

Abstract
Serine acetyltransferase is a key enzyme in the sulfur assimilation pathway of bacteria and plants, and is known to form a bienzyme complex with O-acetylserine sulfhydrylase, the last enzyme in the cysteine biosynthetic pathway. The biological function of the complex and the mechanism of reciprocal regulation of the constituent enzymes are still poorly understood. In this work the effect of complex formation on the O-acetylserine sulfhydrylase active site has been investigated exploiting the fluorescence properties of pyridoxal 5′-phosphate, which are sensitive to the cofactor microenvironment and to conformational changes within the protein matrix. The results indicate that both serine acetyltransferase and its C-terminal decapeptide bind to the α-carboxyl subsite of O-acetylserine sulfhydrylase, triggering a transition from an open to a closed conformation. This finding suggests that serine acetyltransferase can inhibit O-acetylserine sulfhydrylase catalytic activity with a double mechanism, the competition with O-acetylserine for binding to the enzyme active site and the stabilization of a closed conformation that is less accessible to the natural substrate.
Keywords: serine acetyltransferase, O-acetylserine sulfhydrylase, cysteine synthase, fluorescence, protein dynamics, enzymes, conformational changes
Inorganic sulfur assimilation, reduction, and incorporation into L-cysteine are accomplished in bacteria and plants via a branched biosynthetic pathway (Scheme 1 ▶). O-acetylserine sulfhydrylase (OASS; encoded by cysK) is at the point of convergence of the two branches, the assimilatory sulfate reduction pathway that is devoted to sulfide production, and that of L-serine activation via an O-acetylation reaction catalysed by serine acetyltransferase (SAT; encoded by cysE) (Kredich 1992). Genes coding for proteins involved in sulfate transport, reduction, and incorporation into L-cysteine belong to the cysteine regulon (Kredich 1996) whose transcription, except for cysE, is under the control of the transcriptional activator CysB (Kredich 1992). OASS and SAT play a fundamental role in the control of the cysteine biosynthetic pathway in response to variations of sulfur availability. The final product L-cysteine inhibits competitively SAT (Kredich and Tomkins 1966; Hindson 2003; Johnson et al. 2004), whereas sulfur limitation leads to the accumulation of OASS substrate, O-acetylserine (OAS). This metabolite rapidly converts to N-acetylserine, which functions as an inducer of the transcription of the cysteine regulon (Kredich 1992). In plants distinct isoforms of OASS and SAT characterize the different subcellular compartments (e.g., mitochondria, plastids, and cytosol) (Ruffet et al. 1995; Hell et al. 2002), whereas in bacteria only one isoform of SAT and two isoforms of OASS, OASS-A, and OASS-B (cysM) are known (Tai and Cook 2000). OASS-A from Salmonella typhimurium is a homodimer whose structure has been solved in different conformations, i.e., in the absence and presence of substrate analogs and allosteric effectors (Burkhard et al. 1998, 1999, 2000). The three-dimensional structure of SAT has recently been solved for the Escherichia coli (Pye et al. 2004) and Haemophilus influenzae enzymes (Gorman and Shapiro 2004; Olsen et al. 2004), revealing a hexameric quaternary organization.
Scheme 1.

The pathway of cysteine biosynthesis in bacteria and its regulation (adapted from Kredich 1992, 1996). Black thick lines connect proteins that can form bi-enzymatic complexes. The abbreviations used are: OAS, O-acetylserine; NAS, N-acetylserine; CysB, regulatory protein controlling the transcription of the cysteine regulon (Kredich 1996; Tyrrell et al. 1997); APS; adenosine 5′-phosphosulfate; PAPS, 3′-phosphoadenosine 5′-phosphosulfate. OAS converts spontaneously to NAS at neutral pH (Flavin and Slaughter 1965).
Despite reports of an excess of OASS over SAT, varying among subcellular locations and developmental stage in plants (Ruffet et al. 1994; Hell et al. 2002), it is well established that OASS and SAT interact to form a bi-enzyme complex known as cysteine synthase, both in bacteria (Becker et al. 1969; Kredich et al. 1969; Mino et al. 1999, 2000b) and plants (Bogdanova and Hell 1997; Droux et al. 1998; Wirtz et al. 2001). OASS is also involved in complex formation with ATP sulfurylase, whose function is to activate sulfate for the subsequent reduction reactions (Scheme 1 ▶). Complex formation leads to a 1000-fold increase in the rate of ATP sulfurylase activity (Wei et al. 2002). Early works reported that in S. typhimurium only 5% of OASS activity was involved in complex formation with SAT (Kredich and Tomkins 1967), even though this should be considered a conservative estimate due to the known susceptibility of SAT to cold and proteolytic degradation (Mino et al. 2001). The stability of the complex is related to sulfur availability in the cell, with OAS accumulation leading to complex dissociation, an event that is prevented by the presence of sulfide (Kredich et al. 1969; Cook and Wedding 1977; Droux et al. 1998; Mino et al. 1999). One of the better characterized effects of complex formation is the alteration of the kinetic properties of the two enzymes. In bacteria and plants the activity of OASS-A in the complex is reduced with respect to the free enzyme, with a four- to fivefold increase in KM and a twofold decrease in kcat in E. coli (Mino et al. 2000b) and S. typhimurium (Kredich et al. 1969), and a sevenfold increase in KM and a 60-fold decrease in kcat in Arabidopsis thaliana (Droux et al. 1998). The effect on SAT activity, when detected, is of much smaller magnitude, with an about twofold reduction of both KM and kcat (Droux et al. 1998). Recently, yeast two-hybrid system (Bogdanova and Hell 1997) and inhibition studies (Mino et al. 2000a) have pointed out the role of the SAT C-terminal sequence in the interaction with OASS. In particular, both the entire SAT and the decapeptide corresponding to its C-terminal sequence inhibit competitively the binding of OAS to OASS (Mino et al. 2000a), suggesting a direct interaction of SAT with the OASS active site. SAT lacking 20 C-terminal amino acids has proven to be unable to form the cysteine synthase complex (Mino et al. 1999, 2000a). The biological role of the complex and the details of the reciprocal regulation of the constituent enzymes are still a matter of debate. Among other hypotheses, channelling of OAS from SAT to OASS has been discarded based on the original work by Cook and Wedding (1977), where the release of the intermediate has been well documented. In bacteria sequestration of SAT in the cysteine synthase complex seems to have a protective role against cold inactivation and proteolysis (Mino et al. 2001), whereas in plants a possible role of the complex as a metabolic sensor of sulfur supply has been proposed (Hell and Hillebrand 2001; Hell et al. 2002).
In order to gain insight into the mechanism of reciprocal regulation in the complex, we have investigated the effect of intact SAT and the SAT C-terminal decapeptide binding to HiOASS on the fluorescence emission properties of the coenzyme pyridoxal 5′-phosphate (PLP). PLP fluorescence is sensitive both to local changes in the chromophore microenvironment and, by virtue of Förster energy transfer from Trp50 located in the N-terminal domain of OASS (Benci et al. 1999a), to large-scale conformational dynamics involving variations in tryptophan to PLP distance and reciprocal orientation. Tryptophan and PLP fluorescence of StOASS has been previously exploited (Benci et al. 1997, 1999a) to investigate the open-to-closed transition, which underlies OASS catalysis and regulation (McClure and Cook 1994; Burkhard et al. 1999).
Results
The spectroscopic properties of HiOASS-A are very similar to those of the enzyme from S. typhimurium (Becker et al. 1969; Cook and Wedding 1976; Cook et al. 1992; McClure and Cook 1994; Strambini et al. 1996; Benci et al. 1997, 1999b), with an absorption band centered at 412 nm indicative of a PLP molecule in an internal aldimine linkage with the active site lysine residue (Fig. 1A ▶). Excitation of tryptophans (Fig. 1B ▶) results in the direct emission at 344 nm of the aromatic residues and to a longer wavelength emission at 509 nm, which was attributed to energy transfer from tryptophan( s) to the enolimine tautomer of the internal aldimine (Strambini et al. 1996), that isomerizes in the excited state to the ketoenamine tautomer emitting at 509 nm (Benci et al. 1999b). Direct excitation of the cofactor at 412 nm generates an emission spectrum centred at 509 nm (Fig. 1C ▶). Addition of a molar excess of SAT to OASS causes pronounced changes in the emission spectra upon excitation at either 298 nm or 412 nm (Fig. 2A–B ▶). Besides a large increase in the emission at 344 nm upon excitation at 298 nm, due to the contribution of tryptophan residues of SAT, an increase in the emission at 509 nm is observed, with a concomitant blue-shift of the emission maximum from 509 nm to 493 nm (Fig. 2A ▶). A similar effect is observed by measuring directly the cofactor emission upon excitation at 412 nm, with an increase in the fluorescence emission and a blue shift of about 15 nm (Fig. 2B ▶). The increase in the fluorescence emission intensity and the magnitude of the blue-shift are proportional to the concentration of SAT. However, the fluorescence signal associated to the low OASS concentration required for equilibrium measurements is below the sensitivity limit of the instrument and hampers the accurate estimate of the dissociation constant for the complex. However, the dissociation constant is estimated to be lower than 2 nM, a value that should be compared to an inhibition constant of 0.15 nM, obtained for the E. coli complex (Mino et al. 2000a).
Figure 1.

(A) Absorbance spectrum of a solution containing 14 μM HiOASS, 100 mM HEPES (pH 7), at 20°C. (B) Emission spectrum upon excitation at 298 nm (slitex=5nm, slitem=5 nm) of a solution containing 200 nM HiOASS, 100 mM HEPES (pH 7), at 20°C. (C)Emission spectrum upon excitation at 412 nm (slitex=5 nm, slitem=5 nm) of a solution containing 200 nM HiOASS, 100 mM HEPES (pH 7), at 20°C.
Figure 2.

Binding of SAT and P10 to HiOASS. Experiments were carried out at 20 ± 0.5°C. (A) Fluorescence emission spectra upon excitation at 298 nm (slitex=5 nm, slitem=5 nm) of a solution containing 200 nM HiOASS, 100 mM HEPES (pH 7), in the absence (solid line) and presence (dashed line) of 900 nM HiSAT. (B) Fluorescence emission spectra upon excitation at 412 nm (slitex=5 nm, slitem=5 nm) of a solution containing 200 nM HiOASS, 100 mM HEPES (pH 7), in the absence (solid line) and presence (dashed line) of 900 nM HiSAT. (C) Fluorescence emission spectra upon excitation at 298 nm (slitex=2.5 nm, slitem=2.5 nm) of a solution containing 7 μM HiOASS, 100 mM HEPES (pH 7), in the absence (solid line) and presence (dashed line) of 25 μM P10. (D) Fluorescence emission changes at 500 nm upon excitation at 412 nm (slitex=7 nm, slitem=7 nm) as a function of P10 concentration. The solution contains 60 nM HiOASS, 100m MHEPES (pH 7). The solid line through data points represents the fitting to Equation 1, with Kdiss=581 ± 72 nM. The dashed line through data points represents the fitting to Equation 2, with Kdiss=515 ± 29 nM, n=0.94 ± 0.02 and h=1.27 ± 0.07.
The SAT C-terminal decapeptide, GIDDGMNLNI (P10), elicits an effect similar to that of the entire protein on OASS spectroscopic properties. The emission spectrum upon excitation at 412 nm shows an increase in intensity and a blue-shift of about 15 nanometers (data not shown). The emission spectrum upon excitation at 298 nm shows no changes in the intensity of the major peak at 344 nm, while a significant increase in the long wavelength emission leads to a decrease of the 344 nm to 500 nm ratio from about 7.5 in the unligated protein to 3.2 in the presence of a molar excess of the peptide (Fig. 2C ▶). A comparable effect is obtained by treatment of HiOASS with acetate, which produces a large increase and a 15 nm blue-shift of the ketoenamine tautomer emission band, both upon excitation at 298 nm and 412 nm (data not shown). The dependence on P10 concentration of the emission intensity at 500 nm upon excitation at 412 nm gives a hyperbolic curve that can be fitted to an isothermal equation with a dissociation constant of 581 ± 72 nM (Fig. 2D ▶). The inhibition constant for the E. coli complex was reported to be 130 nM (Mino et al. 2000a). Analysis of the data with the Hill equation (Equation 2) improves the fitting and gives a dissociation constant of 515 ± 29 nM, a number of binding sites (n) of 0.94 ± 0.02 and a Hill coefficient (h) of 1.27 ± 0.07. Evidence of cooperativity in OASS is limited to some enzymes from plants, such as Spinacia oleracea (Rolland et al. 1996) and Datura innoxia (Kuske et al. 1994).
The specificity of the interaction of P10 with HiOASS was evaluated by control experiments with a scrambled peptide, DNLINGIDMG (S10). S10 at concentrations up to 600 μM does not elicit any significant effect on the emission properties of HiOASS (data not shown), indicating absence of a tight binding.
Stoichiometric titrations of HiOASS with either HiSAT or P10 (Fig. 3 ▶), give a stoichiometric ratio of 1.43 and 0.95, respectively, i.e., two dimers of OASS form a complex with a hexamer of SAT and one P10 binds to one subunit of OASS. The same stoichiometry has already been suggested, based on size-exclusion chromatography data, for the complexes of E. coli (Mino et al. 2001) and S. typhimurium (Kredich et al. 1969). Addition of a molar excess of P10 to OASS saturated with SAT gives no further spectral changes (data not shown), indicating that there are no sites on OASS available for P10 binding.
Figure 3.

Stoichiometric titrations of HiOASS with HiSAT and P10. Experiments were carried out at 20 ± 0.5°C. (A) Titration of a solution containing 2 μM HiOASS, 100 mM HEPES (pH 7), with HiSAT. Data points represent the emission intensity at 500 nm upon excitation at 412 nm (slitex=3 nm, slitem=3 nm). (B) Titration of a solution containing 7 μM HiOASS, 100 mM HEPES (pH 7), with P10. Data points represent the emission intensity at 500 nm upon excitation at 412 nm (slitex=2.5 nm, slitem=2.5 nm).
Sequences of SAT from Gammaproteobacteria are overall highly similar, except for the C-terminal regions, which display a higher degree of variability (Fig. 4 ▶). In particular, HiSAT and StSAT amino acid sequences are 84% similar and 69% identical, but only three out of the last 10 residues of HiSAT are identical in StSAT. The remaining residues do not share any sequence similarity or similar physicochemical properties. To test whether the HiSAT C-terminal sequence could interact with StOASS active site, we investigated the binding of the H. influenzae C-terminal decapeptide to StOASS. P10 is able to elicit similar effects on HiOASS and StOASS fluorescence emission spectra (Fig. 5 ▶). A saturating concentration of the peptide generates a large increase and a blue-shift of the coenzyme emission band upon excitation at 298 nm or at 412 nm (Fig. 5A–B ▶). Whereas the blueshift is comparable with that elicited on HiOASS, the increase in the fluorescence emission intensity is significantly higher. This can be attributed to the different tautomer distribution in the two enzymes. By fitting the dependence on P10 concentration of the emission intensity at 500 nm upon excitation at 412 nm a dissociation constant of 972 ± 62 nM was obtained (Fig. 5C ▶), similar to the Kdiss for the HiOASS/decapeptide complex. In the case of S. typhimurium enzyme the analysis of the data with the Hill equation for a cooperative system does not improve the quality of the fitting. Furthermore, HiSAT is able to bind to StOASS (Fig. 5D ▶), and, even though a dissociation constant for the complex cannot be obtained due to sensitivity limitations, the strength of the interaction seems to be in a similar range as that of the HiSAT/HiOASS complex.
Figure 4.

Amino acid sequence alignment of bacterial SATs. Identical residues have a black background, while residues with similar physicochemical properties are boxed. HiSAT, SAT from H. influenzae; StSAT, SAT from S. typhimurium; EcSAT, SAT from E. coli; YpSAT, SAT from Yersinia pestis; PlSAT, SAT from Photorhabdus luminescens; ErSAT, SAT from Erwinia carotovora; VcSAT, SAT from Vibrio cholerae; PpSAT, SAT from Photobacterium profundum; MsSAT, SAT from Mannheimia succiniciproducens; HsSAT, SAT from Haemophilus somnus; HdSAT, SAT from Haemophilus ducrey; ApSAT, SAT from Actinobacillus pleuropneumonie. Secondary structure elements are indicated above the alignment for HiSAT (PDB ID 1SST).
Figure 5.

Binding of HiSAT and P10 to StOASS. Experiments were carried out at 20 ± 0.5°C. (A) Fluorescence emission spectra upon excitation at 298 nm (slitex=2 nm, slitem=2 nm) of a solution containing 7 μM OASS, 100 mM HEPES (pH 7) in the absence (solid line) and presence (dashed line) of 25 μM P10. (B) Fluorescence emission spectra upon excitation at 412 nm (slitex=2 nm, slitem=2 nm) of a solution containing 7 μM OASS, 100 mM HEPES (pH 7) in the absence (solid line) and presence (dashed line) of 25 μM P10. (C) Fluorescence emission changes at 500 nm upon excitation at 412 nm as a function of P10 concentration. The solution contains 165 nM StOASS, 100 mM HEPES (pH 7). The solid line through data points represents the fitting to Equation 1. The calculated Kdiss is 972 ± 62 nM. (D) Fluorescence emission spectra upon excitation at 412 nm (slitex=3 nm, slitem=3 nm) of a solution containing 10 μM StOASS, 100 mM HEPES (pH 7) in the absence (solid line) and presence (dashed line) of 59 μM HiSAT.
In bacteria, only one isoform of SAT is known, whereas there are two isoforms of OASS: OASS-A and OASS-B. The latter, involved in sulfur assimilation under anaerobic conditions, is still poorly characterized (Tai et al. 1993). Early data, based on chromatographic coelution of enzyme activities, suggested that only OASS-A could interact with SAT (Becker and Tomkins 1969), but no further studies were carried out in order to directly determine a possible interaction between OASS-B and SAT. For this reason we investigated whether P10 could bind to OASS-B. Up to a concentration of about 1 mM, P10 does not cause on StOASS-B any of the spectroscopic changes observed for HiOASS-A and StOASS-A, upon excitation at either 298 nm or 412 nm (data not shown). These results indicate that P10 does not interact with OASS-B.
Discussion
SAT and OASS from bacteria and plants are known to form a bienzyme complex, which in plants is organelle-specific (Kredich and Tomkins 1966; Kredich et al. 1969; Smith and Thompson 1971; Rolland et al. 1993; Ruffet et al. 1994, 1995; Saito et al. 1995; Bogdanova and Hell 1997; Droux et al. 1998; Noji et al. 1998; Jost et al. 2000; Mino et al. 2000, 2001; Wirtz et al. 2001). In bacteria, only one gene encoding for SAT (cysE) and two genes encoding for the A and B isoforms of OASS (cysK and cysM, respectively) are known (Kredich 1996). Both in bacteria and plants the complex dissociates into its components in the presence of OAS, whereas it is stable in the presence of sulfide (Kredich et al. 1969). In E. coli and S. typhimurium complex formation has no effect on SAT activity, whereas it is associated with an increase in the Michaelis constant for OAS and a reduction in the kcat for the forward reaction of OASS (Kredich et al. 1969; Mino et al. 2000b). Complex formation between SAT and OASS from A. thaliana is also associated with the transition from a cooperative to a noncooperative kinetic model for OASS catalytic activity (Droux et al. 1998). The effect of SAT on OASS activity and the instability of the complex in the presence of OAS suggested a possible effect of SAT on OASS active site structure and/or dynamics. Experimental evidence indicated that the C-terminal sequence of SAT can bind to OASS active site (Mino et al. 2000a,b). Recently, the structure of HiOASS in complex with the C-terminal decapeptide of SAT has been solved (Huang et al. 2005), proving that SAT binding to OASS involves the anchoring of the SAT C-terminal sequence in the OASS active site. Unfortunately, neither the three-dimensional structure of native HiOASS alone or in complex with HiSAT, nor the details of the conformational events that accompany binding of P10 to OASS have been characterized. The fluorescence emission properties of S. typhimurium OASS have proven to be sensitive to the dynamics of the protein structure (McClure and Cook 1994; Benci et al. 1997). Here, they have been exploited to gain insight into conformational changes taking place on HiOASS upon binding of SAT or P10.
The binding of either SAT or P10 has comparable effects on cofactor fluorescence properties, with a three- to fourfold increase in the emission intensity and a concomitant blue-shift of the emission maximum (Fig. 2A–C ▶). A similar increase in the long-wavelength emission band upon excitation at 298 nm was observed for StOASS-A upon addition of acetate and L-cysteine, and was found to be pH-dependent (McClure and Cook 1994). Binding of L-cysteine, or other substrate analogs, as L-serine and methionine, is followed by the formation of the external aldimine that is, in turn, associated with a conformational change from an open to closed structure (Schnackerz et al. 1995). Structural data indicate that the conformational change might be triggered by binding of the α-carboxylate of the amino acid substrate to an α-carboxyl subsite in the enzyme active site (Burkhard et al. 1999). The α-carboxylate forms a strong hydrogen-bonding network with residues Pro67, Thr68, Asn69, and Gly70, which are part of the so-called “asparagine-loop.” The conformational change causes an increase in the lifetime of the excited state of the PLP Schiff base, which is apparent in the increase in the emission intensity of the ketoenamine tautomer (Schnackerz et al. 1995). Acetate, which cannot form the external Schiff base, is believed to abortively occupy the α-carboxyl subsite of OASS, triggering conformational changes similar to those associated with the transition from the internal to the external aldimine of PLP (McClure and Cook 1994; Schnackerz et al. 1995; Burkhard et al. 1999). Saturating amounts of SAT, P10 or acetate cause a comparable blue-shift of about 15 nm of the emission peak maximum of HiOASS, whereas the increase in emission intensity is slightly less pronounced upon the addition of SAT or P10 compared to the addition of acetate. These findings strongly support the hypothesis that SAT or P10 binding to OASS is able to trigger the same dynamic events associated with the transition from the open to the closed conformation. Our results agree with the known effects of cysteine synthase complex formation on OASS kinetic parameters (Kredich et al. 1969; Droux et al. 1998; Mino et al. 2000b), and show that binding of SAT to the OASS active site, besides exerting a competitive inhibition with respect to OAS, stabilizes a protein conformation with a reduced accessibility of the active site to its natural substrate (Burkhard et al. 1999). Recently, crystallographic studies (Huang et al. 2005) have demonstrated that P10 binds to the OASS active site with its C-terminal α-carboxylate fitting in the same position occupied by the α-carboxylate of L-methionine in external aldimine linkage with PLP (Burkhard et al. 1999). The observation that HiSAT C-terminal peptide can bind to StOASS with similar affinity to that measured for HiOASS suggests that conserved residues could play a key role in complex formation. The last residue, Ile267 in HiSAT, is a conserved residue in SAT sequences from Gammaproteobacteria (Fig. 4 ▶), and is involved in a network of interactions with OASS (Huang et al. 2005). In the crystal structure only electron density corresponding to the last four residues of the decapeptide is visible, with only Asn266 and the C-terminal residue Ile267 interacting with the OASS active site. Ile267 probably plays a fundamental role in establishing the interactions that drive complex formation, occupying the site where the lateral chain and α-carboxyl group of the substrate bind in the external aldimine intermediate (Burkhard et al. 1999; Huang et al. 2005). In the presence of the inhibitor L-cysteine the C-terminal region of SAT, which is normally unstructured, folds in a short α-helix and hampers binding of acetylCoA and the subsequent processing of L-cysteine (Olsen et al. 2004). In the inhibited structure the binding pocket of the adenine moiety of acetylCoA is occupied by Phe256 (Olsen et al. 2004), which appears to be a conserved residue in the C-terminal sequences of Gammaproteobacteria (Fig. 4 ▶). These observations point to a dual functional role for the C-terminal “mobile arm” of SAT: an intrasteric inhibitor of SAT catalytic activity and an effector of the OASS active site.
Previous reports on E. coli and S. typhimurium proteins using sedimentation equilibrium experiments (Kredich et al. 1969;Mino et al. 2001) suggested that a hexamer of SAT should bind two dimers of OASS. More recently, it has been suggested that each hexamer of SAT could bind up to six dimers of OASS, one for each C-terminal domain of the protein (Pye et al. 2004). This is the first report of a direct determination of the stoichiometry of binding of SAT to OASS, clearly indicating that two dimers of OASS bind to one hexamer of SAT. Spectroscopic changes at saturating concentrations of either SAT or P10 are comparable, suggesting similar dynamic events taking place with both ligands. Furthermore, titration of OASS with P10 in the presence of stoichiometric amounts of SAT does not induce any spectral change, indicating that there are no OASS active sites available for P10 binding. This observation supports the quaternary geometry of the cysteine synthase complex proposed by Hindson et al. (2000), and is compatible with two different scenarios (Scheme 2 ▶): a SAT trimer binds to only one OASS active site, triggering the closure of the other site through allosteric communication (Scheme 2A ▶), or SAT is able to occupy, or at least to physically restrict access to, both OASS active sites (Scheme 2B ▶). The latter configuration could be allowed by the position of the mobile C-terminal arms of SAT (Olsen et al. 2004). OASS-A plays a key role in the control of the cysteine biosynthetic pathway both directly through the formation of regulatory bi-enzymatic complexes with SAT and ATP sulfurylase and indirectly via its substrate OAS and its product L-cysteine that function as reporters of sulfur supply to the cell and can modulate the activity and the expression of the enzymes of the cysteine regulon (Scheme 1 ▶). The modulation of cysteine biosynthesis is thus achieved through a complex mechanism encompassing transcriptional control, activity regulation via feedback inhibition, and protein– protein interaction. The strength of the OASS–SAT complex is three orders of magnitude higher than that of OASS–ATP sulfurylase, suggesting that the relative abundance and affinity of the proteins involved in complexes could represent a further level of regulation. Many aspects of the organization of the cysteine biosynthetic pathway await further investigations, as for example a probable organization of some proteins in multienzyme complexes (Wei et al. 2002) and the role and regulation of the B isozyme of OASS.
Scheme 2.

Hypothetical quaternary geometry of the cysteine synthase complex. Open ellipse, SAT subunit; gray shape, OASS subunit. SAT C-terminal mobile arms are represented as thick lines.
Materials and methods
Proteins
Recombinant serine acetyltransferase (HiSAT) and O-acetylserine sulfhydrylase (HiOASS) from H. influenzae were expressed and purified as previously described (Olsen et al. 2004; Huang et al. 2005). OASS stock solutions were stored at −80°C in 20 mM HEPES buffer, 50 mM NaCl (pH 7.5). SAT stock solutions were stored at −80°C in 20 mMTris buffer, 50 mM NaCl, 10% glycerol (pH 7.5). The extinction coefficient at 280 nm of HiSAT, calculated from the tabulated extinction coefficients of the aromatic residues (Edelhoch 1967), is 28,710 M−1 cm−1. The extinction coefficient at 412 nm of HiOASS, 7600 M−1 cm−1, was calculated from the amount of PLP released upon alkali-induced denaturation of the protein fully saturated with PLP (Peterson and Sober 1954). Because chloride ions are known to be allosteric effectors of OASS (Burkhard et al. 2000; Tai et al. 2001), proteins stock solutions were diluted with or dialyzed against HEPES buffer to reduce the chloride ion concentration.
Recombinant OASS-A from S. typhimurium (StOASS-A) was expressed in E. coli cells NK3 (Hulanicka et al. 1986) transformed with the pCKM3 expression vector, as previously described (Tai et al. 1993). The cells were disrupted by sonication in the presence of 0.2 mMPLP. The crude extract was treated with streptomycin sulfate and ammonium sulfate as previously described (Hara et al. 1990). The solution containing OASS-A was desalted by dialysis and loaded on a DEAE FF column (Amersham Biosciences). OASS-A was eluted with a linear gradient from 0 to 0.5 M NaCl in 10 mM HEPES (pH 8). Fractions containing OASS-A were concentrated and loaded on an Ultrogel AcA44 column (Sigma-Aldrich) and eluted with a buffer containing 10 mM HEPES, 200 mM NaCl (pH 8). Fractions with an absorbance ratio (A280/A412) ≤3.4 were pooled, dialyzed against 10 mM HEPES (pH 8), and stored at −80°C. OASS-B from S. typhimurium (StOASS-B) was expressed and purified in the recombinant form (P.F. Cook, unpubl.).
Peptides
The decapeptide (P10) corresponding to the C-terminal sequence of HiSAT (GIDDGMNLNI) and the corresponding scrambled peptide (DNLINGIDMG) were synthesized with a Biosystems 433A peptide synthesizer (Perkin-Elmer) and subsequently purified by HPLC. The peptides were dissolved in 25 mM HEPES buffer (pH7.9) and dialyzed O/N against the same buffer using a Spectra/Por dialysis membrane (Spectrum Laboratories, Inc.) with a 500-Da MW cutoff. The peptide solutions were stored at −80°C. The concentration of the peptides was calculated using published methods (Gratzer 1989). The calculated extinction coefficient at 210 nm was 19,961 M−1 cm−1.
Chemicals and buffers
Chemicals, purchased from Sigma-Aldrich, were of the best available quality and were used without further purification. Experiments were carried out in 100 mM HEPES buffer (pH 7), at 20 ± 0.5°C.
Absorption and steady-state fluorescence measurements
Absorption spectra were collected with a Cary 400 Scan spectrophotometer (Varian Inc.) using quartz microcuvettes. The cell-holder temperature was kept constant via a circulating water bath. Fluorescence measurements were carried out using a FluoroMax-3 fluorometer (HORIBA), equipped with a thermostated cell-holder. Emission spectra upon excitation at 298 nm were collected between 310 nm and 550 nm. Spectra upon excitation at 412 nm were collected between 425 nm and 650 nm. Absorption and fluorescence spectra were corrected for the buffer contribution.
Data analysis
The dependence on decapeptide concentration of fluorescence emission at 500 nm upon excitation at 412 nm was fitted to a binding isotherm for noninteracting sites
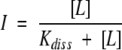 |
(1) |
or to the equation for a cooperative binding (Hill equation)
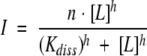 |
(2) |
where I is the signal intensity at a given ligand concentration, [L] is the decapeptide concentration, Kdiss is the dissociation constant of the complex, n is the number of binding sites, and h is the Hill coefficient.
Multiple sequence alignments
SAT aminoacid sequences from representative Gammaproteobacteria, retrieved by protein–protein Blast search (Altschul et al. 1997) of nonredundant sequence databases, were aligned using the ClustalW program (Thompson et al. 1994) set at the default parameters. The sequence conservation pattern was visualized using the programme ESPript (Gouet et al. 1999).
Acknowledgments
The financial support from the Italian Ministry of Education, University and Research (COFIN 2003, to A.M.) and the National Institute for the Physics of Matter (FIRB Nanotechnology 2003, to A.M.) is acknowledged. We gratefully acknowledge Dr. Riccardo Percudani for helpful assistance with multiple sequence alignments.
Abbreviations
SAT, serine acetyltransferase
OASS, O-acetylserine sulfhydrylase
HiOASS/HiSAT, OASS and SAT from H. influenzae
StOASS/StSAT, OASS and SAT from S. typhimurium
PLP, pyridoxal 5′-phosphate
OAS, O-acetylserine
P10, decapeptide corresponding to the last 10 residues of HiSAT
S10, scrambled decapeptide
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051492805.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, M.A. and Tomkins, G.M. 1969. Pleiotrophy in a cysteine-requiring mutant of Salmonella typhimurium resulting from altered protein– protein interaction. J. Biol. Chem. 244 6023–6030. [PubMed] [Google Scholar]
- Becker, M.A., Kredich, N.M., and Tomkins, G.M. 1969. The purification and characterization of O-acetylserine sulfhydrylase-A from Salmonella typhimurium. J. Biol. Chem. 244 2418–2427. [PubMed] [Google Scholar]
- Benci, S., Vaccari, S., Mozzarelli, A., and Cook, P.F. 1997. Time-resolved fluorescence of O-acetylserine sulfhydrylase catalytic intermediates. Biochemistry 36 15419–15427. [DOI] [PubMed] [Google Scholar]
- Benci, S., Bettati, S., Vaccari, S., Schianchi, G., Mozzarelli, A., and Cook, P.F. 1999a. Conformational probes of O-acetylserine sulfhydrylase: Fluorescence of tryptophans 50 and 161. J. Photochem. Photobiol. B-Biol. 48 17–26. [Google Scholar]
- Benci, S., Vaccari, S., Mozzarelli, A., and Cook, P.F. 1999b. Time-resolved fluorescence of O-acetylserine sulfhydrylase. Biochim. Biophys. Acta 1429 317–330. [DOI] [PubMed] [Google Scholar]
- Bogdanova, N. and Hell, R. 1997. Cysteine synthesis in plants: Protein– protein interactions of serine acetyltransferase from Arabidopsis thaliana. Plant J. 11 251–262. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Rao, G.S., Hohenester, E., Schnackerz, K.D., Cook, P.F., and Jansonius, J.N. 1998. Three-dimensional structure of O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 283 121–133. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Tai, C.H., Ristroph, C.M., Cook, P.F., and Jansonius, J.N. 1999. Ligand binding induces a large conformational change in O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 291 941–953. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Tai, C.H., Jansonius, J.N., and Cook, P.F. 2000. Identification of an allosteric anion-binding site on O-acetylserine sulfhydrylase: structure of the enzyme with chloride bound. J. Mol. Biol. 303 279–286. [DOI] [PubMed] [Google Scholar]
- Cook, P.F. and Wedding, R.T. 1976. A reaction mechanism from steady state kinetic studies for O-acetylserine sulfhydrylase from Salmonella typhimurium LT-2. J. Biol. Chem. 251 2023–2029. [PubMed] [Google Scholar]
- ———. 1977. Initial kinetic characterization of the multienzyme complex, cysteine synthetase. Arch. Biochem. Biophys. 178 293–302.319757 [Google Scholar]
- Cook, P.F., Hara, S., Nalabolu, S., and Schnackerz, K.D. 1992. pH dependence of the absorbance and 31P NMR spectra of O-acetylserine sulfhydrylase in the absence and presence of O-acetyl-L-serine. Biochemistry 31 2298–2303. [DOI] [PubMed] [Google Scholar]
- Droux, M., Ruffet, M.L., Douce, R., and Job, D. 1998. Interactions between serine acetyltransferase and O-acetylserine (thiol) lyase in higher plants—Structural and kinetic properties of the free and bound enzymes. Eur. J. Biochem. 255 235–245. [DOI] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6 1948–1954. [DOI] [PubMed] [Google Scholar]
- Flavin, M. and Slaughter, C. 1965. Synthesis of the succinic ester of homoserine, a new intermediate in the bacterial biosynthesis of methionine. Biochemistry 4 1370–1375. [DOI] [PubMed] [Google Scholar]
- Gorman, J. and Shapiro, L. 2004. Structure of serine acetyltransferase from Haemophilus influenzae Rd. Acta Crystallogr. D Biol. Crystallogr. 60 1600–1605. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D.I., and Metoz, F. 1999. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 15 305–308. [DOI] [PubMed] [Google Scholar]
- Gratzer, W.B. 1989. Spectrophotometric determination of protein concentration in the short-wavelength ultraviolet. In Practical handbook of biochemistry and molecular biology (ed. G.D. Fasman), p. 567. CRC Press, Boca Raton, FL.
- Hara, S., Payne, M.A., Schnackerz, K.D., and Cook, P.F. 1990. A rapid purification procedure and computer-assisted sulfide ion selective electrode assay for O-acetylserine sulfhydrylase from Salmonella typhimurium. Protein Expr. Purif. 1 70–76. [DOI] [PubMed] [Google Scholar]
- Hell, R. and Hillebrand, H. 2001. Plant concepts for mineral acquisition and allocation. Curr. Opin. Biotechnol. 12 161–168. [DOI] [PubMed] [Google Scholar]
- Hell, R., Jost, R., Berkowitz, O., and Wirtz, M. 2002. Molecular and biochemical analysis of the enzymes of cysteine biosynthesis in the plant Arabidopsis thaliana. Amino Acids 22 245–257. [DOI] [PubMed] [Google Scholar]
- Hindson, V.J. 2003. Serine acetyltransferase of Escherichia coli: Substrate specificity and feedback control by cysteine. Biochem. J. 375 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson, V.J., Moody, P.C.E., Rowe, A.J., and Shaw, W.V. 2000. Serine acetyltransferase from Escherichia coli is a dimer of trimers. J. Biol. Chem. 275 461–466. [DOI] [PubMed] [Google Scholar]
- Huang, B., Vetting, M.W., and Roderick, S.L. 2005. The active site of O-acetylserine sulfhydrylase is the anchor point for bienzyme complex formation with serine acetyltransferase. J. Bacteriol. 187 3201–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulanicka, M.D., Garrett, C., Jagura-Burdzy, G., and Kredich, N.M. 1986. Cloning and characterization of the cysAMK region of Salmonella typhimurium. J. Bacteriol. 168 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, C.M., Huang, B., Roderick, S.L., and Cook, P.F. 2004. Kinetic mechanism of the serine acetyltransferase from Haemophilus influenzae. Arch. Biochem. Biophys. 429 115–122. [DOI] [PubMed] [Google Scholar]
- Jost, R., Berkowitz, O., Wirtz, M., Hopkins, L., Hawkesford, M.J., and Hell, R. 2000. Genomic and functional characterization of the oas gene family encoding O-acetylserine (thiol) lyases, enzymes catalyzing the final step in cysteine biosynthesis in Arabidopsis thaliana. Gene 253 237–247. [DOI] [PubMed] [Google Scholar]
- Kredich, N.M. 1992. The molecular basis for positive regulation of cys promoters in Salmonella typhimurium and Escherichia coli. Mol. Microbiol. 6 2747–2753. [DOI] [PubMed] [Google Scholar]
- Kredich, N.M. 1996. Biosynthesis of cysteine. ln Escherichia coli and Salmonella (ed. F.C. Neidhardt), pp. 514–527. ASM Press, Washington, DC.
- Kredich, N.M. and Tomkins, G.M. 1966. The enzymic synthesis of L-cysteine in Escherichia coli and Salmonella typhimurium. J. Biol. Chem. 241 4955–4965. [PubMed] [Google Scholar]
- ———. 1967. The biosynthesis of L-cysteine in Escherichia coli and Salmonella typhimurium by a multifunctional enzyme complex. In Organizational biosynthesis (eds. H.J. Vogel, J.O. Lampen, and V. Brysonk), pp. 189–198. Academic Press, New York.
- Kredich, N.M., Becker, M.A., and Tomkins, G.M. 1969. Purification and characterization of cysteine synthetase, a bifunctional protein complex, from Salmonella typhimurium. J. Biol. Chem. 244 2428–2439. [PubMed] [Google Scholar]
- Kuske, C.R., Ticknor, L.O., Guzman, E., Gurley, L.R., Valdez, J.G., Thompson, M.E., and Jackson, P.J. 1994. Purification and characterization of O-acetylserine sulfhydrylase isoenzymes from Datura innoxia. J. Biol. Chem. 269 6223–6232. [PubMed] [Google Scholar]
- McClure, Jr., G.D. and Cook, P.F. 1994. Product binding to the alpha-carboxyl subsite results in a conformational change at the active site of O-acetylserine sulfhydrylase-A: Evidence from fluorescence spectroscopy. Biochemistry 33 1674–1683. [DOI] [PubMed] [Google Scholar]
- Mino, K., Yamanoue, T., Sakiyama, T., Eisaki, N., Matsuyama, A., and Nakanishi, K. 1999. Purification and characterization of serine acetyltransferase from Escherichia coli partially truncated at the C-terminal region. Biosci. Biotechnol. Biochem. 63 168–179. [DOI] [PubMed] [Google Scholar]
- Mino, K., Hiraoka, K., Imamura, K., Sakiyama, T., Eisaki, N., Matsuyama, A., and Nakanishi, K. 2000a. Characteristics of serine acetyltransferase from Escherichia coli deleting different lengths of amino acid residues from the C-terminus. Biosci. Biotechnol. Biochem. 64 1874–1880. [DOI] [PubMed] [Google Scholar]
- Mino, K., Yamanoue, T., Sakiyama, T., Eisaki, N., Matsuyama, A., and Nakanishi, K. 2000b. Effects of bienzyme complex formation of cysteine synthetase from Escherichia coli on some properties and kinetics. Biosci. Biotechnol. Biochem. 64 1628–1640. [DOI] [PubMed] [Google Scholar]
- Mino, K., Imamura, K., Sakiyama, T., Eisaki, N., Matsuyama, A., and Nakanishi, K. 2001. Increase in the stability of serine acetyltransferase from Escherichia coli against cold inactivation and proteolysis by forming a bienzyme complex. Biosci. Biotechnol. Biochem. 65 865–874. [DOI] [PubMed] [Google Scholar]
- Noji, M., Inoue, K., Kimura, N., Gouda, A., and Saito, K. 1998. Isoform-dependent differences in feedback regulation and subcellular localization of serine acetyltransferase involved in cysteine biosynthesis from Arabidopsis thaliana. J. Biol. Chem. 273 32739–32745. [DOI] [PubMed] [Google Scholar]
- Olsen, L.R., Huang, B., Vetting, M.W., and Roderick, S.L. 2004. Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Biochemistry 43 6013–6019. [DOI] [PubMed] [Google Scholar]
- Peterson, E.A. and Sober, H.A. 1954. Preparation of crystalline phosphorylated derivatives of vitamin B6. J. Am. Chem. Soc. 76 169–175. [Google Scholar]
- Pye, V.E., Tingey, A.P., Robson, R.L., and Moody, P.C. 2004. The structure and mechanism of serine acetyltransferase from Escherichia coli. J. Biol. Chem. 279 40729–40736. [DOI] [PubMed] [Google Scholar]
- Rolland, N., Droux, M., Lebrun, M., and Douce, R. 1993. O-acetylserine (thiol)lyase from spinach (Spinacia oleracea L.) leaf: cDNA cloning, characterization, and overexpression in Escherichia coli of the chloroplast isoform. Arch. Biochem. Biophys. 300 213–222. [DOI] [PubMed] [Google Scholar]
- Rolland, N., Ruffet, M.L., Job, D., Douce, R., and Droux, M. 1996. Spinach chloroplast O-acetylserine (thiol)-lyase exhibits two catalytically non-equivalent pyridoxal-5′-phosphate-containing active sites. Eur. J. Biochem. 236 272–282. [DOI] [PubMed] [Google Scholar]
- Ruffet, M.L., Droux, M., and Douce, R. 1994. Purification and kinetic properties of serine acetyltransferase free of O-acetylserine(thiol)lyase from spinach chloroplasts. Plant Physiol. 104 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffet, M.L., Lebrun, M., Droux, M., and Douce, R. 1995. Subcellular distribution of serine acetyltransferase from Pisum sativum and characterization of an Arabidopsis thaliana putative cytosolic isoform. Eur. J. Biochem. 227 500–509. [DOI] [PubMed] [Google Scholar]
- Saito, K., Yokoyama, H., Noji, M., and Murakoshi, I. 1995. Molecular cloning and characterization of a plant serine acetyltransferase playing a regulatory role in cysteine biosynthesis from watermelon. J. Biol. Chem. 270 16321–16326. [DOI] [PubMed] [Google Scholar]
- Schnackerz, K.D., Tai, C.H., Simmons III, J.W., Jacobson, T.M., Rao, G.S., and Cook, P.F. 1995. Identification and spectral characterization of the external aldimine of the O-acetylserine sulfhydrylase reaction. Biochemistry 34 12152–12160. [DOI] [PubMed] [Google Scholar]
- Smith, I.K. and Thompson, J.F. 1971. Purification and characterization of L-serine transacetylase and O-acetyl-L-serine sulfhydrylase from kidney bean seedlings (Phaseolus vulgaris). Biochim. Biophys. Acta 227 288–295. [DOI] [PubMed] [Google Scholar]
- Strambini, G.B., Cioni, P., and Cook, P.F. 1996. Tryptophan luminescence as a probe of enzyme conformation along the O-acetylserine sulfhydrylase reaction pathway. Biochemistry 35 8392–8400. [DOI] [PubMed] [Google Scholar]
- Tai, C.H. and Cook, P.F. 2000. O-acetylserine sulfhydrylase. Adv. Enzymol. Relat. Areas Mol. Biol. 74 185–234. [DOI] [PubMed] [Google Scholar]
- Tai, C.H., Nalabolu, S.R., Jacobson, T.M., Minter, D.E., and Cook, P.F. 1993. Kinetic mechanisms of the A and B isozymes of O-acetylserine sulfhydrylase from Salmonella typhimurium LT-2 using the natural and alternative reactants. Biochemistry 32 6433–6442. [DOI] [PubMed] [Google Scholar]
- Tai, C.H., Burkhard, P., Gani, D., Jenn, T., Johnson, C., and Cook, P.F. 2001. Characterization of the allosteric anion-binding site of O-acetylserine sulfhydrylase. Biochemistry 40 7446–7452. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell, R., Verschueren, K.H., Dodson, E.J., Murshudov, G.N., Addy, C., and Wilkinson, A.J. 1997. The structure of the cofactor-binding fragment of the LysR family member, CysB: A familiar fold with a surprising subunit arrangement. Structure 5 1017–1032. [DOI] [PubMed] [Google Scholar]
- Wei, J., Tang, Q.X., Varlamova, O., Roche, C., Lee, R., and Leyh, T.S. 2002. Cysteine biosynthetic enzymes are the pieces of a metabolic energy pump. Biochemistry 41 8493–8498. [DOI] [PubMed] [Google Scholar]
- Wirtz, M., Berkowitz, O., Droux, M., and Hell, R. 2001. The cysteine synthase complex from plants. Mitochondrial serine acetyltransferase from Arabidopsis thaliana carries a bifunctional domain for catalysis and protein–protein interaction. Eur. J. Biochem. 268 686–693. [DOI] [PubMed] [Google Scholar]