

Abstract
Telomerase is a ribonucleoprotein complex responsible for extending the ends of eukaryotic chromosomes. Structural and biophysical studies of this enzyme have been limited by the inability to produce large amounts of recombinant protein. Here we perform a high-throughput screen to map regions of the Tetrahymena thermophila TERT (Telomerase Reverse Transcriptase) protein that are overexpressed in a soluble form in Escherichia coli using a GFP-fusion system. Many of the soluble protein domains identified do not coincide with domains inferred from multiple sequence alignment, so screening for fluorescent colonies provided information not otherwise readily obtained. The method revealed an essential, independently folded N-terminal domain that was expressed and purified with high yield and found to be suitable for structural analysis. These results provide a tool for future structural and biophysical studies of TERT.
Keywords: telomerase, GFP, high throughput, TERT
Telomerase is a ribonucleoprotein enzyme responsible for the addition of telomeric DNA repeats to the ends of linear chromosomes. Minimally, active telomerase is composed of a protein subunit as well as a large RNA subunit that includes a template sequence for DNA synthesis (Greider and Blackburn 1989). The enzymatic protein subunit, TERT, shares regions of sequence similarity with the reverse transcriptase family of proteins in its C-terminal half (Lingner et al. 1997). Indeed, mutation of conserved aspartic acid residues postulated by sequence alignment to be in the active site of DNA synthesis renders the enzyme inactive. The N-terminal half of TERT contains at least four regions of sequences that are conserved among all TERT homologs identified to date (Friedman and Cech 1999; Xia et al. 2000). Interestingly, these regions do not show sequence similarity to any other known protein sequences, although they are essential for telomerase activity in vitro and in vivo.
The functions of these N-terminal regions have recently begun to be explored. The results of several studies suggest that the QFP, CP, and T motifs (Fig. 1 ▶) of TERT homologs from Tetrahymena thermophila, Saccharomyces cerevisiae, and humans are necessary for specific interaction with the telomerase RNA subunit (Friedman and Cech 1999; Bryan et al. 2000b; Armbruster et al. 2001; O’Connor et al. 2005). Several functions have been proposed for the conserved region present near the N terminus of TERT, called the GQ motif (Xia et al. 2000), including recruitment of a telomerase regulatory protein Est3p and nonspecific nucleic acid binding in budding yeast (Xia et al. 2000; Friedman et al. 2003), interactions with telomeric DNA and telomerase RNA in humans (Lee et al. 2003; Moriarty et al. 2004), and interactions with telomerase RNA in T. thermophila (O’Connor et al. 2005). Despite these recent advances by functional studies, knowledge of the boundaries of the true structured domains of the TERT protein remains elusive.
Figure 1.
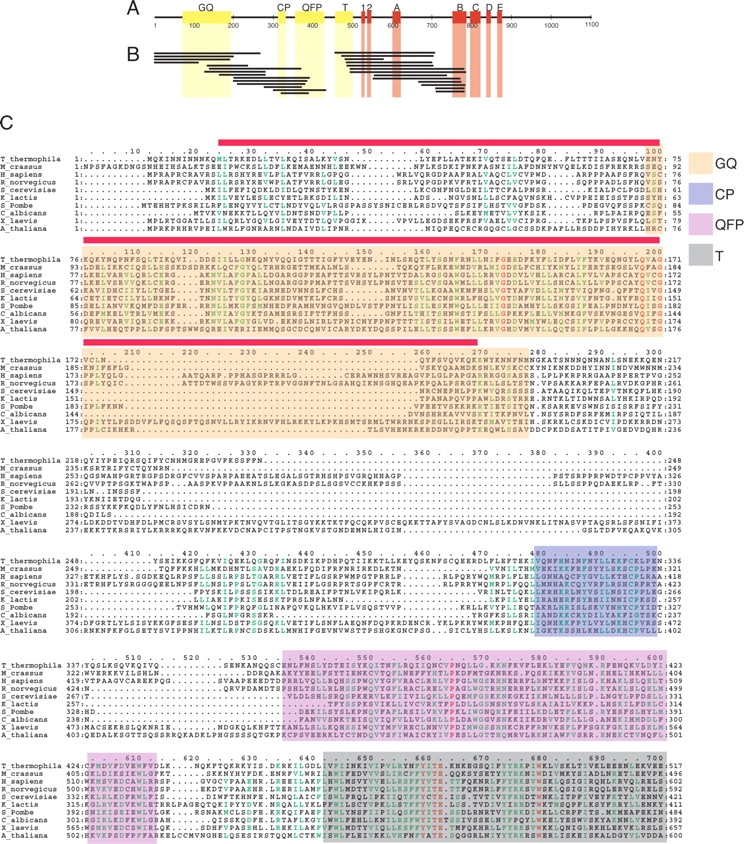
Results ofGFP-fusion high-throughput screening. (A) Conserved sequences in the T. thermophila TERT protein. Red blocks represent regions with similarity to the catalytic reverse transcriptase motifs (Lingner et al. 1997) while yellow blocks represent conservedN-terminal sequences only found in TERT homologs (Xia et al. 2000). Numbers represent amino acids starting at the N terminus. (B) Regions of T. thermophila TERT that can be expressed as a soluble GFP-fusion protein in E. coli as determined from random screening method. Exact sequences can be found in Table 1. (C)Multiple sequence alignment of the N-terminal half of TERT proteins containing the GQ, CP, QFP, and T motifs generated by ClustalW (Chenna et al. 2003). Similar residues are colored in green and identical residues in red. Regions of sequence similarity (represented by yellow blocks in Fig. 1A) as determined by Hidden Markov Model analysis (Xia et al. 2000) are shaded as indicated. The sequence of TERT-GQ2 is indicated by a red line above the alignment.
This lack of structural information persists despite the fact that the Euplotes aediculatus and S. cerevisiae TERT protein sequences were discovered years ago (Lingner et al. 1997) and homologs have now been identified in over 40 organisms. Furthermore, since the regions of TERT outside of the conserved reverse transcriptase motifs show no sequence similarity to other protein sequences, the structure of most of TERT remains unknown. One significant challenge to understanding TERT structure has been the inability to overexpress and purify recombinant forms of this enzyme with a high yield. Full-length TERT proteins have been successfully expressed and isolated from insect cells (Masutomi et al. 2000; Mikuni et al. 2002), budding yeast (Bachand and Autexier 1999), HeLa cells (Schnapp et al. 1998), and in vitro transcription/translation systems (Weinrich et al. 1997). However, these techniques have delivered low yields due to either low expression or poor solubility of the overexpressed protein, making structural studies difficult. Here we use high-throughput methods to map regions of T. thermophila TERT that can be overexpressed in Escherichia coli in a soluble form and purified to homogeneity. Further, we demonstrate that a region of TERT near the very N terminus that is essential for catalytic activity constitutes an independently folded protein domain of 20.5 kDa as demonstrated by limited proteolytic digestion and NMR spectroscopy.
Results
It seemed reasonable that TERT proteins might be made up of one or more independently folded domains, because mutations in conserved sequence blocks disrupt discrete functions (Kelleher et al. 2002) and by analogy to other DNA polymerases that employ accessory domains that impart activities specific to each individual enzyme (Sousa 1996). Expression and purification of these domains in a recombinant system would provide a key tool for studying their structure and function. Our initial attempts to identify fragments of the TERT protein that could be expressed in a soluble, stable form in E. coli and purified with a yield high enough for structural studies failed to produce any useful constructs. These constructs were based on multiple sequence alignments of various TERT homologs (Fig. 1C ▶) (Friedman and Cech 1999; Xia et al. 2000). However, the large size of the TERT proteins (~120 kDa) and the weak sequence conservation among family members (generally only around 20% pair wise sequence identity) made it difficult to predict which regions of the protein constitute well-folded domains.
In order to circumvent these problems, we employed high-throughput screening of random fragments of the T. thermophila TERT protein using a GFP-fusion solubility reporter (Waldo et al. 1999; Kawasaki and Inagaki 2001). Random fragments of the TERT gene of size 600–3400 base pairs were generated by random-tagged PCR (Grothues et al. 1993) and inserted into the pProGFP expression vector upstream of the GFP gene (Kawasaki and Inagaki 2001). E. coli colonies overexpressing soluble TERT-GFP fusion proteins are expected to display strong GFP fluorescence when a standard agar plate is illuminated with a 366 nm UV lamp. Although only 1 in 18 colonies are expected to express the GFP-fusion protein in frame, this method allows for the examination of thousands of constructs in only a few days. An additional advantage of this high-throughput system is that soluble expression of TERT-GFP fusion proteins is monitored in vivo, thus avoiding the laborious steps needed to separate and analyze insoluble and soluble fractions of each construct, as is required of other high-throughput systems (Knaust and Nordlund 2001).
Figure 2A ▶ shows an example of E. coli colonies expressing the TERT-GFP fusion library after irradiation with UV light. From this library, fluorescent colonies were selected for expression tests as shown in Figure 2, B and C ▶. Colonies 1–10 represent a subset of the brightly fluorescent colonies obtained, while colony 11 represents a nonfluorescent control colony. Approximately 10,000 colonies were screened, and ~1.0% showed a strong fluorescence signal. Colonies displaying a strong fluorescent signal were selected for expression tests in 24-well tissue culture plates in 0.5-mL cultures. Addition of IPTG to the cultures resulted in the clear overexpression of fusion proteins for most fluorescent colonies as visualized by SDS-PAGE analysis (Fig. 2C ▶). We found that approximately 1 in 15 fluorescent colonies did not result in the clear overexpression of a fusion protein, perhaps due to post-translational degradation of the fusion protein or the exposure of an internal cryptic ribosome-binding site within the TERT sequence.
Figure 2.

Expression of fluorescent colonies. (A) Example library plate irradiated with 366 nm UV light. Approximately 1% of colonies show strong fluorescence. (B) Examples of colonies selected for expression tests. Colonies 1–10 are brightly fluorescent while colony 11 represents a control colony that is not fluorescent. Fluorescence was visualized after exposing the plate to 366 nm UV light. (C) SDS-PAGE analysis of GFP-fusion protein expression of selected colonies. Asterisks are located to the left of the band representing overexpressed GFP-fusion proteins in each lane. Numbers above each lane correspond to the colony shown in B. Numbers to the left indicate protein molecular weight markers in kDa.
Plasmid DNA was isolated and sequenced from 27 colonies overexpressing fusion proteins larger than 35 kDa in size. Figure 1B ▶ and Table 1 summarize the results of this screen. As expected, several of the sequences obtained in the screen were found to have mutations near the N or C terminus of the construct, due to the random nature of the PCR primers used (Table 1) (Kawasaki and Inagaki 2001). Numerous constructs were found spanning the fingers and part of the palm subdomains of the reverse transcriptase motifs (motifs 1, 2, A, and B) and the region of TERT containing the N-terminal GQ, CP, and QFP motifs. Interestingly, no continuous construct spanning all three of these N-terminal regions that was highly overexpressed was recovered, suggesting that this region may be folded into multiple domains. Although the number of colonies screened was theoretically not sufficient to include every possible fragment of TERT, this method did allow us to examine the expression and solubility of many constructs in only a few days. However, we cannot rule out that sequencing more fluorescent colonies would identify other soluble regions of TERT or that other regions of T. thermophila TERT might require different expression conditions (expression hosts, fusion tags, etc.) for production of soluble protein.
Table 1.
Regions of T. thermophila TERT identified as expressed and soluble by high-throughput screening
| Clone | Residues | Mutations |
| 1 | 1–268 | |
| 2 | 1–201 | |
| 3 | 1–192 | |
| 4 | 1–112 | |
| 5 | 133–237 | |
| 6 | 137–371 | E370Q |
| 7 | 128–370 | |
| 8 | 162–283 | V169A |
| 9 | 200–394 | N202S |
| 10 | 199–388 | |
| 11 | 208–382 | |
| 12 | 277–372 | P278L, D279E, H280R, Y367H, D368A, I371P |
| 13 | 321–390 | |
| 14 | 301–435 | |
| 15 | 464–709 | |
| 16 | 470–703 | Q702K |
| 17 | 482–578 | |
| 18 | 484–701 | |
| 19 | 484–704 | Q702P |
| 20 | 493–781 | |
| 21 | 493–783 | C782Y |
| 22 | 551–773 | S753G |
| 23 | 550–737 | K736N |
| 24 | 664–772 | N666D, S753G |
| 25 | 673–782 | |
| 26 | 711–782 | |
| 27 | 709–781 |
While performing the expression tests, we noticed that all of the sequences obtained that spanned the GQ motif (Table 1, colonies 1, 2, 3, 4, and 5) were overexpressed to an exceptionally high level. We therefore focused on this region of TERT in order to validate that the high-throughput assay was indeed identifying regions of the protein that could be expressed in a soluble form and purified with a high yield. TERT 2–191 (hereafter referred to as TERT-GQ) was subcloned into pET11a with an N-terminal histidine tag and purified to homogeneity by Ni+2 affinity, ion exchange, and gel filtration chromatography. MALDI mass spectrometry confirmed that the proper construct was expressed and purified. Yields of 30 mg of pure protein per liter of E. coli culture were reproducibly obtained when expression was performed overnight at 18°C after induction with low levels of IPTG (see Materials and Methods), thus validating the high-throughput assay as a means of discovering soluble regions of TERT produced in E. coli.
It is conceivable that the random fragment screen might produce protein constructs that are soluble but aremade up of multiple structured domains or that are not folded. To address these concerns, we subjected TERT-GQ to limited proteolysis by Lys-C and analyzed the products by SDS-PAGE (Fig. 3 ▶). Lys-C digestion produced two fragments of approximately 20 kDa with only the smaller being stable at high Lys-C concentrations (Fig. 3 ▶, cf. lanes 2 and 3). MALDI mass spectrometry analysis showed that there were actually four stable species present after digestion with Lys-C having masses of 21,378, 21,256, 20,461, and 20,332 kDa. The masses correspond to protein fragments consisting of amino acids 4–185, 4–184, 13–185, and 13– 184, respectively. Notably, K18 and K182 are protected from proteolysis by Lys-C under our experimental conditions, suggesting that the boundaries for a folded region of this polypeptide lie between residues 13 and 18 at the N terminus and either residue 183 or 184 at the C terminus.
Figure 3.
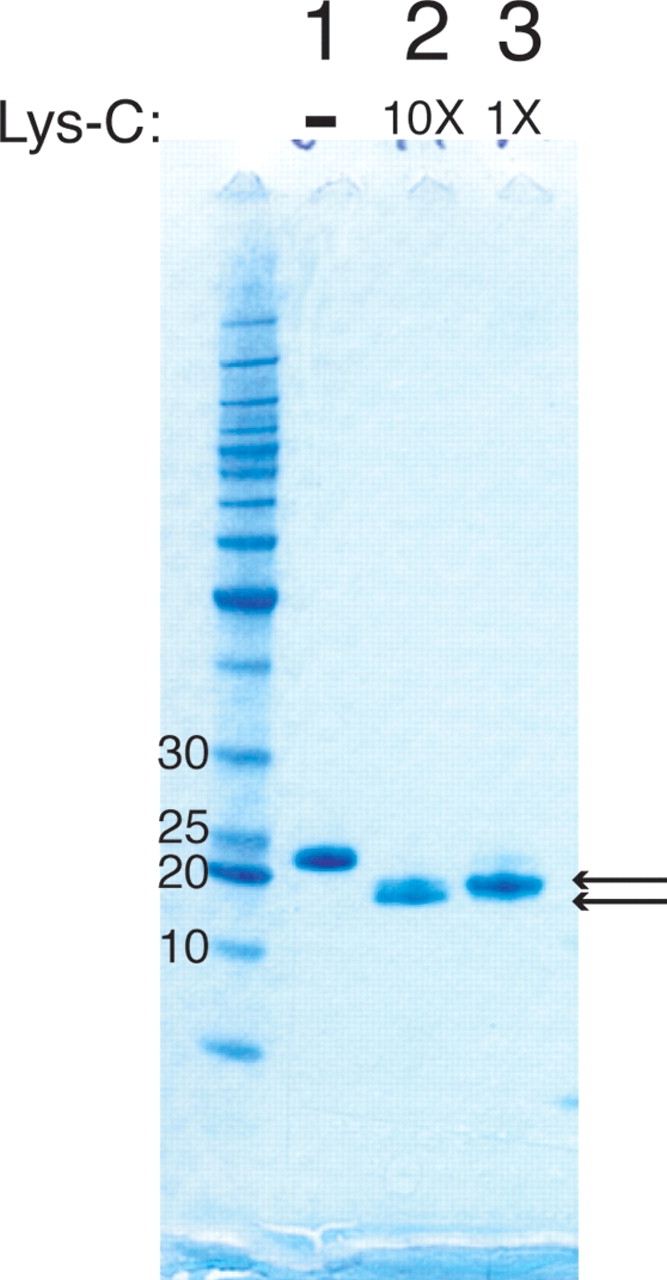
Limited proteolysis of recombinant TERT-GQ. SDS-PAGE analysis of digestion products are shown for undigested recombinant TERT-GQ (lane 1) and after digestion with two concentrations of Lys- C (lanes 2,3). The product shown in lane 2 was digested with 10-fold more Lys-C than the product shown in lane 3. Arrows point to stable digestion products.
To test whether these recombinant proteins were properly folded, we expressed and purified a new construct corresponding to residues 13–184 (hereafter referred to as TERT-GQ2) and subjected it to two-dimensional NMR analysis (Fig. 4 ▶). The 1H-15N 2D HSQC spectrum of 15N-labeled TERT-GQ2 showed good resonance dispersion, suggesting this recombinant construct was indeed a folded domain. Heteronuclear NMR analysis determined that only 5 of 171 residues of TERT-GQ2 were grossly disordered (data not shown). Based on this, we conclude that there is a stable, independently folded domain in the N terminus of T. thermophila TERT protein corresponding to residues 13–184.
Figure 4.

1H-15N HSQC spectrum of 800 μM TERT-GQ2 in 20 mM MES (pH 6.5), 25 mM NaCl, 1 mM DTT, 5% D2O at 30°C.
A mutant version of TERT lacking the TERT-GQ (residues 192–1117, TERT ΔGQ) was tested for catalytic activity in order to determine if TERT-GQ is an essential protein domain. Full-length TERT or TERT ΔGQ was expressed in vitro in rabbit reticulocyte lysates in the presence of in vitro transcribed telomerase RNA and assayed for the ability to extend the telomeric oligonucleotide (TTGGGG)3. As seen in Figure 5 ▶, only the full-length protein shows the ladder of DNA repeats characteristic of telomerase activity, while no activity is detectable for TERT ΔGQ. Thus, we conclude that the TERT-GQ domain is essential for telomerase activity in vitro.
Figure 5.
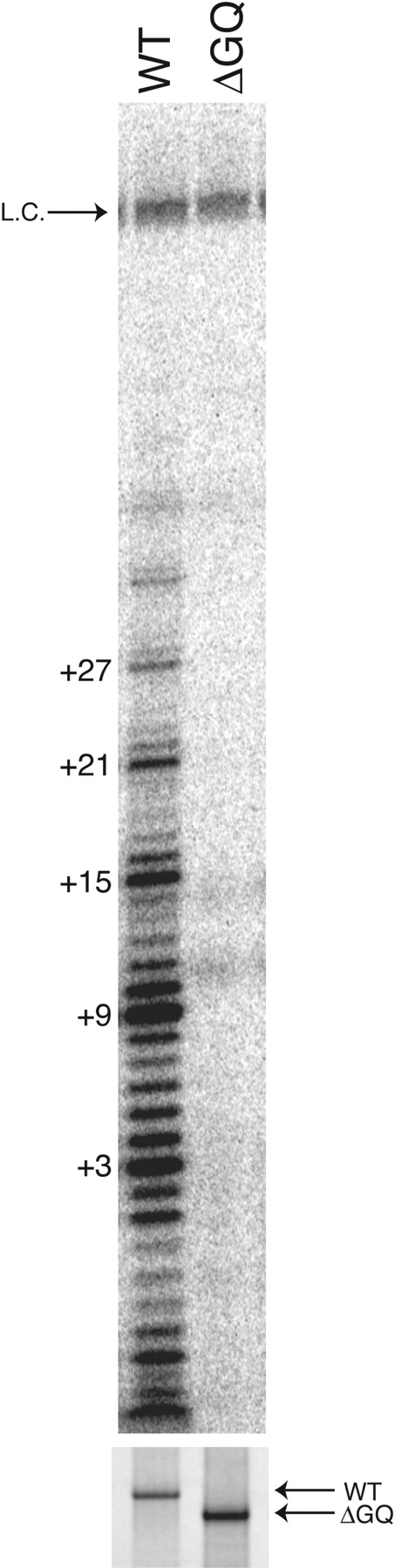
TERT-GQ is required for telomerase activity. Telomerase activity assay for full-length TERT (WT) and TERT-ΔGQ (ΔGQ). Numbers to the left indicate the number of nucleotides added to the primer sequence by telomerase. “L.C.” corresponds to the position of the 100 mer DNA oligo loading control. The bottom panel shows SDS-PAGE analysis of WT and TERT-GQ proteins pulled down from the in vitro transcription/translation reaction.
Discussion
The results presented here suggest that there is an independently folded protein domain near the very N terminus of T. thermophila TERT protein. Our multiple sequence alignment analysis (Fig. 1C ▶) is in agreement with a previous study that suggests the boundaries of the GQ motif defined by sequence similarity fall between residues 73 and 193 of T. thermophila TERT (Xia et al. 2000). However, the limited proteolytic digestion and NMR spectroscopy data described here suggest that the actual folded protein domain extends far N-terminal to the conserved GQ motif, starting at approximately residue 13, in spite of the fact that this N-terminal extension has only a few weakly conserved residues. Interestingly, the starting and ending points of most of the sequences returned from the high-throughput screen do not correspond very closely with the locations of conserved sequences based on multiple sequence alignments (Fig. 1B, C ▶). This may in part be due to the low levels of sequence similarity found between TERT homologs. Thus, the results presented here provide information regarding the domain structure of TERT that was previously unavailable from conventional methods.
Our demonstration that the GQ motif is part of a protein domain not dependent upon further C-terminal sequences for folding is consistent with previous studies on the human homolog. In these experiments, it was demonstrated that production of TERT fragments containing the GQ motif in trans with a GQ deletion mutant can rescue TERT activity when TERT is assembled in the rabbit reticulate lysate in vitro transcription/translation system (Beattie et al. 2001; Moriarty et al. 2004).
In this report, we have provided a map of regions of the T. thermophila TERT protein that can be highly overexpressed and purified to homogeneity from E. coli using the GFP-fusion solubility reporter as a high-throughput tool. This powerful technique, which combines the generation of random protein fragments with the GFP-fusion solubility reporter (Waldo et al. 1999), has to our knowledge been applied to only one other protein sequence, the mouse Vav protein (Kawasaki and Inagaki 2001). The random nature of the protein constructs generated during the screening is particularly suited for a protein like TERT that has only limited sequence similarity among family members, because it generates protein constructs for expression that are unbiased by weak multiple sequence alignments or limited biochemical data. The results of this screen will provide a tool for future structural and biochemical analysis of the TERT protein.
Materials and methods
GFP solubility screen
The template sequence for random PCR amplification was made by amplifying the T. thermophila TERT gene (Bryan et al. 2000a) using the primers ggaattcatggggaaaaaacaccaccaccaccaccacatgcagaaaattaac and atagttagcggccgcttagttggtattctgctg and gel purifying the product on a 1% agarose gel. Random tagged PCR was performed using two PCR steps with primer A (gaccatggattacgccaagcttNNNNNNNNNNNNNNN) used in the first reaction and primer B (gaccatggattacgccaagctt) for the second reaction (where N represents a random mixture of the four nucleotides) (Kawasaki and Inagaki 2001). Each round of PCR was performed in triplicate and pooled at the end. The first PCR reaction was completed in 25 μL total volume with 100 ng of the template, 100 pmol primer A, 0.2 U TaqHifi (Invitrogen), and 0.2 mM each dNTP. This reaction mixture was heated to 95°C for 2 min, followed by 10 cycles of 94°C for 1 min, 40°C for 3 min, and 68°C for 3 min, followed by an extension at 68°C for 10 min. PCR products were purified by Qiaquick PCR purification kit (Qiagen) following the manufacturer’s instructions. The second round of PCR was performed in 50 μL total volume with the first PCR products, 0.2 mM each dNTP, 500 pmol primer B, and 0.4 U TaqHifi. The reaction mixture was heated to 95°C for 2 min, followed by 30 cycles of 94°C for 1 min, 55°C for 1 min, 68°C for 1.5 min, and an elongation step of 68°C for 10 min. PCR products were pooled and purified by the Qiaquick PCR purification kit. Digestions were performed by adding 30 U HindIII to the PCR products and incubating them at 37°C for 16 h, followed by purification by 1.0% agarose gel. Fragments from 600–3400 nt were cut out of the gel and eluted using QIAEXII gel extraction kit (Qiagen). Fragments were ligated into HindIII-digested pProGFP (Kawasaki and Inagaki 2001) and transformed into E. coli XL1-Blue (Stratagene) by electroporation and grown for 37°C for 16 h. Fluorescent colonies were detected by irradiation with a 366 nm UV light and selected for expression test in 24-well linbro plates. For expression tests, 5 μL of a saturated overnight growth of each fluorescent colony was transferred to a fresh 0.5 mL LB culture and incubated with shaking at 37°C for 3 h before induction with 1 mM IPTG and reduction of the temperature to 30°C. A 100-μL portion of each culture was harvested by centrifugation and resuspended in 25 μL of 4× NuPage LDS sample buffer (Invitrogen), heated to 95°C for 5 min, and loaded onto a 4%–12% SDS-PAGE gel (Invitrogen). Plasmids from colonies that showed strong overexpression of fusion proteins greater than 35 kDa were purified and sent for DNA sequencing.
Protein expression and purification
TERT-GQ (residues 2–191) was subcloned into the NdeI and BamHI sites of pET11a using the primer pair ggaattccatatgaaaaaacaccatcaccatcaccatcagaaaattaacaacatt and cgggatccttagttgtttttataccattt. The first primer builds the sequence MKK HHHHHH into the N terminus of the protein sequence to aid in purification. TERT-GQ2 (residues 13–184) was subcloned into the NdeI and BamHI sites of pET28a using the oligos ggaattccatatgctgacccgcaaa and cgggatccttatttctgtttcacctggac. All protein constructs were expressed in E. coli BL21(DE3) (Stratagene) by growing the cells at 37°C to an OD600 of 0.6, after which IPTG was added to 0.1 mM and the temperature dropped to 18°C. Cells were harvested by centrifugation 18 h after induction. Cell pellets were resuspended in buffer A (50 mM sodium phosphate [pH 8.0], 500 mM NaCl, 10 mM imidazole, 10% glycerol, 1 mM PMSF, 5 mM β-mercaptoethanol) with 0.1 mg/mL lysozyme and lysed by brief sonication. Cleared lysates were passed over a 5 mL Ni-NTA resin and washed with 200 mL of Buffer A. Bound proteins were eluted in 50mM sodium phosphate (pH 8.0), 250 mMNaCl, 250 mM imidazole, and 10% glycerol (Buffer B).
TERT-GQ was further purified by a Sephadex-SP cation exchange column (Amersham Biosciences) in a buffer of 20 mM MES (pH 6.0) with a gradient of NaCl from 0 to 1.0 M. TERT-GQ typically eluted at a NaCl concentration of 750 mM. Fractions from the SP column were pooled and concentrated in a spin concentrator (Millipore) and further purified on a Superdex 75 gel filtration column (Amersham Biosciences) in 20 mM MES (pH 6.0), 250 mM NaCl.
TERT-GQ2 was purified by Ni-NTA affinity chromatography as described above for TERT-GQ. Fractions from the Ni- NTA column were treated with 100 U of thrombin (Amersham Biosciences) in order to cleave the histidine tag and dialyzed against 1 L of a solution of 50 mM sodium phosphate (pH 8.0), 150 mM NaCl, 2 mM DTT at 4°C for 18 h. Uncut product and thrombin were removed by Ni-NTA (Qiagen) and Benzamadine sepharose (Amersham Biosciences) chromatography. Final purification was performed on a Superdex 75 gel filtration column in 50 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM DTT.
15N-labeled TERT-GQ2 was prepared as above except that the protein was expressed in minimal media supplemented with (15NH4)2SO4 (1.5 g/L) (Aldrich).
Limited proteolysis
TERT-GQ was concentrated to 10 mg/mL and dialyzed extensively against water to remove all buffers and salts to aid in downstream mass spectrometry analysis. For digestion, 20 or 2 ng of Lys-C enzyme (Promega) was added to 10 μL of the protein solution and incubated at 37°C for 3 h. Following digestion, products were analyzed by 4%–12% SDS-PAGE (Invitrogen) or MALDI mass spectometry using a Voyager- DE STR mass spectrometer (PerSeptive Biosystems).
NMR spectroscopy
NMR data were collected on a Varian INOVA 500MHz spectrometer equipped with a Nalorac HCN triple resonance Z-gradient probe on 15N-labeled 800 μM TERT-GQ2 samples in 20 mM MES (pH 6.5), 25 mMNaCl, 1 mMDTT, 5%D2O at 30°C. Varian BioPack pulse sequences were used with minor modifications. Spectra were processed using NMRPipe (Delaglio et al. 1995) and analyzed with NMR View (Johnson and Blevins 1994).
Telomerase activity assay
Gene sequences for TERT (Bryan et al. 2000a) and TERTΔGQ (residues 192–1117) were subcloned into pET28a and expressed in the TNT rabbit reticulocyte lysate system (Promega) in 250 μL volume with 2.5 μg of plasmid DNA, 200 μL TNT Quick Master mix, 10 μL PCR enhancer, 5 pmol in vitro transcribed telomerase RNA (Bryan et al. 2000a), 10 μM 35S-methionine (10 mCi/mL), and 20 μM cold methionine, and incubated at 30°C for 3 h. The telomerase complex was pulled down by incubating the reaction with 100 μL of T7-tag antibody agarose beads (Novagen) for 1 h at 4°C and washing three times with wash buffer included in the T7-tag antibody agarose bead kit and then three times in telomerase activity buffer composed of 50 mM Tris-HCl (pH 8.3), 1.25 mM MgCl2, 5 mM DTT, and 30% glycerol before being stored at −20°C; 2 μL of each pull-down reaction were examined for protein content by running on a 4%–12% Bis-Tris (Novagen) SDS-PAGE gel.
Activity assays were performed by incubating 10 μL of T7 pull-down beads in 20 μL of 1 μM DNA oligo (TTGGGG)3, 50 mM Tris-HCl (pH 8.3), 1.25 mM MgCl2, 5 mM DTT, 30% glycerol, 0.1 mM dTTP, 8.75 μM dGTP, 1.25 μM α32P-dGTP for 1 h at 30°C. Reactions were stopped by adding 100 μL of stop solution containing 3.6 M ammonium acetate, 1 mg/mL glycogen and a 32P-labeled 100mer DNA oligo that acts as a loading control. DNA products were ethanol precipitated and run out on a 10% polyacrylamide, 7 M urea, 1× TBE sequencing gel for analysis.
Acknowledgments
We thank Johnny Croy for help with NMR spectroscopy, Masato Kawasaki and Fuyuhiko Inagaki for helpful advice and the gift of pProGFP, the University of Colorado DNA sequencing facility for the sequencing of numerous clones, David King for advice on mass spectrometry and limited proteolysis, and Lin Chen for helpful discussions and critical reading of the manuscript. S.J. is a Damon Runyon fellow supported by the Damon Runyon Cancer Research Foundation (DRG-#1821-04).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051532105.
References
- Armbruster, B.N., Banik, S.S., Guo, C., Smith, A.C., and Counter, C.M. 2001. N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol. Cell. Biol. 21 7775–7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachand, F. and Autexier, C. 1999. Functional reconstitution of human telomerase expressed in Saccharomyces cerevisiae. J. Biol. Chem. 274 38027–38031. [DOI] [PubMed] [Google Scholar]
- Beattie, T.L., Zhou, W., Robinson, M.O., and Harrington, L. 2001. Functional multimerization of the human telomerase reverse transcriptase. Mol. Cell. Biol. 21 6151–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan, T.M., Goodrich, K.J., and Cech, T.R. 2000a. A mutant of Tetrahymena telomerase reverse transcriptase with increased processivity. J. Biol. Chem. 275 24199–24207. [DOI] [PubMed] [Google Scholar]
- ———. 2000b. Telomerase RNA bound by protein motifs specific to telomerase reverse transcriptase. Mol. Cell. 6 493–499. [DOI] [PubMed] [Google Scholar]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T.J., Higgins, D.G., and Thompson, J.D. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Friedman, K.L. and Cech, T.R. 1999. Essential functions of amino-terminal domains in the yeast telomerase catalytic subunit revealed by selection for viable mutants. Genes & Dev. 13 2863–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, K.L., Heit, J.J., Long, D.M., and Cech, T.R. 2003. N-terminal domain of yeast telomerase reverse transcriptase: Recruitment of Est3p to the telomerase complex. Mol. Biol. Cell 14 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider, C.W. and Blackburn, E.H. 1989. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337 331–337. [DOI] [PubMed] [Google Scholar]
- Grothues, D., Cantor, C.R., and Smith, C.L. 1993. PCR amplification of megabase DNA with tagged random primers (T-PCR). Nucleic Acids Res. 21 1321–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRview: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Kawasaki, M. and Inagaki, F. 2001. Random PCR-based screening for soluble domains using green fluorescent protein. Biochem. Biophys. Res. Commun. 280 842–844. [DOI] [PubMed] [Google Scholar]
- Kelleher, C., Teixeira, M.T., Forstemann, K., and Lingner, J. 2002. Telomerase: Biochemical considerations for enzyme and substrate. Trends Biochem. Sci. 27 572–579. [DOI] [PubMed] [Google Scholar]
- Knaust, R.K. and Nordlund, P. 2001. Screening for soluble expression of recombinant proteins in a 96-well format. Anal. Biochem. 297 79–85. [DOI] [PubMed] [Google Scholar]
- Lee, S.R., Wong, J.M., and Collins, K. 2003. Human telomerase reverse transcriptase motifs required for elongation of a telomeric substrate. J. Biol. Chem. 278 52531–52536. [DOI] [PubMed] [Google Scholar]
- Lingner, J., Hughes, T.R., Shevchenko, A., Mann, M., Lundblad, V., and Cech, T.R. 1997. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276 561–567. [DOI] [PubMed] [Google Scholar]
- Masutomi, K., Kaneko, S., Hayashi, N., Yamashita, T., Shirota, Y., Kobayashi, K., and Murakami, S. 2000. Telomerase activity reconstituted in vitro with purified human telomerase reverse transcriptase and human telomerase RNA component. J. Biol. Chem. 275 22568–22573. [DOI] [PubMed] [Google Scholar]
- Mikuni, O., Trager, J.B., Ackerly,H.,Weinrich, S.L., Asai, A., Yamashita, Y., Mizukami, T., and Anazawa, H. 2002. Reconstitution of telomerase activity utilizing human catalytic subunit expressed in insect cells. Biochem. Biophys. Res. Commun. 298 144–150. [DOI] [PubMed] [Google Scholar]
- Moriarty, T.J., Marie-Egyptienne, D.T., and Autexier, C. 2004. Functional organization of repeat addition processivity and DNA synthesis determinants in the human telomerase multimer. Mol. Cell. Biol. 24 3720–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor, C.M., Lai, C.K., and Collins, K. 2005. Two purified domains of telomerase reverse transcriptase reconstitute sequence-specific interactions with RNA. J. Biol. Chem. 17: 17533–17539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnapp, G., Rodi, H.P., Rettig, W.J., Schnapp, A., and Damm, K. 1998. One-step affinity purification protocol for human telomerase. Nucleic Acids Res. 26 3311–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa, R. 1996. Structural and mechanistic relationships between nucleic acid polymerases. Trends Biochem. Sci. 21 186–190. [PubMed] [Google Scholar]
- Waldo, G.S., Standish, B.M., Berendzen, J., and Terwilliger, T.C. 1999. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 17 691–695. [DOI] [PubMed] [Google Scholar]
- Weinrich, S.L., Pruzan, R., Ma, L., Ouellette, M., Tesmer, V.M., Holt, S.E., Bodnar, A.G., Lichtsteiner, S., Kim, N.W., Trager, J.B., et al. 1997. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 17 498–502. [DOI] [PubMed] [Google Scholar]
- Xia, J., Peng, Y., Mian, I.S., and Lue, N.F. 2000. Identification of functionally important domains in the N-terminal region of telomerase reverse transcriptase. Mol. Cell. Biol. 20 5196–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]