

Abstract
Affibodies are a novel class of binding proteins selected from phagemid libraries of the Z domain from staphylococcal protein A. The ZSPA-1 affibody was selected as a binder to protein A, and it binds the parental Z domain with micromolar affinity. In earlier work we determined the structure of the Z:ZSPA-1 complex and noted that ZSPA-1 in the free state exhibits several properties characteristic of a molten globule. Here we present a more detailed biophysical investigation of ZSPA-1 and four ZSPA-1 mutants with the objective to understand these properties. The characterization includes thermal and chemical denaturation profiles, ANS binding assays, size exclusion chromatography, isothermal titration calorimetry, and an investigation of structure and dynamics by NMR. The NMR characterization of ZSPA-1 was facilitated by the finding that trimethylamine N-oxide (TMAO) stabilizes the molten globule conformation in favor of the fully unfolded state. All data taken together lead us to conclude the following: (1) The topology of the molten globule conformation of free ZSPA-1 is similar to that of the fully folded structure in the Z-bound state; (2) the extensive mutations in helices 1 and 2 destabilize these without affecting the intrinsic stability of helix 3; (3) stabilization and reduced aggregation can be achieved by replacing mutated residues in ZSPA-1 with the corresponding wild-type Z residues. This stabilization is better correlated to changes in helix propensity than to an expected increase in polar versus nonpolar surface area of the fully folded state.
Keywords: protein engineering, affibody, protein stability, osmolyte, NMR spectroscopy
Several biochemical and biotechnological applications rely on proteins with specific recognition properties. Antibodies have for a long time been, and are still in most cases, the obvious choice, but recent combinatorial approaches have resulted in several novel kinds of affinity proteins (Nygren and Uhlén 1997). One class of such specific binding proteins is called affibodies. These are based on the 58-residue scaffold of the Z domain—a one-domain staphylococcal protein A (SPA) analog (Nilsson et al. 1987). The Z domain possesses several properties suitable for an artificial binding protein: it is small, soluble, stable, and easily produced with common expression systems. Affibodies are selected from phagemid libraries of Z-domain constructs in which 13 solvent accessible residues are subjected to combinatorial mutagenesis (Nord et al. 1997). The residues to be randomized were chosen based on the interaction surface for the Z-domain interaction with the Fc fragment from IgG (Deisenhofer 1981). Specific binders are then obtained by phage-display screening against the desired target protein. The methodology has resulted in the identification of several affibodies with high specificity for the applied panning target (Hansson et al. 1999; Nord et al. 2001; Rönnmark et al. 2002; Sandström et al. 2003).
We have previously determined the solution structure of the complex between the ZSPA-1 affibody, which was selected using SPA as a panning target (Eklund et al. 2002), and the Z domain (Wahlberg et al. 2003). During this work we also realized that the free state of ZSPA-1 is more dynamic than expected and that it shows many of the characteristics of a molten globule: The 1H–15N heteronuclear single quantum correlation (15N-HSQC) NMR spectrum displays poor resonance dispersion and substantial line-broadening; the far-UV circular dichroism (CD) spectrum reveals a decreased helix content compared with the Z domain; the thermal melting profile shows a lower melting temperature and less cooperative melting; and there is binding and enhanced fluorescence of the hydrophobic dye 8-anilino-1-naphtalenesulfonic acid (ANS) in the presence of ZSPA-1. The term “molten globule” is here used as a description of a flexible state with nativelike secondary structure but less defined tertiary structure (Dobson 1994; Ptitsyn 1995; Redfield 1999) without considering if it is a folding intermediate or not. We also noted, from the concentration dependence of the CD spectrum and based on size exclusion chromatography (SEC) experiments, that some kind of self-association might be involved when the protein concentration is increased. Nevertheless, the ZSPA-1 affibody binds its target with micromolar affinity and has shown to be very useful in several biotechnological applications (Gräslund et al. 2002; Falk et al. 2003; Eklund et al. 2004).
Because the binding process forces ZSPA-1 to adopt an energetically less favorable state, the coupled folding and binding mechanism presumably means that some of the interaction energy gained upon complex formation is lost. This would not be the case for an affibody with a stably folded scaffold. We present in this study a more detailed biophysical investigation of ZSPA-1 with the objective to, on a molecular level, understand how and why the mutations have caused the very different behavior of this affibody compared with the parental Z domain. We have also characterized four ZSPA-1 mutants made by changing certain residues back to the amino acids of the wild-type Z domain. The mutants were chosen primarily to reduce the exposed hydrophobic surface of the folded state. The V11N mutation was chosen because the V11 side chain does not seem to be involved in the Z interaction (Wahlberg et al. 2003), and the F32Q, W35K, and the double (F32Q/W35K) mutations were made because the NMR characterization of ZSPA-1 indicated increased dynamics in that region.
Working with marginally stable proteins increases the demand for finding the right experimental conditions. Several reports have shown that certain small, naturally occurring, organic compounds, called osmolytes, are able to shift the folding equilibrium in favor of the folded state (Baskakov and Bolen 1998; Bolen and Baskakov 2001; Celinski and Scholtz 2002). Here, we have made use of trimethyl-amine N-oxide (TMAO) to facilitate the NMR analysis and the investigation of the dynamics in ZSPA-1.
Results
We present results consistent with the model shown in Scheme 1 ▶.
Scheme 1.

The dominant species at normal conditions is the molten globule (MG) state. The compact folded state is only stable when ZSPA-1 is bound to its target protein. It is possible that it is present also without the binding partner, but then it is energetically equal to other conformations in the MG state. The unfolded state is favored by increased temperature or denaturant concentration, whereas addition of TMAO promotes the MG state. Increased protein concentration enhances the formation of a soluble but aggregated state, which is accompanied by loss of helical structure.
Stability
A common feature of molten globules is unfolding with reduced cooperativity (Schulman et al. 1997). We tested this possibility by monitoring the stability of ZSPA-1 against chemical denaturation by guanidine hydrochloride (GuHCl; Fig. 1A,B ▶). We observe slightly different behavior depending on the method that is used to monitor the process. Using the mean residue ellipticity (measured by CD at 222 nm), the denaturant concentration at which 50% of the protein is unfolded (D50%) is 0.9 M, whereas tryptophan fluorescence shows D50% for 0.6 M GuHCl (Fig. 1A ▶). This means that unfolding by chemical denaturation is not a two-state process and that one or more additional state(s) are present during the unfolding pathway. The CD signal is retained at higher denaturant concentrations than the tryptophan fluorescence, suggesting that the intermediate state(s) retains secondary structure while losing its tertiary structure. This is consistent with the molten globule character of the protein. Considering the small difference between the two unfolding curves, and that the tryptophan is not located in the core of the protein and therefore is not a sensitive probe of unfolding of, for instance, residual secondary structure, we believe that we are looking at a noncooperative folding process rather than the accumulation of stable intermediates.
Figure 1.
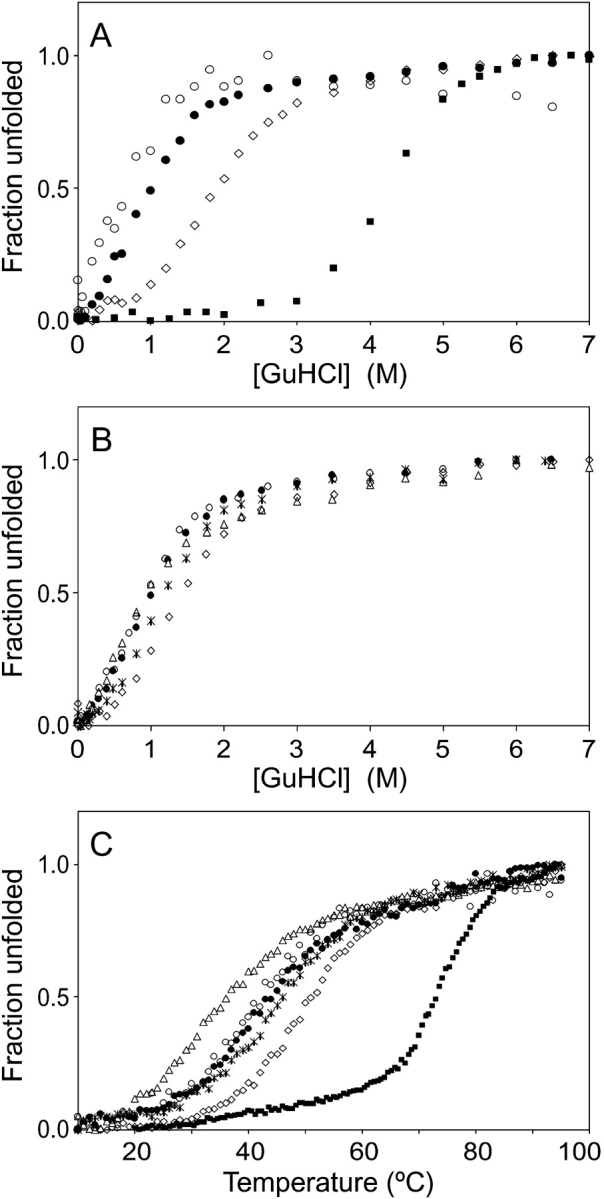
Stability measurements of the Z domain, ZSPA-1, and the ZSPA-1 mutants. (A) Chemical denaturation of the Z domain (squares) and ZSPA-1 (solid circles) monitored by CD at 222 nm and of ZSPA-1 (open circles) monitored by fluorescence at 350 nm. (Diamonds) Denaturation of ZSPA-1 in the presence of 1 M TMAO monitored by CD. (B) Chemical denaturation of ZSPA-1 (solid circles), F32Q (diamonds), W35K (triangles), F32Q/W35K (stars), and V11N (open circles) monitored by CD at 222 nm. (C) Thermal denaturation of the Z domain (squares), ZSPA-1 (solid circles), F32Q (diamonds), W35K (triangles), F32Q/W35K (stars), and V11N (open circles) monitored by CD at 208 nm.
The chemical denaturation experiments confirm the decreased stability of ZSPA-1 compared with the Z domain as previously seen by thermal melting (Fig. 1C ▶; Wahlberg et al. 2003). The D50% point occurs at four times higher GuHCl concentration for the Z domain than for ZSPA-1 (Fig. 1A ▶). The reduced helix content in ZSPA-1 compared with the Z domain (mean residue ellipticity of −22,000 deg cm−1 dmole−1 compared with −30,000 deg cm−1 dmole−1 at 208 nm) can be explained either by a permanent loss in helicity or by transient helix unfolding in the MG state. We prefer the latter explanation because we see strong signs of all three helices being present, as discussed below. The addition of 1 M TMAO results in a stabilization of ZSPA-1 (Fig. 1A ▶).
Self-association
In concentrations approaching the millimolar range, the CD spectrum of ZSPA-1 changes dramatically: the amplitude of the signal decreases and the minimum at 208 nm disappears (Wahlberg et al. 2003). The concentration dependence indicates self-association during which ZSPA-1 loses helicity. SEC contributes further evidence for the presence of oligomeric states. Loaded at 2 mM concentration, ZSPA-1 elutes at a smaller elution volume than expected and with a highly asymmetric peak (Fig. 2A ▶); both features are characteristic for self-associating proteins (see, e.g., Hansson et al. 2001). The decreased elution volume means that the average molecular size is larger, whereas the tailing indicates that there is a rapid equilibrium between species of different size. The SEC experiment performed at 0.1 mM protein shows that both these features are strongly concentration-dependent, a fact that further strengthens the aggregation hypothesis. We also note that the addition of TMAO does not change this behavior, even though TMAO shifts the unfolding equilibrium in favor of the MG state. From these data we conclude that the MG state of ZSPA-1 is involved in the aggregation equilibrium (Scheme 1 ▶), but it is not possible to exclude that the denatured state is also involved.
Figure 2.

Size exclusion chromatograms of (A) Z domain (solid squares), Z domain together with 13.7-kDa ribonuclease A (open squares), ZSPA-1 loaded at 2 mM (solid circles), and ZSPA-1 loaded at 0.1 mM (open circles) and (B) the ZSPA-1 mutants V11N (open circles), F32Q (diamonds), W35K (triangles), and F32Q/W35K (stars) loaded at 1 mM. The protein absorbance was monitored at 215 nm (W35K and F32Q/W35K) or at 280 nm (the other proteins).
Structural characterization of the molten globule
Searching for conditions more suitable for NMR studies, we found that addition of 1 M to 1.6 M of TMAO greatly improved the appearance of the 15N-HSQC NMR spectrum (Fig. 3A,C ▶). This is primarily seen as sharper resonances while the dispersion does not change. The improvement achieved by the osmolyte made it possible to acquire NOESY, TOCSY, and triple resonance experiments and, at least partially, assign the 15N-HSQC of ZSPA-1. Assignments could be made for all expected backbone amide resonances except K7, E8, L9, R14, I31, F32, and W35. Assignment of the chemical shifts for the backbone atoms (N, HN, Cα, Hα, and C′) also provided the secondary shifts, which are good indicators of secondary structure (Fig. 4A ▶; Wishart and Sykes 1994). The results are consistent with all three helices being present in the MG state, and even though there are some gaps in the assigned sequence, there are no data contradicting this conclusion. The presence of the helices is further strengthened by observation of the characteristic nuclear Overhauser effect (NOE) patterns of helical secondary structure. The NOESY spectrum was also searched for long-range NOEs present in the Z-bound state of ZSPA-1, and several such NOEs were identified. The topology of ZSPA-1 in the MG state is therefore similar to the structure of ZSPA-1 determined in complex with the Z domain. However, it is not possible to conclude if the NOE connectivities originate from a single conformation or if they represent the average of several rapidly interconverting conformations.
Figure 3.

15N-HSQC NMR spectra for (A) ZSPA-1, (B) F32Q, (C) ZSPA-1 in the presence of 1 M TMAO and (D) W35K. All spectra were recorded at 30°C in 20 mM potassium phosphate buffer (pH 5.6).
Figure 4.
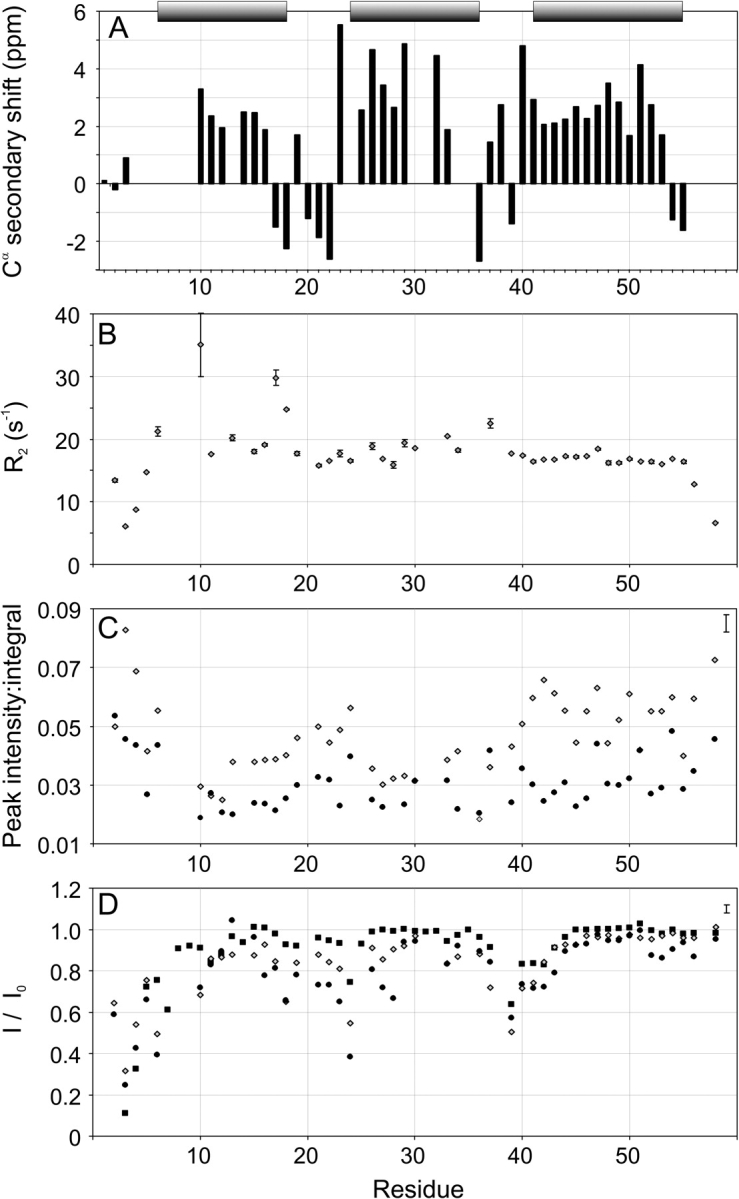
NMR data for ZSPA-1. (A) Secondary Cα chemical shifts (Wishart and Sykes 1994) of ZSPA-1 in 1.6 M TMAO. The bars indicate the positions of the helices in the Z-bound ZSPA-1 structure (Wahlberg et al. 2003). (B) Transverse relaxation rates (R2) of ZSPA-1 in 1.6 M TMAO. (C) Peak intensity:integral ratios of resonances in the 15N-HSQC spectrum of ZSPA-1 with (diamonds) and without (circles) 1 M TMAO. (D) Saturation transfer protection of the Z domain (squares) and ZSPA-1 with (diamonds) and without (circles) 1 M TMAO. Typical error bars for data in C and D are shown in the upper right corners.
Dynamics
Assignment of the 15N-HSQC spectrum allowed us to investigate the backbone dynamics on different time scales (Farrow et al. 1994; Palmer 2001). The results are summarized in Figure 4, B–D ▶. The most obvious manifestation of dynamics is the broadening of several resonances, indicating conformational interconversion on the microsecond-to-millisecond time scales. A comparison of the peak intensity and integral of the HSQC resonances (Fig. 4C ▶) shows that backbone resonances from helices 1 and 2 experience the largest broadening. The linewidth data were complemented with 15N transverse relaxation (R2) measurements (Fig. 4B ▶). The variation of R2 values along the peptide backbone is consistent with increased dynamics in helices 1 and 2 as compared with helix 3. In particular, S10, V17, and T18 in helix 1 show very high relaxation rates. High R2 values can also be expected for other resonances in helices 1 and 2 for which R2 could not be measured because of poor signal-to-noise ratios. The overall R2 level appears to be twice as high for ZSPA-1 compared with the Z domain (data not shown). This is most likely an effect of averaging of oligomeric states.
We observe a decrease in backbone amide hydrogen protection in saturation transfer experiments, which report on dynamics on the millisecond-to-second time scales, in helices 1 and 2 (Fig. 4D ▶). The saturation transfer experiments also show that the overall backbone amide protection is slightly lower in ZSPA-1 than it is in the Z domain. Steady-state 1H–15N NOE data (data not shown) do not give any indication of excessive dynamics in ZSPA-1 on the picosecond-to-nanosecond time scales, except for the dynamics in N- and C-terminal regions that are observed in most proteins.
The role of TMAO
According to the effects of osmolytes on protein structure and stability, presented, for instance, in Bolen and Baskakov (2001), it should be possible to study ZSPA-1 in the presence of TMAO without affecting structural properties of the stabilized state. We have made use of this to investigate the properties of the MG state. However, to confirm that the osmolyte shifts the folding equilibrium but does not change the structure, we compared data for ZSPA-1 with and without TMAO. For the NMR data this was possible because the 15N-HSQC of ZSPA-1 (without TMAO) could be assigned from a series of experiments with gradually decreased TMAO concentration. The chemical shifts are almost identical with and without TMAO, indicating that the chemical environment of the amide resonances has not changed. The line broadening is increased (Fig. 4C ▶), and the protection of amide hydrogens is decreased (Fig. 4D ▶) by reduced TMAO concentration. These results agree well with a higher fraction denatured protein present in the sample at lower TMAO concentrations. We also notice that ZSPA-1 shows similar ANS-binding and SEC behavior with and without TMAO. The CD spectrum shows a small increase in signal amplitude and denaturation profiles indicate a stabilization with TMAO present (Fig. 1A ▶). Both these results are consistent with an increased population of the MG state.
Binding and stability of the mutants
To investigate the effect of certain side-chain replacements in the selected ZSPA-1 affibody, we made four mutants of ZSPA-1 in which these residues were replaced with those of the wild-type Z domain. The mutants were characterized by a standard set of experiments, consisting of the far-UV CD spectrum, thermal and chemical denaturation, ANS-binding, SEC, the 15N-HSQC NMR spectrum, and Z-domain binding investigated by isothermal titration calorimetry (ITC).
Thermal and chemical denaturation experiments show that the F32Q mutation makes ZSPA-1 more stable, whereas the W35K and V11N mutations decrease the stability (Fig. 1B,C ▶). The stability of the F32Q/W35K double mutant is intermediate to those of F32Q and W35K. The differences in stability are small; all D50% values for chemical denaturation fall within a range of 0.6 M. The denaturation profiles have the same appearance as that of ZSPA-1, indicating that the folding process is not fully cooperative.
The CD spectra (data not shown) of the mutants are almost identical to that of ZSPA-1, except for W35K, which is already partially unfolded at 20°C. ANS binding experiments (Fig. 5 ▶) show decreased ANS fluorescence compared with ZSPA-1 for all the mutants except V11N, indicating that the structures of the folded (MG) states are more compact than for ZSPA-1.
Figure 5.

Fluorescence of ANS (dotted line) and ANS in samples containing the Z domain (squares), ZSPA-1 (solid circles), F32Q (diamonds), W35K (triangles), F32Q/W35K (stars), and V11N (open circles).
The SEC data for the F32 and W35 mutants suggest that these proteins do not aggregate (Fig. 2C ▶); the peaks are symmetric, and the elution volumes are not concentration-dependent. The shifts in elution volume compared with the Z domain can be explained by the rapid unfolding equilibrium, which makes the average size of the proteins slightly larger. The V11N mutant behaves almost identical to ZSPA-1 in SEC experiments.
The trend seen in the unfolding experiments is confirmed by 15N-HSQC spectra; whereas F32Q looks more structured than ZSPA-1 (Fig. 3B ▶), the W35K spectrum displays many of the characteristics of an unfolded protein (Fig. 3D ▶). The spectrum of the F32Q/W35K double mutant (data not shown) is similar to that of F32Q, which confirms that this mutation is able to confer an increased structural stability.
ITC measurements (Fig. 6 ▶) were used to determine a dissociation constant (Kd) of 1.8 μM for the ZSPA-1:Z do-main interaction. All four mutants were found to bind with a lower affinity. The stability data for the F32Q mutant taken together give the impression that the affibody can be stabilized by this mutation. However, the mutation also reduces the binding affinity for the Z domain by a factor of 10 (Fig. 6 ▶). The V11N mutation causes a smaller, but still significant, twofold decrease in binding affinity. The W35K and the F32Q/W35K double mutant do not show any Z-domain binding at all, and this illustrates the importance of the tryptophan for the interaction (Wahlberg et al. 2003).
Figure 6.

ITC measurements of the thermodynamics of the Z:affibody complex formation for ZSPA-1 (solid squares), V11N (diamonds), and F32Q (triangles) and the reference experiment of Z domain titrated into buffer (open squares). Values of Kd obtained from the fitted data are 1.8 μM (ZSPA-1), 4.6 μM (V11N), and 14.3 μM (F32Q).
Discussion
Why does ZSPA-1 behave like a molten globule?
The extensive mutations made to create ZSPA-1 have changed two major properties of the three-helix bundle. First, there is no longer a unique stably folded low-energy conformation, and second, the free energy difference between the folded (MG) and unfolded states is decreased. We here attempt to explain these effects on a molecular level.
The structural characterization of ZSPA-1 shows that the topology of the MG state is similar to the Z-domain-bound state and that the correct core packing is, at least transiently, present. Still, the NMR spectrum suffers from broadening, indicating interconversions between several conformations, the CD spectrum shows a lower degree of helicity than in the Z domain, and the protein binds ANS. This behavior agrees well with a so-called highly ordered molten globule (Dobson 1994; Redfield et al. 1994). The 13 mutations have thus not caused any extensive structural rearrangements, just minor disturbances. Our results show that the altered properties are located to helices 1 and 2 while the third helix is not affected. This might seem obvious, but there was an evident possibility that any disturbance in the mutated helices also would destabilize helix 3. Compiling some of the dynamics data into one figure (Fig. 7 ▶), we obtain a picture suggesting that the increased mobility is localized to the beginning of helix 1 and around residues F32 and W35 in helix 2. The increased flexibility in the helices also affects the stability of the core packing, making the whole protein more dynamic. TMAO acts, as in many other cases, to promote the ”folded” state—in our case this is the molten globule ensemble—but is not able to stabilize a completely folded protein.
Figure 7.
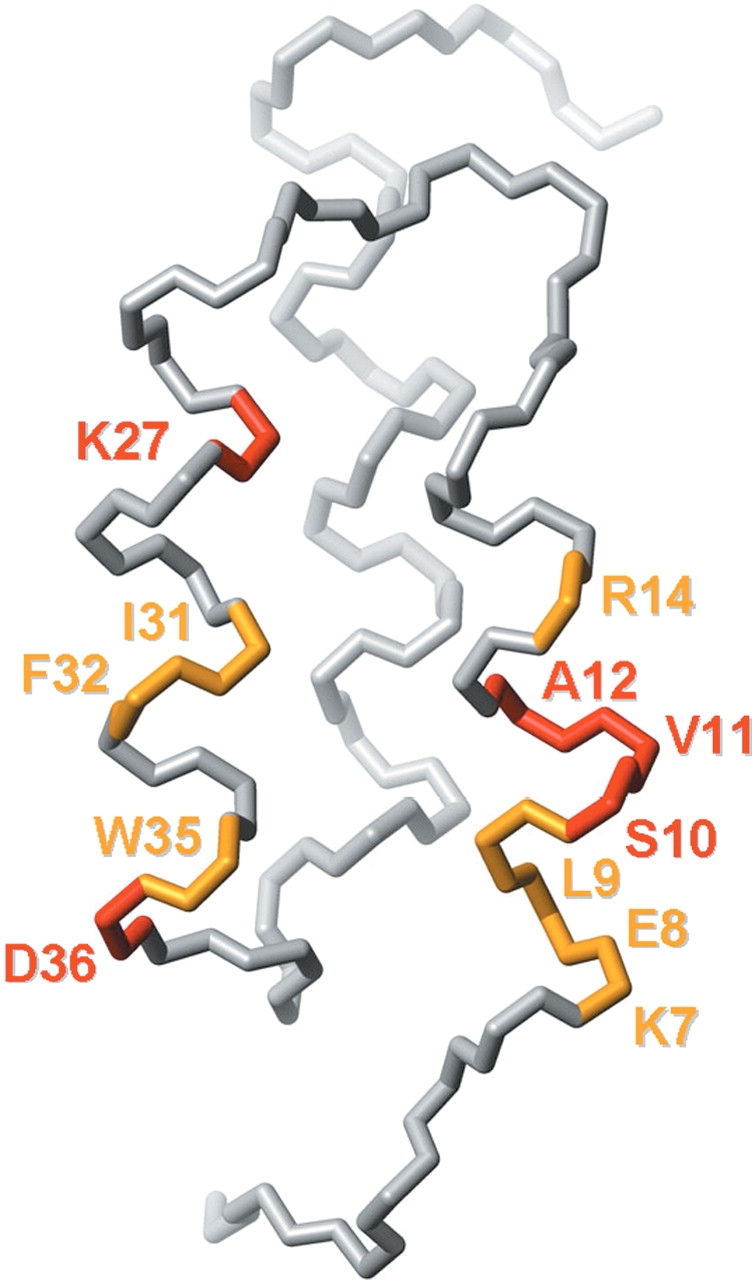
Backbone trace illustrating dynamic parts of ZSPA-1. Residues colored in yellow could not be assigned in the 15N-HSQC of ZSPA-1 in 1 M or 1.6 M TMAO. Residues colored in red have a peak intensity:integral ratio of <0.03 in 1 M TMAO. The figure was created using MOLMOL (Koradi et al. 1996).
Effect of the mutations
The structure of ZSPA-1 in complex with the Z domain suggests that the polar:nonpolar surface area ratio of 36%:64% can be unfavorable for folding because the average ratio in soluble globular proteins is 43%:57% (Miller et al. 1987). The V11N mutant was made to test if it is enough to reduce the amount of exposed hydrophobic surface to increase the stability. Because the NMR structure (Wahlberg et al. 2003) indicated that this residue was not directly involved in the Z-domain interaction, a mutation back to the original asparagine should reduce the exposed hydrophobic surface but not affect the binding. Our results show that this is not the case. No significant increase in stability is seen, and the binding, in fact, becomes weaker. This implies either that the V11 side chain somehow is “selected” as important for the binding, even if it is not in contact with the target in the fully formed complex, or that an asparagine in this position is exceptionally unfavorable for the interaction.
The NMR characterization of ZSPA-1 focused our interest on the large aromatic side chains in helix 2. F32 and W35 seem to be very important for the interaction but could also destabilize the free protein because they are exposed at the surface. Three mutants were made to investigate the role of these residues: F32Q, W35K, and the double mutant F32Q/ W35K. The binding studies confirm our conclusions: the Z-domain affinity is seriously reduced for the F32Q mutant and not detectable for the other two. Surprisingly, we find that the substitution of W35 with the lysine present in the wild-type Z domain does not stabilize the affibody. This indicates that some of the other mutations made in ZSPA-1 are not compatible with the lysine side chain in this position. For example, the distance between this lysine and the positively charged R14 could theoretically be as short as 3.5 Å. F32Q seems to stabilize the affibody in several aspects, even together with W35K. One can suspect that the F32–K35 interaction is responsible for the decreased stability of W35K, because F32 is the closest mutated neighbor to W35, but then one should expect to see some synergistic effects for the double mutant (i.e., a greatly improved stability for both removing F32 and the Phe–Lys interaction). Instead, it seems as if the double mutant assumes properties that are the average of those of F32Q and W35K.
Exposed hydrophobic surface versus helix propensity
The mutants were designed based on the hypothesis that the introduction of hydrophobic side chains on the exposed surface of ZSPA-1 is the primary reason for the decreased stability. Our results, however, indicate a more intricate picture, because we have seen that just removing exposed hydrophobic surface does not change the stability and behavior, as in the V11N mutant. We then subjected the sequences of ZSPA-1, the Z domain, and the ZSPA-1 mutants to several secondary-structure prediction routines (Combet et al. 2000) and these reveal a pattern in which the key issue seems to be the helix propensity of the sequence (Fig. 8 ▶). While the helices in the Z domain are predicted by all methods, the results for ZSPA-1 are more divergent. The programs seem to suggest extended strand structures for both helix 1 and helix 2, which actually is the predominant secondary structure in aggregated proteins (Yang et al. 2003). Whether this suggestion is true or not, it is obvious that the mutations introduced in the three-helix bundle decrease the helix propensity.
Figure 8.

Secondary structure prediction for the Z domain, ZSPA-1, and the ZSPA-1 mutants using the secondary consensus prediction method (Combet et al. 2000); h, α-helix; e, extended strand; c, random coil; ?, ambiguous state. The nature of the residues (open circles for polar and solid circles for nonpolar) according to Kyte and Doolittle (1982) are indicated above and below the sequences of the Z domain and ZSPA-1, respectively. Underlined residues indicate helices in the structure of the Z:ZSPA-1 complex.
Secondary-structure prediction data for the ZSPA-1 mutants are also consistent with our results. The helix propensity of the V11N mutant is very similar to that of ZSPA-1, and it shows almost identical stability. The F32Q and the double mutant (F32Q/W35K) seem to greatly improve the propensity for formation of helix 2, and our experiments confirm an increased stability. Comparing the change in hydrophobicity (Kyte and Doolittle 1982) further strengthens the argument: the decrease in hydrophobicity is slightly larger for V11N than for F32Q, but for the amount of stabilization it is the other way around. If the hydrophobicity were the key issue, then F32Q/W35K should be the most stable mutant, but this is obviously not the case.
Several articles have suggested that certain binary patterns of polar and nonpolar amino acids specify α-helical regions in proteins (Kamtekar et al. 1993; Xiong et al. 1995; Wei et al. 2003). Applying this classification on the residues in the Z domain and ZSPA-1 (Fig. 8 ▶) makes it easier to understand how the mutations affect the helix propensity. F32Q shortens the four-residue-long hydrophobic sequence (29–32) and makes it more suitable for α-helix formation. V11N, on the other hand, does not significantly change the pattern, and W35K does not even change the classification of that residue. These observations suggest that changes in secondary structure propensity have a larger effect on stability than surface hydrophobicity.
Implications for protein engineering
Already initially, we suspected that it would be difficult to simultaneously optimize ZSPA-1 for both stability and high Z-domain affinity, and this fact becomes very obvious from the characterization of the ZSPA-1 mutants. The mechanism by which ZSPA-1 recognizes its target is apparently complex, as evidenced by the behavior of the V11N mutant. There is also a possibility that some flexibility is needed for ZSPA-1 to find the right position in the complex. Induced fit is seen for the Z domain upon complex formation (Wahlberg et al. 2003), and it is reasonable to suspect that ZSPA-1 must experience a similar rearrangement. A stabilized structure might in that case reduce the affinity for the target. It is also important to note that not only is strong affinity required of the affibody, but that specificity is maybe even more important. Considering this together with our results, flexibility seems to play an important role in how ZSPA-1 recognizes its target. Whether or not this is a general property of affibodies remains to be investigated.
This leads us to the important question: how representative is ZSPA-1 as an affibody? The size of the phage-display library from which it was selected was small (4 × 107) compared with the total number of possibilities (~1017). Even though the Z:ZSPA-1 binding is not weak, affibodies with much lower dissociation constants (Kd) have been found for other targets (Gunneriusson et al. 1999; Nord et al. 2001). In addition, the selection of affibodies toward domains of protein A, corresponding to their ancestral scaffold structure, could involve restrictions regarding addressable epitopes of the target protein (Eklund et al. 2002). These facts suggest that the ZSPA-1 affibody might not be very representative for affibodies in general. Nevertheless, it still provides an interesting system for studying protein stability and molecular recognition by coupled folding.
We have in this work shown that to obtain an increased stability for ZSPA-1, it is not sufficient to consider the amount of exposed hydrophobic surface. Rather, our data support that the overall stability of the ZSPA-1 affibody is related to the helix propensities of the introduced substitutions. Considering these results, it appears that either rational design on a very detailed level or further selections from a secondary phage-display library are needed to obtain a completely optimized variant of the ZSPA-1 affibody.
Materials and methods
Protein preparation
Samples of the Z domain and ZSPA-1 were prepared as described in Lendel et al. (2002). The mutations were introduced in the ZSPA-1-containing pET28a+ vectors using the Quickchange Mutagenesis Kit (Stratagene) and oligonucleotides from Invitrogen. The proteins were produced and purified in the same way as the Z domain and ZSPA-1 with exception for the mutants without any SPA binding (W35K and F32Q/W35K). For those, the affinity purification step was replaced with salt precipitation: Ammonium sulfate was added to the cell lysate (in 25 mM Tris-HCl at pH 8.0, 1 mM EDTA, 200 mM NaCl) until 55% saturation in the first step and 80% saturation in the second step, and each step was followed by centrifugation. The recovered pellet from the second step was resolved in the buffer used for the following gel filtration step (Lendel et al. 2002).
Concentration determination for the mutants without any tryptophan residues (W35K and W35K/F32Q) was performed by combining the peptide backbone absorbance at 190 nm and intensity measurements of Coomassie-stained SDS gels. For the absorbance measurements, an extinction coefficient of 1 × 104 M−1 cm−1 per residue was used. The SDS images were analyzed with software from BioRad, and the concentrations were measured relative to a ZSPA-1 sample of known concentration.
Optical spectroscopy
CD was measured on a JASCO J-810 spectropolarimeter equipped with a Peltier temperature control system. Denaturation studies were performed in the temperature region 10°–95°C using samples containing 15 μM to 30 μM protein in 20 mM potassium phosphate buffer (pH 5.6). Chemical denaturation experiments were performed at 20°C with GuHCl concentrations ranging from 0 M to 7 M and the same protein concentrations and buffer.
Tryptophan and ANS fluorescence was monitored in 20 mM potassium phosphate buffer (pH 5.6), at room temperature using a CARY Eclipse fluorescence spectrometer (Varian). Tryptophan was excited at 280 nm, and emission was monitored at 350 nm. ANS was excited at 350 nm, and emission spectra were recorded for 400–650 nm. Protein concentrations were 40 μM for the ANS experiments and 20 μM for the unfolding experiments. The ANS (Sigma) concentration was 50 μM.
The fraction of unfolded protein was calculated as (S − Smin)/(Smax − Smin), where S is the measured signal (CD or fluorescence) and Smax and Smin are the maximum and minimum signals in the experimental series.
NMR
NMR samples of ZSPA-1 typically contained 1 mM 15N- or 13C,15N-labeled protein in 20 mM potassium phosphate buffer (pH 5.6) with 10% D2O and 0 M to 1.6 M TMAO. Data were acquired at 30°C on Bruker Avance 500 MHz and 600 MHz spectrometers. Spectral widths for 15N-HSQC experiments used for the dynamics measurements were between 7000 and 8000 Hz for 1H and 2200 Hz for 15N, with data set sizes of 2048 × 256 complex data points and 8 or 16 scans per t1 point.
For the assignment of ZSPA-1 in TMAO we used 15N-TOCSY, 15N-NOESY, HNCA, HNCO, 15N-HSQC, and 13C-HSQC experiments (Cavanagh et al. 1996). A titration from 1.6 M down to 0 M TMAO was used to transfer the 15N-HSQC assignments to the spectrum of ZSPA-1 in the absence of TMAO. Data were processed with NMRPIPE (Delaglio et al. 1995) and analyzed using Ansig for Windows (Helgstrand et al. 2000).
Saturation transfer from solvent to the protein amide hydrogens (Forsén and Hoffman 1963) was measured at 600 MHz using 25-Hz presaturation of the water signal during 1.3 sec prior to the excitation pulse of the 15N-HSQC sequence (Skelton et al. 1992). The results were compared with a reference experiment acquired without presaturation. Resonance peaks were integrated using built-in XWINNMR (Bruker) routines, and the ratio between the peak integrals of the experiments recorded with and without pre-saturation was calculated.
Qualitative measurements of line broadening were obtained by dividing the maximum peak intensities in the 15N-HSQC spectrum with the peak volumes. These operations were performed using Ansig for Windows.
The 15N relaxation data were measured on the 600-MHz spectrometer, and XWINNMR was used for data processing. The measurements were performed using CPMG (R2) and steady-state {1H}–15N NOE experiments as described in Farrow et al. (1994). The steady-state NOE experiment was recorded with a 5-sec 1H saturation pulse train, and a 5-sec recycle delay was used in the control experiment. These experiments were recorded, interleaved, and repeated once. The CPMG delay in the R2 experiments, that is, the time between refocusing pulses, was 450 μsec with sampling times T of 16.0, 32.0, 47.9, 63.9, 79.9, 95.9, 111.9, and 159.8 msec. One data set (T = 47.9 msec) was duplicated. The recycle delay was 1.7 sec. Resonances were integrated using built-in XWINNMR routines. The R2 data were fitted to A • exp(−R2 • t) using Matlab (Math Works Inc.). Experimental errors due to random noise were obtained from duplicate experiments and error propagation simulated using the Monte Carlo method. The errors in the steady-state NOE experiment were obtained from duplicate experiments.
ITC
ITC was performed with a MCS-ITC titration microcalorimeter (MicroCal Inc.). All solutions were degassed before the experiments. Protein samples used in each experiment were dialyzed against the same batch of buffer (20 mM potassium phosphate at pH 5.7) to minimize artifacts due to differences in buffer composition. In a typical experiment, Z domain was titrated into ZSPA-1 or mutant protein in the cell using a 250-μL syringe. Each titration consisted of a 2-μL preliminary injection followed by 25 subsequent 10-μL injections. Measurements were performed at 30°C. Concentrations of the Z domain were 300 μM and 380 μM when titrated into 30 μM of ZSPA-1 or 32 μM of V11N solutions, respectively. And 800 μM of the Z domain was used for titration into 55 μM of F32Q. Data were corrected for the small heat effects observed in control injections of Z domain into buffer. To obtain dissociation constants, binding isotherms were fitted to a single set of identical sites model in the Origin software (MicroCal Inc.).
SEC
Analytical SEC was performed using a Hiload Superdex 30 16/60 column together with an ÄKTA purifier system (Amersham Biosciences). The flow was 1 mL/min, and the absorbance was monitored at 280 nm, or 215 nm for proteins without any tryptophan residues. Samples of 1–2 mL were loaded and eluted with a 20 mM sodium phosphate, 150 mM sodium chloride buffer (pH 7.2). Experiments were also performed in the presence of 1.5 M TMAO. Ribonuclease A (Amersham Biosciences) was used as a molecular weight standard (13.7 kDa).
Acknowledgments
This work was supported by the Swedish Research Council (Vetenskapsrådet) and by the Knut and Alice Wallenberg Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
15N-HSQC, 1H-15N heteronuclear single quantum correlation
ANS, 8-anilino-1-naphtalenesulfonic acid
CD, circular dichroism
D50%, denaturant concentration at which 50% of the protein is unfolded
GuHCl, guanidine hydrochloride
ITC, isothermal titration calorimetry
NMR, nuclear magnetic resonance spectroscopy
NOE, nuclear Over-hauser effect
SEC, size exclusion chromatography
SPA, staphylococcal protein A
TMAO, trimethylamine N-oxide.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04728604.
References
- Baskakov, I.V. and Bolen, D.W. 1998. Forcing thermodynamically unfolded proteins to fold. J. Biol. Chem. 273 4831–4834. [DOI] [PubMed] [Google Scholar]
- Bolen, D.W. and Baskakov, I.V. 2001. The osmophobic effect: Natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 310 955–963. [DOI] [PubMed] [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer III, A.G., and Skelton, N.J. 1996. Protein NMR spectroscopy: Principles and practice. Academic Press, New York.
- Celinski, S.A. and Scholtz, J.M. 2002. Osmolyte effects on helix formation in peptides and stability of coiled-coils. Protein Sci. 11 2048–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combet, C., Blanchet, C., Geourjon, C., and Deléage, G. 2000. NPS@: Network protein sequence analysis. TIBS 25 147–150. [DOI] [PubMed] [Google Scholar]
- Deisenhofer, J. 1981. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphyloccocus aureus at 2.9- and 2.8-Å resolution. Biochemistry 20 2361–2370. [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRpipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M. 1994. Solid evidence for molten globules. Curr. Biol. 4 636–640. [DOI] [PubMed] [Google Scholar]
- Eklund, M., Axelsson, L., Uhlén, M., and Nygren, P.-Å. 2002. Anti-idiotypic protein domains selected from protein A-based affibody libraries. Proteins 48 454–462. [DOI] [PubMed] [Google Scholar]
- Eklund, M., Sandström, K., Teeri, T.T., and Nygren, P.-Å. 2004. Site-specific and reversible anchoring of active proteins onto cellulose using a cellulosome-like complex. J. Biotechnol. 109 277–286. [DOI] [PubMed] [Google Scholar]
- Falk, R., Agaton, C., Kiesler, E., Jin, S., Wieslander, L., Visa, N., Hober, S., and Ståhl, S. 2003. An improved dual-expression concept, generating high-quality antibodies for proteomics research. Biotechnol. Appl. Biochem. 38 231–239. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Forsén, S. and Hoffman, R.A. 1963. A new method for the study of moderately rapid chemical exchange rates employing nuclear magnetic double resonance. Acta Chem. Scand. 17 1787–1788. [Google Scholar]
- Gräslund, S., Eklund, M., Falk, R., Uhlén, M., Nygren, P.-Å., and Ståhl, S. 2002. A novel affinity gene fusion system allowing protein A-based recovery of non-immunoglobulin gene products. J. Biotechnol. 99 41–50. [DOI] [PubMed] [Google Scholar]
- Gunneriusson, E., Nord, K., Uhlén, M., and Nygren, P.-Å. 1999. Affinity maturation of a Taq DNA polymerase specific affibody by helix shuffling. Protein Eng. 12 873–878. [DOI] [PubMed] [Google Scholar]
- Hansson, M., Ringdahl, J., Robert, A., Power, U., Goetsch, L., Nguyen, T., Uhlén, M., Ståhl, S., and Nygren, P.-Å. 1999. An in vitro selected binding protein (affibody) shows conformation-dependent recognition of the respiratory syncytial virus (RSV) G protein. Immunotechnology 4 237–252. [DOI] [PubMed] [Google Scholar]
- Hansson, H., Okoh, M.P., Smith, C.I.E., Vihinen, M., and Härd, T. 2001. Intermolecular interactions between the SH3 domain and the proline-rich TH region of Bruton’s tyrosine kinase. FEBS Lett. 489 67–70. [DOI] [PubMed] [Google Scholar]
- Helgstrand, M., Kraulis, P., Allard, P., and Härd, T. 2000. Ansig for Windows: An interactive computer program for semiautomatic assignment of protein NMR spectra. J. Biomol. NMR 18 329–336. [DOI] [PubMed] [Google Scholar]
- Kamtekar, S., Schiffer, J.M., Xiong, H., Babik, J.M., and Hecht, M.H. 1993. Protein design by binary patterning of polar and nonpolar amino acids. Science 262 1680–1685. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Kyte, J. and Doolittle, R.F. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157 105–132. [DOI] [PubMed] [Google Scholar]
- Lendel, C., Wahlberg, E., Berglund, H., Eklund, M., Nygren, P.-Å., and Härd, T. 2002. 1H, 13C and 15N resonance assignments of an affibody–target complex. J. Biomol. NMR 24 271–272. [DOI] [PubMed] [Google Scholar]
- Miller, S., Janin, J., Lesk, A.M., and Chothia, C. 1987. Interior and surface of monomeric proteins. J. Mol. Biol. 196 641–656. [DOI] [PubMed] [Google Scholar]
- Nilsson, B., Moks, T., Jansson, B., Abrahmsén, L., Elmblad, A., Holmgren, E., Henrichson, C., Jones, T.A., and Uhlén, M. 1987. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1 107–113. [DOI] [PubMed] [Google Scholar]
- Nord, K., Gunneriusson, E., Ringdahl, J., Ståhl, S., Uhlén, M., and Nygren, P.-Å. 1997. Binding proteins selected from combinatorial libraries of an 3-helical bacterial receptor domain. Nat. Biotechnol. 15 772–777. [DOI] [PubMed] [Google Scholar]
- Nord, K., Nord, O., Uhlén, M., Kelley, B., Ljungqvist, C., and Nygren, P.-Å. 2001. Recombinant human factor VIII-specific affinity ligands selected from phage-displayed combinatorial libraries of protein A. Eur. J. Biochem. 268 4269–4277. [DOI] [PubMed] [Google Scholar]
- Nygren, P.-Å. and Uhlén, M. 1997. Scaffolds for engineering novel binding sites in proteins. Curr. Opin. Struct. Biol. 7 463–469. [DOI] [PubMed] [Google Scholar]
- Palmer III, A.G. 2001. NMR probes of molecular dynamics: Overview and comparison with other techniques. Annu. Rev. Biophys. Biomol. Struct. 30 129–155. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. 1995. Structures of folding intermediates. Curr. Opin. Struc. Biol. 5 74–78. [DOI] [PubMed] [Google Scholar]
- Redfield, C. 1999. Molten globules. Curr. Biol. 9 R313. [DOI] [PubMed] [Google Scholar]
- Redfield, C., Smith, R.A.G., and Dobson, C.M. 1994. Structural characterization of a highly-ordered ‘molten globule’ at low pH. Nat. Struct. Biol. 1 23–29. [DOI] [PubMed] [Google Scholar]
- Rönnmark, J., Grönlund, H., Uhlén, M., and Nygren, P.-Å. 2002. Human immunoglobulin A (IgA)-specific ligands from combinatorial engineering of protein A. Eur. J. Biochem. 269 2647–2655. [DOI] [PubMed] [Google Scholar]
- Sandström, K., Xu, Z., Forsberg, G., and Nygren, P.-Å. 2003. Inhibition of the CD28–CD80 co-stimulation signal by a CD28-binding ligand developed by combinatorial protein engineering. Protein Eng. 6 691–697. [DOI] [PubMed] [Google Scholar]
- Schulman, B.A., Kim, P.S., Dobson, C.M., and Redfield, C. 1997. A residue-specific NMR view of the non-cooperative unfolding of a molten globule. Nat. Struct. Biol. 4 630–634. [DOI] [PubMed] [Google Scholar]
- Skelton, N.J., Kördel, J., Akke, M., and Chazin, W.J. 1992. Nuclear magnetic resonance studies of the internal dynamics in apo, (Cd2+)1 and (Ca2+)2 calbindin D9k. The rates of amide proton exchange with solvent. J. Mol. Biol. 227 1100–1117. [DOI] [PubMed] [Google Scholar]
- Wahlberg, E., Lendel, C., Helgstrand, M., Allard, P., Dincbas-Renqvist, V., Hedqvist, A., Berglund, H., Nygren, P.-Å., and Härd, T. 2003. An affibody in complex with a target protein: Structure and coupled folding. Proc. Natl. Acad. Sci. 100 3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Y., Liu, T., Sazinsky, S.L., Moffet, D.A., Pelczer, I., and Hecht, M.H. 2003. Stably folded de novo proteins from a designed combinatorial library. Protein Sci. 12 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4 171–180. [DOI] [PubMed] [Google Scholar]
- Xiong, H., Buckwalter, B.L., Shieh, H.-M., and Hecht, M.H. 1995. Periodicity of polar and nonpolar amino acids is the major determinant of secondary structure in self-assembling oligomeric peptides. Proc. Natl. Acad. Sci. 92 6349–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W.Y., Larios, E., and Gruebele, M. 2003. On the extended 3-conformation propensity of polypeptides at high temperature. J. Am. Chem. Soc. 125 16220–16227. [DOI] [PubMed] [Google Scholar]