. The mutant Q143A had a midpoint of 3.0 M GuHCl for C-td unfolding/ refolding, and Q54A/Q143A had a midpoint of 3.1 M GuHCl. If the partially folded intermediate has a structured C-td and unstructured N-td, in the intermediate conformation the side chain of Gln143 would be in close proximity to hydrophobic domain interface residues of the structured C-td. Substituting this side chain for alanine may have increased the stability of the C-td by eliminating potentially unfavorable interactions between the closely positioned polar and hydrophobic side chains. Two of the other mutants, R79A and Q54A, also displayed intermediate to unfolded transition midpoints slightly greater than that of wild type (Table 1). These differences may be due to slight changes in the m values of the mutants or other effects on the stability of the intermediate that would be difficult to identify without a more detailed description of the conformation.
Double-mutant cycle analysis has been extensively used as a method to evaluate interaction energy between two amino acid side chains in close proximity in the native state conformation of a protein (Horovitz 1996). By comparing the ΔG° for the single and double alanine substitution mutants of Arg79/Met147 and Gln54/ Gln143, it was possible to estimate their interaction energies in the native state. Destabilization of the N-td exhibited by the single mutants R79A (−1.0 kcal/mol) and M147A (−1.1 kcal/mol) summed to equal the destabilization of the N-td exhibited by the double mutant R79A/ M147A (−2.1 kcal/mol). This suggests that the interaction of Arg79 and Met147 does not contribute significantly to overall stability of the native state. In contrast, destabilization of the N-td by the single mutants Q54A (−0.2 kcal/mol) and Q143A (−0.5 kcal/mol) did not sum to equal the destabilization of the N-td by the double mutant Q54A/Q143A (−1.4 kcal/mol). Instead, these side chains displayed a free energy of interaction of ~0.7 kcal/mol. It is important to note that double-mutant cycle analyses are reliable measures of interaction energy only if the native state conformations of the mutant proteins are identical to the wild-type protein. While CD and fluorescence spectroscopy studies suggest that the conformations of peripheral domain interface mutants are similar to wild type, these techniques have relatively low sensitivity and would not likely detect altered domain pairing. Additionally, as seen in Table 1, the error associated with the ΔG° values is high in some 2038 Protein Science, vol. 14 cases (R79A and M147A), which limits the accuracy of calculated free energies of interaction. However, interaction energies for these residues also appear minimal when estimated by transition midpoints, which have much lower error. Together, these results suggest minimal to no interaction energy between these side chains.
Despite the minimal interaction energies of peripheral domain interface side chains, single and double alanine substitutions of these residues resulted in a significant decrease in the native to intermediate ΔG° ranging from 0.2 to 2.1 kcal/mol (Table 1). As described above, the hydrophobic domain interface residues are also critical for maintaining stability of the N-td by precise positioning and burial of hydrophobic surface area (Flaugh et al. 2005). While both the peripheral and hydrophobic domain interface residues act to stabilize the N-td, they likely do so by very different means. Given that the peripheral residues are in close proximity to the interface hydrophobic cluster, we suspect that these side chains function to shield the cluster from solvent. In this way, a decrease in the size and polarity of the side chains by mutation to alanine would cause an unfavorable exposure of the hydrophobic residues to solvent and thus destabilization of the N-td through a loss of favorable domain interface contacts.
Effect of interface substitutions on the kinetic refolding pathway
At 37°C and pH 7.0, wild-type HγD-Crys refolded to a native-like conformation at concentrations of GuHCl >1.0 M. Previous studies found that productive kinetic refolding of wild-type HγD-Crys was best fit to two exponentials with a t1/2 value of 15 sec for the first phase and 190 sec for the second phase (Kosinski-Collins et al. 2004). The structural transitions that corresponded to these two kinetic phases were elucidated by using engineered triple tryptophan mutant proteins (Kosinski-Collins et al. 2004).The triple tryptophan mutant proteins had three of the four native tryptophans of HγD-Crys mutated to phenylalanine, so that refolding of the two domains could be independently monitored by fluorescence spectroscopy. Refolding rates of the two mutants containing tryptophans from the C-td were comparable to the first exponential fit of wild type, with t1/2 values of 30 sec for both mutants. Similarly, refolding rates of the two mutant proteins with tryptophans in the N-td were comparable to the second exponential fit of wild type, with t1/2 values of 190 and 210 sec. Together these data suggest that the wild type protein refolded through a short lived kinetic intermediate, in which the C-td was largely folded and the N-td was largely unfolded (Kosinski-Collins et al. 2004).
Kinetic refolding of the single and double alanine mutants of peripheral interface residues were also best fit to two exponentials, suggesting a similar sequential domain refolding pathway as wild type (Kosinski-Collins et al. 2004). By this model, the C-td of the mutant proteins refolded first followed by the N-td. Under this assumption, refolding of the C-td was not notably affected by mutations of the peripheral interface residues as seen by similar t1/2 values as wild type. Conversely, all of the mutations resulted in a decreased refolding rate for the N-td. These results suggest that, for full-length wild-type HγD-Crys, refolding of the C-td does not depend on correct contacts between peripheral domain interface residues, while refolding of the N-td does. Previous experiments on single amino acid substitutions of hydrophobic domain interface residues also indicated reduced refolding rates for the N-td but not the C-td (Flaugh et al. 2005).
Slow kinetic transformations during protein refolding that occur on a second-to-minute timescale are often attributed to cis-trans proline isomerization (Brandts et al. 1975). Wild-type HγD-Crys has five proline residues, all of which are found in the trans conformation in the native state. Three of the prolines are in the N-td, one is in the peptide linking the two domains, and the final proline is in the C-td. The N-td of HγD-Crys refolds slower than the C-td, suggesting that the slow refolding of the N-td may be due to cis-trans isomerization of prolines in the connecting peptide and the N-td. The data presented here makes this unlikely. If the slow intermediate to native transition in HγD-Crys refolding was dependent on cis-trans proline isomerization, mutations in the domain interface would not be expected to decrease refolding rates to the extent that is observed here. Instead, we suggest that the slow kinetic step of HγD-Crys refolding corresponds to domain pairing accompanied by refolding of the N-td. These results suggest a refolding pathway where the solvent-exposed domain interface of the folded C-td likely acts as a nucleating center for refolding of the N-td (Flaugh et al. 2005).
For the mutant R79A/M147A, kinetic refolding of the N-td occurred with a t1/2 nine times greater than that of wild type. Interestingly, the single and double mutations of Gln54 and Gln143 did not have as significant an effect on the refolding rates as did mutations of Arg79 and Met147. The double mutant Q54A/Q143A had a t1/2 for the intermediate to native transition of 600 sec compared with 190 sec for wild type. Similarly, Q54A and Q143A both had t1/2 values for intermediate to native transition that were increased less than threefold over that for wild type. These results suggest that the side chains of Gln54 and Gln143 are not as critical in the kinetic refolding pathway as are those of Arg79 and Met147. The positioning of Gln54 and Gln143 may occur later during the kinetic pathway after the hydrophobic cluster and Arg79 and Met147 are brought together. By this model, mutations of Gln54 and Gln143 could still have a significant effect on stability without severely altering kinetic refolding behavior, as is observed here.
Domain stability and interactions
The two domains of the β- and γ-crystallins, composed of two Greek-key motifs each, are thought to result from a gene duplication event (Wistow et al. 1983). Folding and stability of full-length β- and γ-crystallins as well as proteins corresponding to their isolated domains have been extensively studied (for review, see Bloemendal et al. 2004). The individual domains of many of the β- and γ-crystallins exhibit distinctly different stabilities. As described above, the C-td of HγD-Crys is more stable than the N-td at pH 7.0 (Kosinski-Collins et al. 2004). In contrast to this, the N-td of BγB-Crys is more stable than the C-td at pH 2.0 (Rudolph et al. 1990; Mayr et al. 1997). This difference is presumably due to a greater number of acidic amino acids on the surface of the C-td of BγB-Crys, which at pH 2.0 destabilizes the domain by charge repulsion. The different domain stabilities exhibited by BγB-Crys and HγD-Crys may be due to discrepancies in experimental conditions and not inherent differences in the proteins.
For some of the β- and γ-crystallins that display differential domain stability, the domains are more stable in the full-length protein than in isolation (Sharma et al. 1990; Mayr et al. 1997; Wieligmann et al. 1999). This suggests that for these proteins, domain interface contacts play a significant role in the stability. Indeed, mutating domain interface residue of BγB-Crys, HβB1-Crys, or HγD-Crys destabilizes the proteins (Palme et al. 1997; Kim et al. 2002; Flaugh et al. 2005). Additional evidence indicates that domain interface interactions are important in determining oligomeric states of these proteins (Hope et al. 1994; Mayr et al. 1994; Trinkl et al. 1994).
Implications for understanding aggregation and cataract
Aged human lenses contain both water soluble and insoluble crystallin. The amount of protein in the water insoluble fraction increases with age and in cataract (Ringens et al. 1982). Analyses of the insoluble crystallin from aged or cataractous lenses have confirmed the presence of covalent damage, including methionine oxidation, and glutamine or asparagine deamidation (Hanson et al. 1998, 2000; Lampi et al. 1998; Ma et al. 1998).
Erroneous protein aggregation is associated with a variety of human diseases, in addition to mature-onset cataract (Sato et al. 1996; Harper et al. 1997; Prusiner 1998; Uversky et al. 2001). A common characteristic of known aggregation processes is polymerization from a partially folded or nonnative conformation (Mitraki and King 1989; Wetzel 1994; Booth et al. 1997; Jiang et al. 2001). In vivo, such partially folded species often represent incompletely folded polypeptide chains released from the ribosome (Mitraki and King 1989; Wetzel 1994). However, this phenomenon cannot explain the presence of aggregation-prone crystallin species in the lens, as crystallin aggregation likely occurs late in life, long after the proteins were initially synthesized. Instead, the aggregation-prone conformations are probably generated by destabilization of the native state and partial unfolding induced by covalent damage. The effects of deamidation on the structure, stability, and other in vitro properties of HβB1-Crys have been comprehensively studied by Lampi and colleagues (Lampi et al. 2001, 2002; Kim et al. 2002; Harms et al. 2004). These studies demonstrated that deamidation can cause altered structure, altered oligomer conformation, and reduced stability of HβB1-Crys, in vitro. These alterations may be significant in mature-onset cataractogenesis.
Some cases of rare juvenile-onset cataracts in humans are associated with single amino acid substitutions of the γ-crystallins. The mutations R14C, P23T, R36S, and R58H of HγD-Crys all result in childhood cataract (Héon et al. 1999; Stephan et al. 1999; Kmoch et al. 2000; Santhiya et al. 2002). Recombinant proteins with the R14C, R36S, and R58H mutations have similar native state conformations and stabilities as those of wild-type HγD-Crys, but do have reduced phase transition barriers in vitro (Pande et al. 2000, 2001; Basak et al. 2003). These mutations probably cause cataract by crystallization (R58H and R36S) and intermolecular disulfide bonding (R14C) of the native molecules in the lenses of affected individuals (Kmoch et al. 2000; Pande et al. 2000, 2001). These processes are unlikely to account for the formation of mature-onset cataracts from aged proteins that are of a wild-type sequence aside from covalent damage. The P23T recombinant mutant of HγD-Crys also had similar stability as that of the wild-type protein but had greatly reduced solubility in vitro, and was hypothesized to cause congenital cataract by precipitation in the lens (Evans et al. 2004). It is unclear how this relates to the protein insolubility found in mature-onset cataract.
The single amino acid substitution, T5P of human γC crystallin (HγC-Crys) is associated with Coppock-like cataract that causes a dust-like opacity in the lens nucleus of newborns (Héon et al. 1999; Santhiya et al. 2002). In contrast to the congenital mutants of HγD-Crys, Fu and Liang (2002) showed that the T5P mutation of HγCCrys alters the native state conformation and destabilizes the protein. Liang (2004) went on to further show that the T5P mutant also had an increased propensity to interact with the lens chaperone, human αA crystallin. The mechanism of inherited cataract for the T5P mutant of HγC-Crys may be due to aggregation caused by the altered structure and decreased stability or alternatively loss of native state interactions with other crystallins due to the altered conformation (Liang 2004). Either way, the mechanism of cataract for the mutant of HγC-Crys may more closely reflect that of mature-onset cataract than the R14C, R36S, and R58H mutants of HγD-Crys.
Our interest in identifying partially structured intermediates during HγD-Crys unfolding/refolding reflects the possibility that such species may be related to aggregation-prone precursors of cataract formation in the eye lens. Given that the peripheral residues Gln54, Gln143, and Met147 are potential sites of covalent damage, partially unfolded intermediates populated as a result of their modification are of particular interest for understanding aggregation and cataract. The results reported here indicate that these interface residues are critical for stability, and thus, covalent damage of the sidechains in aged lenses may result in native state destabilization of HγD-Crys. This would effectively increase the probability of populating the partially unfolded conformer with a folded C-td and unfolded N-td. One model for interactions between such partially unfolded conformations includes domain swapping, a mechanism of protein oligomerization that has been previously described by Liu and Eisenberg (2002). In younger adults these damaged, partially unfolded species would likely be scavenged by α-crystallins. However, at some point these chaperones may become saturated, resulting in the late onset of aggregation reactions from damaged crystallins.
Materials and methods
Mutagenesis, expression, and purification of recombinant HγD-Crys
Site-directed single alanine substitutions of residues Gln54, Arg79, Gln143, and Met147 were constructed by using sitedirected mutagenesis. Primers encoding the respective alanine substitutions (IDT-DNA) were used to amplify a pQE.1 plasmid encoding the HγD-Crys gene with an N-terminal 6-His tag (Kosinski-Collins et al. 2004). The double mutation Q54A/Q143A was constructed by using the primer encoding the Q143A substitution to amplify the Q54A mutant plasmid. The double mutant R79A/M147A was created in an analogous manner, using the M147A primer with the R79A plasmid. DNA sequencing of all constructs was performed to verify the substitutions and to ensure no additional mutations were present (Massachusetts General Hospital).
Recombinant wild-type and mutant HγD-Crys proteins were prepared as previously described (Kosinski-Collins et al. 2004). Briefly, proteins extracted from E. coli cell lysates were purified to >98% homogeneity by affinity chromatography with a Ni-NTA resin (Qiagen).
CD and fluorescence spectroscopy
CD spectra of recombinant mutant proteins were collected with an AVIV model 202 CD spectrometer by using a 0.25-cm pathlength cuvette. The temperature was maintained at 37°C with an internal Peltier thermoelectric temperature controller. The buffer conditions for all experiments were 10 mM sodium phosphate, 5 mM DTT, and 1 mM EDTA (pH 7.0). Protein was present at 100 μg/mL. Protein concentrations were determined by absorbance at 280 nm using an extinction coefficient of 41,040 cm−1 M−1 for wild-type and mutant His-tagged proteins. Spectra were collected from 200–260 nm to monitor secondary structure, and the buffer signal was subtracted from all spectra.
Fluorescence emission spectra were collected at 37°C by using a Hitachi F-4500 fluorimeter equipped with a circulating water bath to control temperature. Intrinsic tryptophan fluorescence was measured in the range of 310–420 nm by using an excitation wavelength of 295 nm. All samples contained 10 μg/mL purified protein in 10 mM sodium phosphate, 5 mM DTT, 1 mM EDTA (pH 7.0), and GuHCl where appropriate. All spectra were corrected for the buffer baseline.
Equilibrium unfolding/refolding
Equilibrium unfolding experiments were carried out by diluting purified mutant proteins to 10 μg/mL into increasing concentrations of GuHCl from 0 to 5.5 M (purchased as an 8.0 M solution from Sigma-Aldrich). Buffer conditions for all unfolding samples were 10 mM sodium phosphate, 5 mM DTT, and 1 mM EDTA (pH 7.0). The unfolding samples were incubated at 37°C for 24 h, by which time equilibrium had been reached.
Equilibrium refolding experiments were carried out by initially preparing an unfolded stock solution containing 100 μg/ mL purified protein in 5.5 M GuHCl. The unfolded stock solution was incubated at 37°C for 6 h and then diluted into refolding buffer to a final protein concentration of 10 μg/mL. Refolding buffer contained 10 mM sodium phosphate, 5 mM DTT, 1 mM EDTA (pH 7.0), and GuHCl from 0.55–5.5 M. The refolding samples were allowed to reach equilibrium by incubation at 37°C for 24 h.
Fluorescence emission spectra were recorded for all unfolding/ refolding samples by using an excitation wavelength of 295 nm and monitoring emission from 310–420 nm. GuHCl concentrations were determined by measuring the refractive indexes of all samples. Data was analyzed by plotting concentration of GuHCl versus fluorescence intensity at 360 nm divided by fluorescence intensity at 320 nm (FI 360/320 nm). The ratio of fluorescence intensities at these wavelengths was chosen for the analysis in order to simultaneously monitor changes in the native and unfolded maxima. Equilibrium unfolding/refolding experiments were performed a minimum of three times for each protein.
Equilibrium unfolding/refolding data were analyzed to determine transition midpoints, ΔG° and m values by fitting to a three-state model by using the method of Clark et al. (1993) with the curve fitting feature of Kaleidagraph (Synergy software). Averages and standard deviations were determined for the parameters of each protein from the fits of three separate experiments.
All transitions were fit to a three-state model described by Equations 2, 3, and 4:
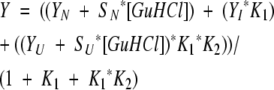 |
(2) |
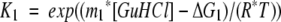 |
(3) |
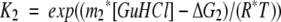 |
(4) |
where Y is the observed FI 360/320 nm signal, YN and YU are the intercepts of the native and unfolded baselines, SN and SU are the slopes of the native and unfolded baselines, and YI is the signal of the intermediate. Additionally, m1 and ΔG1 are the m value and ΔG° for the native to intermediate transition, and m2 and ΔG2 are the m value and ΔG° for the intermediate to unfolded transition. T is temperature in Kelvin, and R is the gas constant in units of kJ/mol/K.
Productive refolding kinetics
Kinetic refolding experiments were carried out by initially preparing unfolded stock solutions of the mutant proteins at 100 μg/mL in 5.5 M GuHCl. The unfolded stock solutions were incubated for 3 h at 37°C to ensure complete unfolding. Refolding buffer containing 10 mM sodium phosphate, 5 mM DTT, and 1 mM EDTA (pH 7.0) was equilibrated at 37°C. Unfolded protein was injected into refolding buffer by using a syringe port injection system to give a final protein concentration of 10 μg/mL. The refolding samples were continuously excited at 295 nm and fluorescence emission monitored at 350 nm for 3 h. The fluorescence spectra of refolded samples were subsequently measured to ensure that the proteins had refolded into a native-like conformation. Kinetic refolding data were fit to one, two, and three exponentials by using the curve fitting feature of Kaleidagraph (Synergy software), and the model with the best fit was determined by inspection. Productive kinetic refolding experiments were performed three times for each protein, from which averages and standard deviations were calculated for the fitted parameters.
Acknowledgments
We would like to thank Ishara Mills and Veronica Zepeda for helpful discussions and technical assistance. This work was supported by NIH grant GM17980, awarded to J.K. S.F. was supported by a Cleo and Paul Schimmel Fellowship.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051460505.
References
- Andley, U.P., Mathur, S., Griest, T.A., and Petrash, J.M. 1996. Cloning, expression, and chaperone-like activity of human αA-crystallin. J. Biol. Chem. 271 31973–31980. [DOI] [PubMed] [Google Scholar]
- Aquilina, J.A., Benesch, J.L., Ding, L.L., Yaron, O., Horwitz, J., and Robinson, C.V. 2005. Subunit exchange of polydisperse proteins: Mass spectrometry reveals consequences of α A-crystallin truncation. J. Biol. Chem. 100 10611–10616. [DOI] [PubMed] [Google Scholar]
- Basak, A., Bateman, O., Slingsby, C., Pande, A., Asherie, N., Ogun, O., Benedek, G.B., and Pande, J. 2003. High-resolution X-ray crystal structures of human γD crystallin (1.25 Å) and the R58H mutant (1.15 Å) associated with aculeiform cataract. J. Mol. Biol. 328 1137–1147. [DOI] [PubMed] [Google Scholar]
- Bateman, O.A., Sarra, R., van Genesen, S.T., Kappe, G., Lubsen, N.H., and Slingsby, C. 2003. The stability of human acidic β-crystallin oligomers and hetero-oligomers. Exp. Eye Res. 77 409–422. [DOI] [PubMed] [Google Scholar]
- Bax, B., Lapatto, R., Nalini, V., Driessen, H., Lindley, P.F., Mahadevan, D., Blundell, T.L., and Slingsby, C. 1990. X-ray analysis of β B2-crystallin and evolution of oligomeric lens proteins. Nature 347 776–780. [DOI] [PubMed] [Google Scholar]
- Bloemendal, H., de Jong, W., Jaenicke, R., Lubsen, N.H., Slingsby, C., and Tardieu, A. 2004. Ageing and vision: Structure, stability and function of lens crystallins. Prog. Biophys. Mol. Biol. 86 407–485. [DOI] [PubMed] [Google Scholar]
- Booth, D.R., Sunde, M., Bellotti, V., Robinson, C.V., Hutchinson, W.L., Fraser, P.E., Hawkins, P.N., Dobson, C.M., Radford, S.E., Blake, C.C., et al. 1997. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 385 787–793. [DOI] [PubMed] [Google Scholar]
- Boyle, D. and Takemoto, L. 1994. Characterization of the α-γ and α-β complex: Evidence for an in vivo functional role of α-crystallin as a molecular chaperone. Exp. Eye Res. 58 9–15. [DOI] [PubMed] [Google Scholar]
- Brandts, J.F., Halvorson, H.R., and Brennan, M. 1975. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 14 4953–4963. [DOI] [PubMed] [Google Scholar]
- Clark, A.C., Sinclair, J.F., and Baldwin, T.O. 1993. Folding of bacterial luciferase involves a non-native heterodimeric intermediate in equilibrium with the native enzyme and the unfolded subunits. J. Biol. Chem. 268 10773–10779. [PubMed] [Google Scholar]
- Delaye, M. and Tardieu, A. 1983. Short-range order of crystallin proteins accounts for eye lens transparency. Nature 302 415–417. [DOI] [PubMed] [Google Scholar]
- Evans, P., Wyatt, K., Wistow, G.J., Bateman, O.A., Wallace, B.A., and Slingsby, C. 2004. The P23T cataract mutation causes loss of solubility of folded γD-crystallin. J. Mol. Biol. 343 435–444. [DOI] [PubMed] [Google Scholar]
- Fernald, R.D. and Wright, S.E. 1983. Maintenance of optical quality during crystalline lens growth. Nature 301 618–620. [DOI] [PubMed] [Google Scholar]
- Flaugh, S.L., Kosinski-Collins, M.S., and King, J. 2005. Contributions of hydrophobic domain interface interactions to the folding and stability of human γD-crystallin. Protein Sci. 14 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L. and Liang, J.J. 2002. Conformational change and destabilization of cataract γC-crystallin T5P mutant. FEBS Lett. 513 213–216. [DOI] [PubMed] [Google Scholar]
- Haley, D.A., Horwitz, J., and Stewart, P.L. 1998. The small heat-shock protein, αB-crystallin, has a variable quaternary structure. J. Mol. Biol. 277 27–35. [DOI] [PubMed] [Google Scholar]
- Hanson, S.R., Smith, D.L., and Smith, J.B. 1998. Deamidation and disulfide bonding in human lens γ-crystallins. Exp. Eye Res. 67 301–312. [DOI] [PubMed] [Google Scholar]
- Hanson, S.R., Hasan, A., Smith, D.L., and Smith, J.B. 2000. The major in vivo modifications of the human water-insoluble lens crystallins are disulfide bonds, deamidation, methionine oxidation and backbone cleavage. Exp. Eye Res. 71 195–207. [DOI] [PubMed] [Google Scholar]
- Harms, M.J., Wilmarth, P.A., Kapfer, D.M., Steel, E.A., David, L.L., Bachinger, H.P., and Lampi, K.J. 2004. Laser light-scattering evidence for an altered association of β B1-crystallin deamidated in the connecting peptide. Protein Sci. 13 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, J.D., Lieber, C.M., and Lansbury Jr., P.T. 1997. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-β protein. Chem. Biol. 4 951–959. [DOI] [PubMed] [Google Scholar]
- Héon, E., Priston, M., Schorderet, D.F., Billingsley, G.D., Girard, P.O., Lubsen, N., and Munier, F.L. 1999. The γ-crystallins and human cataracts: A puzzle made clearer. Am. J. Hum. Genet. 65 1261–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenders, H.J. and Bloemendal, H. 1983. Lens proteins and aging. J. Gerontol. 38 278–286. [DOI] [PubMed] [Google Scholar]
- Hope, J.N., Chen, H.C., and Hejtmancik, J.F. 1994. Aggregation of β A3-crystallin is independent of the specific sequence of the domain connecting peptide. J. Biol. Chem. 269 21141–21145. [PubMed] [Google Scholar]
- Horovitz, A. 1996. Double-mutant cycles: A powerful tool for analyzing protein structure and function. Fold Des. 1 R121–R126. [DOI] [PubMed] [Google Scholar]
- Horwitz, J. 1992. α-Crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. 89 10449–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke, R. 1999. Stability and folding of domain proteins. Prog. Biophys. Mol. Biol. 71 155–241. [DOI] [PubMed] [Google Scholar]
- Jiang, X., Smith, C.S., Petrassi, H.M., Hammarstrom, P., White, J.T., Sacchettini, J.C., and Kelly, J.W. 2001. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry 40 11442–11452. [DOI] [PubMed] [Google Scholar]
- Kim, Y.H., Kapfer, D.M., Boekhorst, J., Lubsen, N.H., Bachinger, H.P., Shearer, T.R., David, L.L., Feix, J.B., and Lampi, K.J. 2002. Deamidation, but not truncation, decreases the urea stability of a lens structural protein, βB1-crystallin. Biochemistry 41 14076–14084. [DOI] [PubMed] [Google Scholar]
- Kmoch, S., Brynda, J., Asfaw, B., Bezouska, K., Novak, P., Rezacova, P., Ondrova, L., Filipec, M., Sedlacek, J., and Elleder, M. 2000. Link between a novel human γD-crystallin allele and a unique cataract phenotype explained by protein crystallography. Hum. Mol. Genet. 9 1779–1786. [DOI] [PubMed] [Google Scholar]
- Kosinski-Collins, M.S. and King, J. 2003. In vitro unfolding, refolding, and polymerization of human γD crystallin, a protein involved in cataract formation. Protein Sci. 12 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski-Collins, M.S., Flaugh, S.L., and King, J. 2004. Probing folding and fluorescence quenching in human γD crystallin Greek key domains using triple tryptophan mutant proteins. Protein Sci. 13 2223–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampi, K.J., Ma, Z., Hanson, S.R.A., Azuma, M., Shih, M., Shearer, T.R., Smith, D.L., Smith, J.B., and David, L.L. 1998. Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp. Eye Res. 67 31–43. [DOI] [PubMed] [Google Scholar]
- Lampi, K.J., Oxford, J.T., Bachinger, H.P., Shearer, T.R., David, L.L., and Kapfer, D.M. 2001. Deamidation of human βB1 alters the elongated structure of the dimer. Exp. Eye Res. 72 279–288. [DOI] [PubMed] [Google Scholar]
- Lampi, K.J., Kim, Y.H., Bachinger, H.P., Boswell, B.A., Lindner, R.A., Carver, J.A., Shearer, T.R., David, L.L., and Kapfer, D.M. 2002. Decreased heat stability and increased chaperone requirement of modified human βB1-crystallins. Mol. Vis. 8 359–366. [PubMed] [Google Scholar]
- Liang, J.J. 2004. Interactions and chaperone function of αA-crystallin with T5P γC-crystallin mutant. Protein Sci. 13 2476–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. and Eisenberg, D. 2002. 3D domain swapping: As domains continue to swap. Protein Sci. 11 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubsen, N.H., Aarts, H.J., and Schoenmakers, J.G. 1988. The evolution of lenticular proteins: The β- and γ-crystallin super gene family. Prog. Biophys. Mol. Biol. 51 47–76. [DOI] [PubMed] [Google Scholar]
- Ma, Z., Hanson, S.R., Lampi, K.J., David, L.L., Smith, D.L., and Smith, J.B. 1998. Age-related changes in human lens crystallins identified by HPLC and mass spectrometry. Exp. Eye Res. 67 21–30. [DOI] [PubMed] [Google Scholar]
- Mayr, E.M., Jaenicke, R., and Glockshuber, R. 1994. Domain interactions and connecting peptides in lens crystallins. J. Mol. Biol. 235 84–88. [DOI] [PubMed] [Google Scholar]
- ———. 1997. The domains in γB-crystallin: Identical fold-different stabilities. J. Mol. Biol. 269 260–269. [DOI] [PubMed] [Google Scholar]
- Mitraki, A. and King, J. 1989. Protein folding intermediates and inclusion body formation. Bio/technology 7 690–697. [Google Scholar]
- Norledge, B.V., Mayr, E.M., Glockshuber, R., Bateman, O.A., Slingsby, C., Jaenicke, R., and Driessen, H.P. 1996. The X-ray structures of two mutant crystallin domains shed light on the evolution of multi-domain proteins. Nat. Struct. Biol. 3 267–274. [DOI] [PubMed] [Google Scholar]
- Oyster, C.W. 1999. The lens and the vitreous. In The human eye: Structure and function, chap. 12. Sinauer Associates, Inc., Sunderland, MA.
- Palme, S., Slingsby, C., and Jaenicke, R. 1997. Mutational analysis of hydrophobic domain interactions in γ B-crystallin from bovine eye lens. Protein Sci. 6 1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande, A., Pande, J., Asherie, N., Lomakin, A., Ogun, O., King, J.A., Lubsen, N.H., Walton, D., and Benedek, G.B. 2000. Molecular basis of a progressive juvenile-onset hereditary cataract. Proc. Natl. Acad. Sci. 97 1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande, A., Pande, J., Asherie, N., Lomakin, A., Ogun, O., King, J., and Benedek, G.B. 2001. Crystal cataracts: Human genetic cataract caused by protein crystallization. Proc. Natl. Acad. Sci. 98 6116–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S.B. 1998. Prions. Proc. Natl. Acad. Sci. 95 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringens, P.J., Hoenders, H.J., and Bloemendal, H. 1982. Effect of aging on the water-soluble and water-insoluble protein pattern in normal human lens. Exp. Eye Res. 34 201–207. [DOI] [PubMed] [Google Scholar]
- Rudolph, R., Siebendritt, R., Nesslauer, G., Sharma, A.K., and Jaenicke, R. 1990. Folding of an all-β protein: Independent domain folding in γ II-crystallin from calf eye lens. Proc. Natl. Acad. Sci. 87 4625–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhiya, S.T., Shyam Manohar, M., Rawlley, D., Vijayalakshmi, P., Namperumalsamy, P., Gopinath, P.M., Loster, J., and Graw, J. 2002. Novel mutations in the γ-crystallin genes cause autosomal dominant congenital cataracts. J. Med. Genet. 39 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, S., Ward, C.L., Krouse, M.E., Wine, J.J., and Kopito, R.R. 1996. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J. Biol. Chem. 271 635–638. [DOI] [PubMed] [Google Scholar]
- Slingsby, C. and Bateman, O.A. 1990. Quaternary interactions in eye lens β-crystallins: Basic and acidic subunits of β-crystallins favor heterologous association. Biochemistry 29 6592–6599. [DOI] [PubMed] [Google Scholar]
- Slingsby, C., Norledge, B., Simpson, A., Bateman, O.A., Wright, G., Driessen, H.P.C., Lindley, P.F., Moss, D.S., and Bax, B. 1997. X-ray diffraction and structure of crystallins. Prog. Retin. Eye Res. 16 3–29. [Google Scholar]
- Stephan, D.A., Gillanders, E., Vanderveen, D., Freas-Lutz, D., Wistow, G., Baxevanis, A.D., Robbins, C.M., VanAuken, A., Quesenberry, M.I., Bailey-Wilson, J., et al. 1999. Progressive juvenile-onset punctate cataracts caused by mutation of the γD-crystallin gene. Proc. Natl. Acad. Sci. 96 1008–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkl, S., Glockshuber, R., and Jaenicke, R. 1994. Dimerization of β B2-crystallin: The role of the linker peptide and the N- and C-terminal extensions. Protein Sci. 3 1392–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V.N., Li, J., and Fink, A.L. 2001. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 276 10737–10744. [DOI] [PubMed] [Google Scholar]
- Wetzel, R. 1994. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 12 193–198. [DOI] [PubMed] [Google Scholar]
- Wieligmann, K., Mayr, E.M., and Jaenicke, R. 1999. Folding and selfassembly of the domains of βB2-crystallin from rat eye lens. J. Mol. Biol. 286 989–994. [DOI] [PubMed] [Google Scholar]
- Wistow, G.J. and Piatigorsky, J. 1988. Lens crystallins: The evolution and expression of proteins for a highly specialized tissue. Annu. Rev. Biochem. 57 479–504. [DOI] [PubMed] [Google Scholar]
- Wistow, G., Turnell, B., Summers, L., Slingsby, C., Moss, D., Miller, L., Lindley, P., and Blundell, T. 1983. X-ray analysis of the eye lens protein γ-II crystallin at 1.9 Å resolution. J. Mol. Biol. 170 175–202. [DOI] [PubMed] [Google Scholar]