

Abstract
GCN2 is the α-subunit of the only translation initiation factor (eIF2α) kinase that appears in all eukaryotes. Its function requires an interaction with GCN1 via the domain at its N-terminus, which is termed the RWD domain after three major RWD-containing proteins: RING finger-containing proteins, WD-repeat-containing proteins, and yeast DEAD (DEXD)-like helicases. In this study, we determined the solution structure of the mouse GCN2 RWD domain using NMR spectroscopy. The structure forms an α + β sandwich fold consisting of two layers: a four-stranded antiparallel β-sheet, and three side-by-side α-helices, with an αββββαα topology. A characteristic YPXXXP motif, which always occurs in RWD domains, forms a stable loop including three consecutive β-turns that overlap with each other by two residues (triple β-turn). As putative binding sites with GCN1, a structure-based alignment allowed the identification of several surface residues in α-helix 3 that are characteristic of the GCN2 RWD domains. Despite the apparent absence of sequence similarity, the RWD structure significantly resembles that of ubiquitin-conjugating enzymes (E2s), with most of the structural differences in the region connecting β-strand 4 and α-helix 3. The structural architecture, including the triple β-turn, is fundamentally common among various RWD domains and E2s, but most of the surface residues on the structure vary. Thus, it appears that the RWD domain is a novel structural domain for protein-binding that plays specific roles in individual RWD-containing proteins.
Keywords: NMR, GI domain, hydrogen bond network, protection factor, protein structure
In eukaryotic cells, protein synthesis is regulated in response to various environmental stresses by phosphorylation of the α-subunit of the translation initiation factor 2 (eIF2α). Among the four eIF2α kinases identified in mammals thus far, only GCN2 is present among various eukaryotes (Fig. 1 ▶), in which it appears to be involved in growth under amino acid starvation conditions (Dever 1999). In the yeast Saccharomyces cerevisiae, GCN2, the only eIF2α kinase, was first described and its function in translational control has been extensively investigated in vivo and in vitro (Hinnebusch and Natarajan 2002). Phosphorylation of eIF2α by GCN2 consequently induces the translation of the GCN4 mRNA, which encodes a transcriptional activator of genes for amino acid biosynthetic enzymes in various pathways in yeast. Although the amino acid biosynthetic pathways are markedly different between yeast and higher eukaryotes, GCN2 homologs were also identified in higher eukaryotes, such as Drosophila melanogaster (Santoyo et al. 1997; Olsen et al. 1998) and Mus musculus (Berlanga et al. 1999; Sood et al. 2000; Zhang et al. 2002). Expression of D. melanogaster GCN2 mRNA is developmentally regulated, and at later stages becomes restricted to the central nervous system (Santoyo et al. 1997; Olsen et al. 1998). A recent study using a Gcn2−/− knockout (loss-of-function) strain of mice demonstrated that GCN2 is required for adaptation to amino acid deprivation in mice (Zhang et al. 2002). However, the physiological function of GCN2 from higher eukaryotes and its role in regulating total or gene-specific translation remain unclear.
Figure 1.
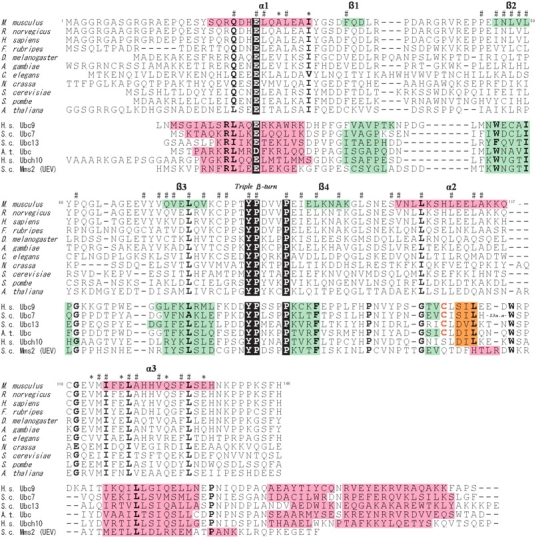
Structure-based sequence alignment of the RWD domain of GCN2 and the E2 family from various eukaryotes. Accession codes used for GCN2 are as follows. NCBI Accession: M. musculus, XP_192908; Rattus norvegicus, XP_230462; Homo sapiens, XP_031612; Fugu rubripes, CAAB01002380; D. melanogaster, AAC13490; Anopheles gambiae, XP_320188; Caenorhabditis elegans, NP_496781; Neurospora crassa, CAA62973; S. cerevisiae, NP_010569; Schizosaccharomyces pombe, NP_595991; Arabidopsis thaliana, CAD30860. Those used for the E2 family are as follows. PDB code: H. sapiens Ubc9, 1U9A; S. cerevisiae Ubc7, 2UCZ; S. cerevisiae Ubc13, 1JAT-A; A. thaliana UBC, 2AAK; H. sapiens Ubch10, 1I7K; S. cerevisiae Mms2, 1JAT-B. According to the secondary structural elements in the PDB files, α-helices, 310-helices, and β-strands are colored pink, orange, and green, respectively. In the RWD domain, invariant residues of the YPXXXP motif, and Glu in the first α-helix (Asp or Glu in E2) are indicated by a black background. Highly conserved residues (>90%) are indicated in bold type. Asterisks indicate putative interaction sites with GCN1, as described in the text. The code # indicate residues with a small solvent-accessible surface area (<10%).
The GCN2 protein has three functionally distinct domains in addition to the kinase domain, and activation of the kinase results from their specific interactions with RNAs and other proteins. According to the findings in the yeast system, uncharged tRNAs that accumulate in amino acid-starved cells bind to a regulatory domain in GCN2 that resembles histidyl-tRNA synthetase (HisRS-related domain; Wek et al. 1995; Zhu et al. 1996). This interaction is thought to induce a conformational change of GCN2 that unmasks the adjacent kinase domain for activation (Dong et al. 2000). The other RNA-binding domain, located at the C-terminus, is required for association with ribosomes and is proposed to facilitate GCN2 dimerization (Ramirez et al. 1991; Zhu and Wek 1998; Qiu et al. 2001). This interaction is necessary for GCN2 activation, and allows monitoring of the uncharged tRNA levels in cells (Sattlegger and Hinnebusch 2000).
Activation of GCN2 further requires binding to GCN1, which forms a stable complex with the ATP-binding cassette protein GCN20, and functions on elongating ribosomes (Vazquez de Aldana et al. 1995; Garcia-Barrio et al. 2000). The N-terminus of GCN2 contains a minimal essential region for interacting with GCN1 (Garcia-Barrio et al. 2000; Kubota et al. 2000, 2001). PSI-BLAST searches (Altschul et al. 1997) initiated with this N-terminal region in GCN2 revealed significant similarity to many RING finger-containing proteins, WD-repeat-containing proteins, yeast DEAD (DEXD)-like helicases, the Impact protein family (a product of an evolutionary conserved gene that is genetically imprinted in mice; Yamada et al. 1999), and a range of hypothetical proteins. Therefore, the newly defined domain was named the RWD domain, after the first three proteins (Doerks et al. 2002), or the GI domain, after the GCN2 and Impact proteins (Kubota et al. 2000). However, little is known about the structure and function of the RWD domains as well as the RWD-containing proteins (Doerks et al. 2002).
In this study, we determined the solution structure of this novel protein-binding domain, the RWD domain, at the N-terminus of mouse GCN2 by heteronuclear NMR methods. This is the first report of an RWD structure, which reveals the structural characteristics of the RWD domains.
Results
Resonance assignments and NMR structure determination
Samples of the 13C/15N-labeled RWD domain, composed of 137 residues, were prepared for structure determination by the cell free protein expression system. The protein sample has tag sequences (14 residues in total) at the N-terminus (GSSGSSGM) and the C-terminus (SGPSSG), which are both derived from the expression vector. NMR resonances were assigned using conventional heteronuclear methods with the 13C/15N-labeled protein. The backbone resonance assignments were complete, with the exception of the amide group of Asn100, and of 10 residues in the tag sequence regions. A best-fit superposition of the ensemble of the 20 lowest energy structures is shown in Figure 2A ▶. The statistics of the structures as well as the distance and torsion angle constraints used for the program CYANA are summarized in Table 1. The root-mean-square deviation (RMSD) from the mean structure was 0.34 ± 0.04 Å for the backbone (N, Cα, C′) atoms, and 0.76 ± 0.06 Å for all heavy (nonproton) atoms in the well-ordered region (residues 20–42, 51–61, and 70–137).
Figure 2.
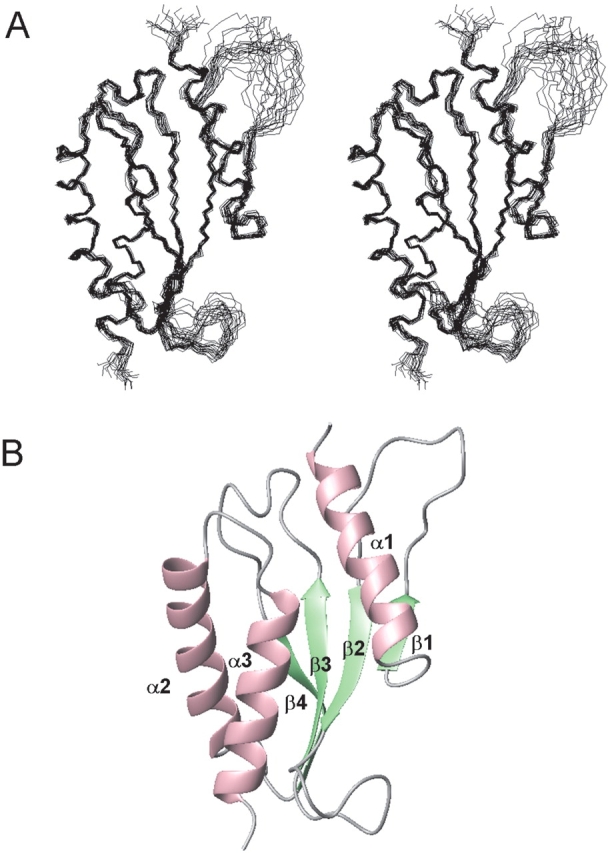
Overall structure of the RWD domain. (A) Stereoview illustrating a trace of the backbone atoms for the ensemble of the 20 lowest energy structures (residues 17–139). (B) Ribbon diagram of the RWD domain in the same view. The α-helices and the β-strands are depicted in pink and green, respectively.
Table 1.
Summary of conformational constraints and statistics of the final 20 best structures of the RWD domain of mouse GCN2
| NOE upper distance restraints | |
| Intraresidual (|i − j| 3 0) | 609 |
| Medium-range (1 ≤ |i − j| ≤ 4) | 959 |
| Long-range (|i − j| > 4) | 589 |
| Total | 2157 |
| Dihedral angle restraints (φ and ψ) | 112 |
| CYANA target function value (Å2) | 1.80 ± 0.18 |
| Number of violations | |
| Distance violations (>0.30 Å) | 0 |
| Dihedral angle violations (>5.0°) | 0 |
| AMBER energies (kcal/mole) | |
| Total | −5121 ± 103 |
| van der Waals | −435 ± 20 |
| Electrostatic | −5813 ± 95 |
| RMSD from ideal geometry | |
| Bond length (Å) | 0.0074 ± 0.0001 |
| Bond angles (°) | 1.87 ± 0.03 |
| Ramachandran plot (%)a | |
| Residues in most favored regions | 81.0 |
| Residues in additional allowed regions | 14.0 |
| Residues in generously allowed regions | 2.9 |
| Residues in disallowed regions | 2.1 |
| RMSD deviation from the averaged coordinates (Å)b | |
| Backbone atoms | 0.34 ± 0.04 |
| Heavy atom | 0.76 ± 0.06 |
a For residues 19–138.
b For residues 20–42, 51–61, and 70–137.
The NMR results show that the RWD domain has an α + β sandwich fold with the N- and C-termini on different sides of the molecule (Fig. 2B ▶). One layer consists of a four-stranded antiparallel βsheet (β1: 38–40, β2: 55–59, β3: 72–77, β4: 91–96), while the other layer consists of three α-helices (α1: 20–33, α2: 103–117, α3: 123–137). These elements are connected in the order of α-β-β-β-β-α-α. Of the nine proline residues, the structure contains one cis-Pro84 in the well-ordered loop connecting β3 and ββ β1/β2 loop, the β2/β3 loop, and the tag sequence regions are not well ordered.
Hydrogen-deuterium (1H/2H) exchange
To obtain more structural information about the RWD domain, we studied the hydrogen-deuterium exchange kinetics of the amide protons that were followed by recording the 1H–15N HSQC spectra. Protection factors estimated from 1H/2H exchange experiments provide a useful measure to evaluate the conformational stability of the backbones of protein molecules (Sivaraman et al. 2001; Chi et al. 2002). The total exchange rates of 48 residues (out of 137) could be unambiguously followed (Fig. 3A ▶). Most of the exchange-protected amide protons belong to the well-determined secondary structure elements, indicating that these protons are involved in regular hydrogen bonds (Fig. 3B ▶). Amide protons with high protection factors are significantly concentrated on β2 and β3 the middle part of α2, and the inside part of α3. It appears that these regions make a stable structural core of the RWD domain, in which many hydrophobic residues are well conserved among species (Fig. 1 ▶). In contrast, almost all of the residues in α1 and α4 have undetectable protection factors, and only half of the residues in α1 show relatively low protection factors, indicating that these regions surrounding the core region are less stable (Fig. 3B ▶). High protection factors are also observed in several residues that are not directly involved in forming a β-sheet or an α-helix, which will be discussed later.
Figure 3.

Protection factors for the amide proton exchange in the RWD domain. (A) Plot of the HN protection factors of main-chain residues. Residues without values disappeared in the first 23 min of exchange, as did the protons from the Asn and Gln side chains. (B) Ribbon diagrams of the RWD domain colored according to the protection factors. The color code for the residues is set according to the protection factors measured: blue, more than 104; and cyan, less than 104. Residues involved in the hydrogen bond network are shown, as described in the text.
Unique triple β-turn
A characteristic feature of the structure is the ordered loop between β3 and β4, where the 80PPTYPDVV87 region can be regarded as a unique triple β-turn. The three reverse turns are consecutively connected such that each β-turn shares two residues with another turn. Thus, the polypeptide backbone undergoes three 180° changes in its direction (Fig. 4A ▶). According to the classification of β-turn types, using four values for the φ and ψ angles of the turn residues i + 1 and i + 2 (Hutchinson and Thornton 1994), the first β-turn (80PPTY83), the second β-turn (82TYPD85), and the third β-turn (84PDVV87) are classified as type I, type VIa, and type VIII, respectively (Table 2). Particularly, the type VIa and type III β-turns are relatively rare and characteristic of the triple β-turn. As in the type VIa β-turn, the second turn contains cis-Pro at the i + 2 position, and the ring of cis-Pro84 is stacked with an aromatic ring of the preceding Tyr83, which is one of the major stabilizing factors of the type VIa β-turn (Yao et al. 1994). It is notable that the Tyr and Pro residues are invariant in the RWD domains (Fig. 1 ▶). As in the type VIII β-turn, the central residues (i + 1, i + 2) in the third turn adopt an αRβ conformation, and the distance between Cα(i) of Pro84 and Cα(i + 3) of Val86 is relatively long (6.3 Å), compared to the other types (3.4~4.5 Å; Chou 2001). In addition, the three residues, Tyr83, Asp85, and Val86, in the triple β-turn exhibit high protection factors, indicating that the triple β-turn forms a stable structure, unlike the other loops in the RWD domain (Fig. 3 ▶). Often, rigid β-turns have a hydrogen bond between the NH of residue i and the CO of residue i + 3 (Chou 2001). Thus, in view of the determined structure, hydrogen bonds would exist between the O of Pro80 and the HN of Tyr83, and between the O of Thr82 and the HN of Asp85 in the triple β-turn (Fig. 4B ▶). The high protection factor of Val86 seems to be due to solvent inaccessibility.
Figure 4.
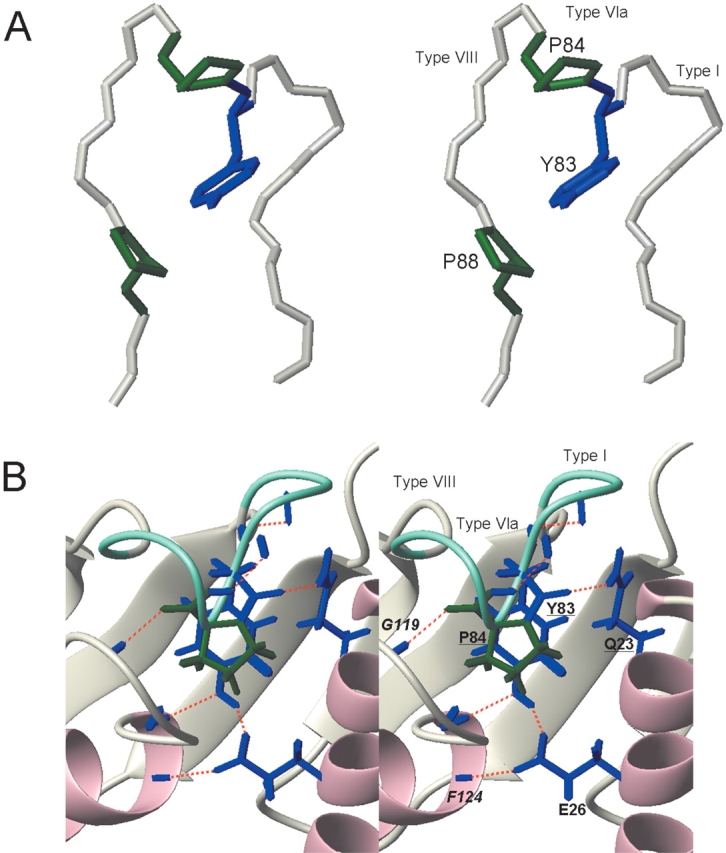
Stereoview of the structure of the triple β-turn. (A) The invariant residues, Tyr83 (blue), Pro84 (green), and Pro88 (green), are indicated. The distances between Cα(i) and Cα(i + 3) in the first, second, and third turns are 5.7 Å, 5.0 Å, and 6.3 Å, respectively. (B) Model of the hydrogen bond network involving the β-triple turn. Residues involved in the hydrogen bond network are shown, as described in the text. Residues indicated by italicized letters show the amide protons or the carbonyl oxygens. Underlined residues indicate those into which mutations were introduced in this study. Hydrogen bonds are depicted by red bars. The triple β-turn is indicated in aquamarine.
Table 2.
φ and ψ angles of triple β-turn
| RWD (80PPTYPDVV87) | Mms2 (UEV) (65GPNYPDSP72) | Ubc9 (E2) (65KDDYPSSP72) | Standard valuesa | |||||
| Proteins seq. | φ | ψ | φ | ψ | φ | ψ | φ | ψ |
| X1 | −65 | 164 | −77 | −175 | −92 | 177 | ||
| X2 | −69 | 3 | −51 | −33 | −70 | −27 | −60 | −30 (I) |
| X3 | −120 | −5 | −101 | 7 | −91 | 17 | −90 | 0 (I) |
| Y4 | −58 | 140 | −69 | 143 | −75 | 138 | −60 | 120 (VIa) |
| P5 | −91 | 12 | −93 | 11 | −94 | 21 | −90 | 0 (VIa) |
| X6 | −60 | −30 | −70 | −34 | −69 | −31 | −60 | −30 (VIII) |
| X7 | −125 | 165 | −107 | 145 | −116 | 152 | −120 | 120 (VIII) |
| X8 | −69 | 140 | −72 | 159 | −82 | 161 | ||
The angles of each residue in the RWD domain are from the average of the 20 structures. Those of the crystal structures were calculated from PDB data as follows: yeast Mms2, PDB code 1JAT-B; mammalian Ubc9, 1U9A.
a Standard values for the types of β-turns are derived from the report by Hutchinson and Thornton (1994). The types are shown by Roman numerals in parentheses. In the classification, normal cutoffs of 30° are used for deviations from standard angles, with one angle allowed to deviate by 45° (Hutchinson and Thornton 1994). In addition, the ω angles of the invariant Pro (in bold) are −4, −15, and −2°, respectively.
The buried Tyr in the triple β-turn
As a result, this triple β-turn allows Tyr83, which is stacked with cis-Pro84 in the second β-turn, to be buried into the core of the protein and thus completely inaccessible to the solvent (Fig. 4 ▶). The NMR data show the characteristic properties of the buried Tyr83 residue in the solution structure. The hydroxyl proton of a buried Tyr frequently makes hydrogen bonds. An unusually downfield-shifted resonance was detected at 13.66 ppm in the 1D NMR spectrum (data not shown). It remained as a singlet when the protein was labeled with 13C/15N, and the NOE cross-peaks were unambiguously observed with the HN protons of Met122 and Ile123 in the 15N-NOESY spectrum, and with the Hα and Hβ of Val121 and the Hɛ2 of Tyr83 in the 13C-NOESY spectrum. Thus, we identified this down-field-shifted signal as the hydroxyl proton of the buried Tyr83. In addition, the chemical shifts of Hδ1 (7.38 ppm) and Hδ2 (6.97 ppm) differ from each other, as do those of Hɛ1 (6.77 ppm) and Hɛ2 (7.26 ppm). These findings indicate that the hydrophobic side-chain packing on Tyr83 is so tight that its aromatic ring can barely flip at 25°C, which would also contribute to the stability of the triple β-turn. It is quite likely that this tight packing involves two invariant Pro residues. One is cis-Pro84, which is stacked with the aromatic ring of Tyr83, and the other is Pro88, which makes van der Waals contacts with the side of the aromatic ring (Fig. 4A ▶). The invariant residues, Tyr83, Pro84, and Pro88, are characteristic of the sequence and structure of the RWD domain, and thus we call this sequence the “YPXXXP motif.”
The core hydrogen bond network
The determined structure supports a model of an internal hydrogen bond network around the triple β-turn (Fig. 4B ▶). The hydroxyl proton of Tyr83 hydrogen bonds with the Oɛ1 of Glu26, probably causing the downfield-shifted resonance of the hydroxyl proton (Fernández et al. 1997). The OH group of Tyr83 interacts with the HN of Met122 and/or the HN of Ile123 at the beginning of α3. The other Oɛ of Glu26 forms a hydrogen bond with the HN of Phe124. The presence of two hydrogen bonds with the amides of Ile123 and Phe124 would be consistent with their downfield-shifted resonances, 9.00 and 10.21 ppm, respectively (Wishart et al. 1991). The amide protons of Met122 and Ile123 show high protection factors (Fig. 3 ▶), probably due to hydrogen bonds and/or solvent inaccessibility.
The hydrogen bonds of Ile123 and Phe124 at the N-terminus of α3 appear to function for N-capping of the α-helix. The term “helix capping” is generally used to de scribe the alternative hydrogen-bonding patterns that satisfy unfilled hydrogen-bonding capacity at the ends of α-helices, and the capping residues flank the α-helix (Aurora and Rose 1998). In the structure of the RWD domain, the N-terminal part of α3 runs against α1, at the angle of 41° in almost the same plane (Fig. 2 ▶). At the point of contact, the amide hydrogens of Ile123 and Phe124 at the N-terminus are satisfied by the proximity of the side-chain hydrogen bond acceptors of Tyr83 and Glu26 in α1, respectively, although the donors and the acceptors are sequentially distant (Fig. 4B ▶).
Additionally, two other hydrogen bonds concerning the residues Tyr83 and Pro84 appear to exist in the structure. One is a hydrogen bond between the O of Tyr83 and the Hɛ of Gln23 in α1, and the other is between the O of Pro84 and the HN of Gly119 in the ordered loop connecting α2 and α3 (Fig. 4B ▶). It seems likely that these hydrogen bonds link the stable triple β-turn to α1 and the α2/β3 loop, respectively, thus contributing to the maintenance and stability of the structure. It is noteworthy that Gln23 and Gly119 are also highly conserved among GCN2 proteins.
Mutational analysis of the hydrogen bond network
To verify the importance of the hydrogen bond network in stabilizing the conformation of the RWD domain, point mutations were introduced at each of the three residues involved, Glu26, Tyr83, and Pro84 (Fig. 4B ▶). We constructed seven RWD domain mutants (E26A, E26D, E26K, E26Q; Y83A, Y83F; P84A) labeled with 15N, and performed 1H–15N HSQC NMR measurements and far-ultraviolet circular dichroism (far-UV CD) measurements. Whereas the resonances were well dispersed in the wild-type RWD domain, all of the NMR spectra of the mutants showed a small number of resonances compared to that of the wild type (Fig. 5A ▶). These results indicate that the mutants do not form a native conformation. This is supported by the CD spectra of all the mutants showing that secondary structure contents significantly decrease (Fig. 5B ▶). Thus, both results indicate that none of these point mutations allow the mutant proteins to form a native conformation. These could be explained well by the effects on the hydrogen bond network, as shown in Figure 4B ▶. For the mutations at Glu26, the results with the E26A and E26K mutants would be due to the disruption of both hydrogen bonds with Tyr83 and Phe124, while that with E26Q would be due to the disruption of either hydrogen bond. The result with E26D would be explained by the difference in the distance between the acceptor and donor. The result of the Y83F mutant would be caused by the lack of the OH group involved in the hydrogen bonds. On the other hand, in the Y83A and P84A mutants, the absence of the Tyr-cis-Pro ring stacking probably does not allow the formation of the triple β-turn. These findings show that the whole hydrogen bond network involving these conserved residues is required for the native conformation of the RWD domain.
Figure 5.
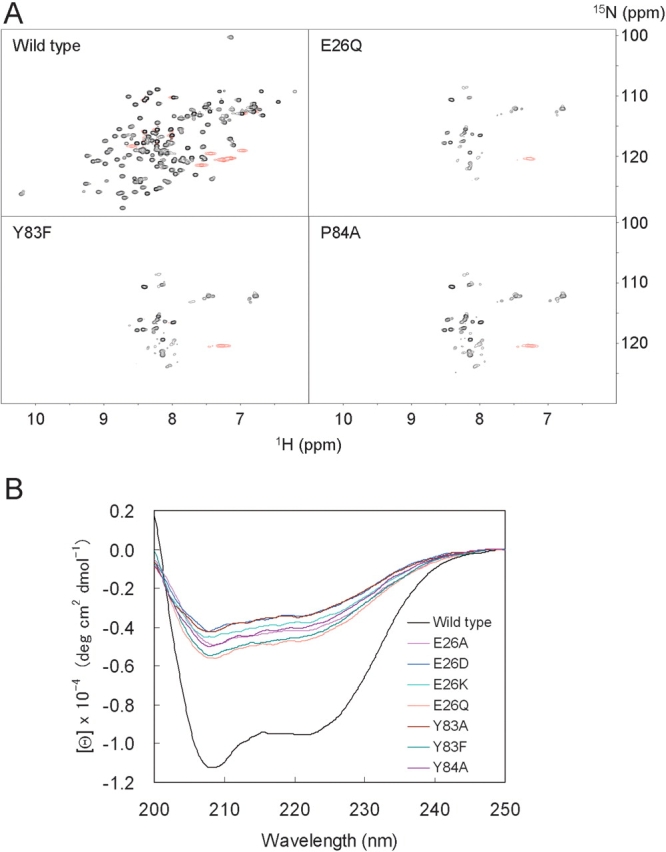
1H–15N HSQC spectra (A) and far-UV CD spectra (B) of the wild-type RWD domain and the mutants. NMR spectra of E26D, E26K, and Y83A mutants are not shown, because they are essentially the same as those of the other mutants.
Discussion
Putative binding surface with GCN1
The protein-binding domain at the N-terminus of mouse GCN2 has an α + β sandwich fold with an α-β-β-β-β-α-α topology, and is characterized by a unique, stable loop (the YPXXXP motif) comprising a triple β-turn involved in hydrogen bonds. Sequence alignments indicated the sequence conservation of a series of key residues (Fig. 1 ▶), suggesting that the RWD domain structure is well conserved among the GCN2 proteins. It was reported that the RWD domain of D. melanogaster GCN2 can interact with the yeast GCN1/ GCN20 complex, suggesting the evolutionary conservation of the mode of the GCN1–GCN2 interaction (Garcia-Barrio et al. 2000). Furthermore, considering the high conservation of the yeast C-terminal GCN1 segment (2064–2382) that interacts with GCN2 (Sattlegger and Hinnebusch 2000; Kubota et al. 2001), the important residues involved in the interaction between GCN1 and GCN2 are presumably well retained throughout eukaryotes. To gain insights into the putative functional residues in the RWD domain of GCN2, we mapped the highly conserved residues onto the RWD structure, to identify surface clusters that may play a functional role. Figure 6 ▶ shows that the highly conserved residues on the surface are concentrated on α1 and α3: Ala29 and Ile33 in α1; and Val121, Glu125, Gln131, and Glu136 in α3. A sequence comparison with other RWD domains from mouse indicated that only the residues in α3 are specific to the RWD domain of GCN2 (Fig. 7 ▶). Thus, these α3 residues are probably involved in the specific interaction with GCN1. The two negatively charged residues in α3, Glu123, and Glu136, are reminiscent of the finding that Arg2259 in GCN1 is required for the interaction with the GCN2 RWD domain in yeast (Sattlegger and Hinnebusch 2000). A specific site, shaped by the two side-by-side helices, may be important for the interaction.
Figure 6.

Surface representation and ribbon diagram of the RWD domain showing the side chains of highly conserved residues. The alignment analysis using the ConSurf server (www/bioinfo.tau.ac.il/ConSurf/; Glaser et al. 2003) assigns a conservation score to each residue (9—conserved, 1—variable). In this surface representation, residues scored as 9 and 8 are colored red and orange, respectively, while those with scores of less than 8 are white. In the ribbon diagram, the side chains of the highly conserved residues are indicated in red, and α1 and α3 are in orange.
Figure 7.

Sequence alignment of the RWD domains of mouse RWD-containing proteins. Domains other than the RWD domain within the protein are indicated in parentheses. The YPXXXP motif, and Glu in the first α-helix are indicated by a black background. Highly conserved residues among the mouse RWD domains (>90%) are indicated in bold type. Asterisks indicate putative interaction sites of the GCN2 RWD domain with GCN1, as described in the text. The code # indicates residues with a small solvent-surface area in the GCN2 RWD domain (<10%).
It was previously reported that the yeast GCN2 mutants with the RWD domain mutations of Glu26, Tyr83, and Pro84 (E26A, E26K, Y83A, and Y83A/P84A) failed to show two-hybrid or in vitro interactions with GCN1, suggesting that these residues were essential for this interaction (the numbering is according to that of the mouse RWD domain; Kubota et al. 2000). However, the mutational analysis in this study shows that the structures of these mutants are quite unstable, and that they do not form a wild-type structure. The findings of the previous report are better explained by the structural destabilization of the mutants, rather than by the lack of interaction sites with GCN1.
Three mouse cDNAs, derived from a single gene (α, β, and γ), encoding different isoforms of GCN2, were cloned (Sood et al. 2000; Zhang et al. 2002). Interestingly, their sequences differ only in the RWD domain region. The α isomer lacks the RWD domain region, while the β and γ isomers contain the complete sequence and the latter half of the RWD domain region, respectively, and they all appear to be differentially expressed. In the γ isomer, the truncated RWD region includes the region from V87 to the last residue, corresponding to β4, α2, and α3 in the full-length RWD domain. The 2D HSQC spectrum was measured with a 15N-labeled truncated RWD domain, and revealed that this region is unstructured (data not shown). Considering that the α and γ isomers presumably lack the interaction sites with GCN1, these isomers may be activated by a GCN1-independent mechanism (Sood et al. 2000; Zhang et al. 2002).
Structural homology to ubiquitin-conjugating enzymes
A 3D structural search using Dali (Holm and Sander 1993) revealed that the RWD domain shares significant structural homology to a ubiquitin conjugating-enzyme (E2), mammalian UBC9 (Tong et al. 1997), and the yeast ubiquitin E2 variant (UEV) protein, Mms2 (VanDemark et al. 2001), with Z-scores of 7.4 and 8.6, and sequence identities of 13% and 17%, respectively. Structure-based sequence alignments (Fig. 1 ▶) as well as structural comparisons between the RWD domains and E2s (Fig. 8 ▶), showed that the common secondary structural elements adopt an α + β sandwich motif with an α-β-β-β-β-α-α topology, although the E2s usually have two additional helices at the C terminus. Both the RWD domains and E2s always possess the YPXXXP motif between β and β4. As seen in the RWD domain of GCN2, the motif in many E2s was found to form a triple β-turn formation, in which the first, second, and third turns are usually types I, Via, and VIII, respectively. To investigate whether the triple β-turn occurs in proteins other than RWD domains or E2s, we did sequence analysis of the YPXXXP motif, and searched for back bone traces of the eight residues (32 atoms) similar to those of the turns in the nonredundant representatives of PDB coordinates. Although the YPXXXP sequence appears in various proteins, significant similarities of the back bone traces were found only in the loops of E2s and their homologs, which always have the motif (data not shown). These findings indicate that the triple β-turn is unique to the RWD domains and E2s in the structures that have been determined so far, and its formation requires not only the YPXXXP motif, but also other elements, including such an internal hydrogen bond network. In addition, the N-capping of α1, by Glu26 in α3, can also be seen in many E2s, where Asp often occupies the position instead of Glu (Fig. 1 ▶). Hence, these important similarities indicate that the E2, UEV, and RWD domains can be classified into a structural group that seems to have originated from a common ancestor. It is noteworthy that these key conserved residues are structural elements, but not interacting elements with other proteins. In contrast to these similarities, the functions of the proteins differ strikingly; E2 is an enzyme, while UEV and the RWD domain function in protein binding. Furthermore, a remarkable structural difference occurs in the α2 region (Fig. 8 ▶). In the RWD domain, the region forms an α-helix composed of 15 residues. In E2s and UEV, however, it forms a long extended stretch, where E2 has the catalytic Cys residue followed by a 310-helix, and UEV lacks the Cys but has a short α-helix. Considering these substantial differences in function and structure, it appears that the RWD domain is a novel structural domain for protein binding.
Figure 8.

Comparison of the RWD domain, UEV, and E2. UEV, yeast Mms2, complexed with Ubc13: PDB code 1JAT-B; and E2, mammalian Ubc9: 1U9A. Shown are the invariant Tyr (blue) and Pro (green) located in the triple β-turn, and the semiconserved Glu or Asp (blue) located in the first α-helix. The E2 active site cysteine is indicated in yellow. The α-helices and the β-strands are depicted in pink and green, respectively, while structural differences among the three proteins are indicated in red.
Concluding remarks
A comparison of the 11 RWD domain sequences from mouse that are now available highlights the conservation of the key Glu residue in the first α-helix and the YPXXXP motif, as well as the conservation of hydrophobic residues presumably involved in forming the core (Fig. 7 ▶). As for the α2 region, the corresponding region in the other RWD domains is predicted to form an α-helix by the PSIPRED method (McGuffin et al. 2000; data not shown). However, the residues that seem to be located on the surface or in the loops tend to vary, depending on the RWD domain. These findings suggest that the structures of the RWD domains are virtually identical to each other, while the binding substrates are different. Intriguingly, the RWD containing proteins often have an E2 homolog domain, RING-finger domains, or WD-repeat domains (Fig. 7 ▶), which often exist in proteins involved in the ubiquitin-mediated pathway (Weissman 2001). It was already reported that the AO7 protein (Q9QZR0), including consecutive RWD and RING finger domains, acts as a substrate for polyubiquitination by binding directly to an E2 enzyme via its RING domain (Lorick et al. 1999). It is tempting to speculate that the RWD domain interacts with an E2 enzyme for polyubiquitination in the same way as UEV binds to E2, so that the complex serves as a binding scaffold for two ubiquitins (VanDemark et al. 2001). Some RWD domains might be implicated in the ubiquitin-mediated pathway. Further studies will be required to determine the functions of the RWD domains as well as the RWD-containing proteins.
Materials and methods
Protein expression and purification
The DNA encoding the RWD domain of mouse GCN2 (Glu17–Lys139) was subcloned by PCR from the mouse full-length cDNA clone with the ID RIKEN cDNA 2900069K12 (Kawai et al. 2001). This DNA fragment was cloned into the expression vector pCR2.1 (Invitrogen) as a fusion with an N-terminal 6-His affinity tag and a TEV protease cleavage site. The 13C/15N-labeled fusion protein was synthesized by the cell free protein expression system, as described elsewhere (Kigawa et al. 1999). The solution was first adsorbed to a HiTrap Chelating column (Amersham Biosciences), which was washed with buffer A (50 mM Tris-HCl buffer [pH 8.0] containing 500 mM sodium chloride and 10 mM imidazole) and eluted with buffer B (50 mM Tris-HCl buffer [pH 8.0] containing 500 mM sodium chloride and 500 mM imidazole). To remove the His-tag, the eluted protein was incubated at 30°C for 1 h with the TEV protease. After dialysis against buffer A without imidazole, the dialysate was mixed with imidazole, to a 10 mM final concentration, and then was applied to a HiTrap Chelating column, which was washed with buffer A. The flow-through fraction was loaded onto a HiTrap Desalting column (Amersham Biosciences) with buffer C (20 mM Tris-HCl buffer [pH 8.0]). The RWD-containing fractions were applied to a HiTrap Q column by a concentration gradient of buffer C and buffer D (20 mM Tris-HCl buffer [pH 8.0] containing 1 M sodium chloride). The RWD-containing fractions were collected, and a protease inhibitor cocktail (Complete [EDTA-free], Roche Applied Science) and DTT (final concentration, 1 mM) were added.
For NMR measurements, the purified protein was concentrated to ~1.0 mM in 1H2O/2H2O (9:1) 20 mM Tris-d11-HCl buffer (pH 7.0) containing 100 mM NaCl, 1 mM 1,4-dl-dithiothreitol-d10 (d-DTT), and 0.02% NaN3. It was stable for at least 6 months, when stored at 4°C.
NMR spectroscopy, structure determination, and analysis
All NMR measurements were performed at 25°C on Bruker AVANCE 700 and AVANCE 800 spectrometers. Sequence-specific backbone assignments were made with the 13C/15N-labeled sample, using standard triple-resonance experiments (Wüthrich 1986; Bax 1994). Assignments of side chains were obtained from HBHACONH, HCCCONNH, CCCONNH, HCCH-TOCSY1, HCCH-COSY, and CCH-TOCSY spectra. 3D 15N- and 13C-edited NOESY spectra with 80- and 40-msec mixing times were used to determine distance restraints. Data sets of 512 (1H) × 30 (15N) × 120 (1H), and 512 (1H) × 38 (13C) × 146 (1H) complex points were recorded for spectra widths of 13.9 ppm × 20.0 ppm × 11.4 ppm, and 13.9 ppm × 32.8 ppm × 11.4 ppm, respectively. The spectra were processed with the program NMRPipe (Delaglio et al. 1995), and the program Kujira (N. Kobayashi, pers. comm.), created on the basis of NMRview (Johnson and Blevins 1994), was employed for optimal visualization and spectral analysis.
Automated NOE cross-peak assignments (Herrmann et al. 2002) and structure calculations with torsion angle dynamics (Güntert et al. 1997) were performed using the software package CYANA1.07 (http://www.guentert.com). Peak lists of the two NOESY spectra were generated as input with the program NMRview (Johnson and Blevins 1994). The input further contained the chemical shift list corresponding to the sequence-specific assignments. Dihedral angle restraints were derived using the program TALOS (Cornilescu et al. 1999). No hydrogen bond constraints were used.
A total of 100 structures were independently calculated. The 20 conformers of the CYANA cycle 7 with the lowest final CYANA target function values were energy-minimized in a water shell with the program OPALp (Koradi et al. 2000), using the AMBER force field (Cornell et al. 1995). The structures were validated using PROCHECK-NMR (Laskowski et al. 1996). The program MOLMOL (Koradi et al. 1996) was used to analyze the resulting 20 energy-minimized conformers and to prepare drawings of the structures.
The atomic coordinates and structure factors (code 1UKX) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics (http://www.rcsb.org).
Mutational analysis
Point mutations were introduced into the vector described above, using the Quickchange site-directed mutagenesis kit (Stratagene). The 15N- labeled wild-type and seven mutant proteins were prepared by the cell-free protein expression system, as described above. Protein samples contained ~0.1 mM protein in the same buffer, as described above. The products were checked by the mass spectrometry analyses (data not shown). Measurements of 1H–15N HSQC spectra were performed at 25°C on a Bruker AVANCE 600 spectrometer with a cryo probehead.
Slow amide proton exchange analysis
The 15N/13C-labeled sample, which was a portion of the sample used for structure determinations, was lyophilized. The exchange reaction was started by dissolving the lyophilized sample in 99.9% 2H2O, to a final concentration of ~0.2 mM protein. Slowly exchanging amide protons were investigated by recording a series of consecutive 1H–15N HSQC spectra every 23 min at 25°C on a Bruker AVANCE 600 spectrometer with the cryo probehead. Rate constants (kex) for amide proton exchange were determined by fitting the time decrease of their corresponding cross-peak volumes to a single exponential decay function. The protection factors (P) for the various amide protons in the protein were thus estimated on the basis of the method reported by Bai et al. (1993), using the equation P = krc/kex, where krc and kex represent the exchange rates of the protein in the random coil and native conformation states, respectively.
CD measurements
CD spectra were recorded on a JASCO J-820 spectropolarimeter, using a quartz cuvette with a 2-mm path length. Spectra between 200 nm and 250 nm were obtained using a scanning speed of 10 nmm/min, a response time of 4.0 sec, and a bandwidth of 1 nm. Measurements were carried out at 20°C with a fixed protein concentration of 10 μM in the same buffer, as described above. After subtraction of a solvent spectrum, data were represented as mean residue ellipticities.
Acknowledgments
We are indebted to Drs. Hideki Hatanaka, Toshio Yamazaki, and Yutaka Muto for valuable suggestions and comments on the structural analysis. We are grateful to Yukiko Fujikura, Yoko Motoda, Miyuki Saito, Yukako Miyata, Atsuo Kobayashi, Noriko Hirakawa, Hajime Inamochi, Masaomi Ikari, Fumiko Hiroyasu, Maki Shibata, Megumi Watanabe, Mizuyu Miyamoto, Miyuki Sato, Mari Hirato, and Satoko Yasuda for their technical assistance. We also thank Dr. Akitsugu Takasu for help with the analysis of the 1H/2H change experiments. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04751804.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora, R. and Rose, G.D. 1998. Helix capping. Protein Sci. 7 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, Y. Milne, S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax, A. 1994. Multidimensional nuclear magnetic resonance methods for protein studies. Curr. Opin. Struct. Biol. 4 738–744. [Google Scholar]
- Berlanga, J.J., Santoyo, J., and De Haro, C. 1999. Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2α kinase. Eur. J. Biochem. 265 754–762. [DOI] [PubMed] [Google Scholar]
- Chi, Y.H., Kumar, T.K., Kathir, K.M., Lin, D.H., Zhu, G., Chiu, I.M., and Yu, C. 2002. Investigation of the structural stability of the human acidic fibro-blast growth factor by hydrogen-deuterium exchange. Biochemistry 41 15350–15359. [DOI] [PubMed] [Google Scholar]
- Chou, K.C. 2001. Prediction of signal peptides using scaled window. Peptides 22 1973–1979. [DOI] [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Cornilescu, G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13 289–302. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Dever, T.E. 1999. Translation initiation: Adept at adapting. Trends Biochem. Sci. 24 398–403. [DOI] [PubMed] [Google Scholar]
- Doerks, T., Copley, R.R., Schultz, J., Ponting, C.P., and Bork, P. 2002. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 12 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, J., Qiu, H., Garcia-Barrio, M., Anderson, J., and Hinnebusch, A.G. 2000. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6 269–279. [DOI] [PubMed] [Google Scholar]
- Fernández, C., Szyperski, T., Bruyère, T., Ramage, P., Mösinger, E., and Wüthrich, K. 1997. NMR solution structure of the pathogenesis-related protein P14a. J. Mol. Biol. 266 576–593. [DOI] [PubMed] [Google Scholar]
- Garcia-Barrio, M., Dong, J., Ufano, S., and Hinnebusch, A.G. 2000. Association of GCN1–GCN20 regulatory complex with the N-terminus of eIF2α kinase GCN2 is required for GCN2 activation. EMBO J. 19 1887–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser, F., Pupko, T., Paz, I., Bell, R.E., Bechor-Shental, D., Martz, E., and Ben-Tal, N. 2003. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19 163–164. [DOI] [PubMed] [Google Scholar]
- Güntert, P., Mumenthaler, C., and Wüthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273 283–298. [DOI] [PubMed] [Google Scholar]
- Herrmann, T., Güntert, P., and Wüthrich, K. 2002. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24 171–189. [DOI] [PubMed] [Google Scholar]
- Hinnebusch, A.G. and Natarajan, K. 2002. Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot. Cell 1 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Hutchinson, E.G. and Thornton, J.M. 1994. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 3 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B. and Blevins, R. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Kawai, J., Shinagawa, A., Shibata, K., Yoshino, M., Itoh, M., Ishii, Y., Arakawa, T., Hara, A., Fukunishi, Y., Konno, H., et al. 2001. Functional annotation of a full-length mouse cDNA collection. Nature 409 685–690. [DOI] [PubMed] [Google Scholar]
- Kigawa, T., Yabuki, T., Yoshida, Y., Tsutsui, M., Ito, Y., Shibata, T., and Yokoyama, S. 1999. Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett. 442 15–19. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Güntert, P. 2000. Point-centered domain decomposition for parallel molecular dynamics simulation. Comput. Phys. Commun. 124 139–147. [Google Scholar]
- Kubota, H., Sakaki, Y., and Ito, T. 2000. GI domain-mediated association of the eukaryotic initiation factor 2α kinase GCN2 with its activator GCN1 is required for general amino acid control in budding yeast. J. Biol. Chem. 275 20243–20246. [DOI] [PubMed] [Google Scholar]
- Kubota, H., Ota, K., Sakaki, Y., and Ito, T. 2001. Budding yeast GCN1 binds the GI domain to activate the eIF2α kinase GCN2. J. Biol. Chem. 276 17591–17596. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Lorick, K.L., Jensen, J.P., Fang, S., Ong, A.M., Hatakeyama, S., and Weissman, A.M. 1999. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. 96 11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin, L.J., Bryson, K., and Jones, D.T. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16 404–405. [DOI] [PubMed] [Google Scholar]
- Olsen, D.S., Jordan, B., Chen, D., Wek, R.C., and Cavener, D.R. 1998. Isolation of the gene encoding the Drosophila melanogaster homolog of the Saccharomyces cerevisiae GCN2 eIF-2α kinase. Genetics 149 1495–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, H., Dong, J., Hu, C., Francklyn, C.S., and Hinnebusch, A.G. 2001. The tRNA-binding moiety in GCN2 contains a dimerization domain that interacts with the kinase domain and is required for tRNA binding and kinase activation. EMBO J. 20 1425–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, M., Wek, R.C., and Hinnebusch, A.G. 1991. Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol. Cell Biol. 11 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoyo, J., Alcalde, J., Méndez, R., Pulido, D., and de Haro, C. 1997. Cloning and characterization of a cDNA encoding a protein synthesis initiation factor-2α (eIF-2α) kinase from Drosophila melanogaster. Homology to yeast GCN2 protein kinase. J. Biol. Chem. 272 12544–12550. [DOI] [PubMed] [Google Scholar]
- Sattlegger, E. and Hinnebusch, A.G. 2000. Separate domains in GCN1 for binding protein kinase GCN2 and ribosomes are required for GCN2 activation in amino acid-starved cells. EMBO J. 19 6622–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman, T., Arrington, C.B., and Robertson, A.D. 2001. Kinetics of unfolding and folding from amide hydrogen exchange in native ubiquitin. Nat. Struct. Biol. 8 331–333. [DOI] [PubMed] [Google Scholar]
- Sood, R., Porter, A.C., Olsen, D.A., Cavener, D.R., and Wek, R.C. 2000. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2α. Genetics 154 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, H., Hateboer, G., Perrakis, A., Bernards, R., and Sixma, T.K. 1997. Crystal structure of murine/human Ubc9 provides insight into the variability of the ubiquitin-conjugating system. J. Biol. Chem. 272 21381–21387. [DOI] [PubMed] [Google Scholar]
- VanDemark, A.P., Hofmann, R.M., Tsui, C., Pickart, C.M., and Wolberger, C. 2001. Molecular insights into polyubiquitin chain assembly: Crystal structure of the Mms2/Ubc13 heterodimer. Cell 105 711–720. [DOI] [PubMed] [Google Scholar]
- Vazquez de Aldana, C.R., Marton, M.J., and Hinnebusch, A.G. 1995. GCN20, a novel ATP binding cassette protein, and GCN1 reside in a complex that mediates activation of the eIF-2 α kinase GCN2 in amino acid-starved cells. EMBO J. 14: 3184–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman, A.M. 2001. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell. Biol. 2 169–178. [DOI] [PubMed] [Google Scholar]
- Wek, S.A., Zhu, S., and Wek, R.C. 1995. The histidyl-tRNA synthetase-related sequence in the eIF-2 α protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell Biol. 15 4497–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222 311–333. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. John Wiley & Sons, Inc., New York.
- Yamada, Y., Hagiwara, Y., Shiokawa, K., Sakaki, Y., and Ito, T. 1999. Spatiotemporal, allelic, and enforced expression of Ximpact, the Xenopus homolog of mouse imprinted gene impact. Biochem. Biophys. Res. Commun 256 162–169. [DOI] [PubMed] [Google Scholar]
- Yao, J., Feher, V.A., Espejo, B.F., Reymond, M.T., Wright, P.E., and Dyson, H.J. 1994. Stabilization of a type VI turn in a family of linear peptides in water solution. J. Mol. Biol. 243 736–753. [DOI] [PubMed] [Google Scholar]
- Zhang, P., McGrath, B.C., Reinert, J., Olsen, D.S., Lei, L., Gill, S., Wek, S.A., Vattem, K.M., Wek, R.C., Kimball, S.R., et al. 2002. The GCN2 eIF2α kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell Biol. 22 6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, S. and Wek, R.C. 1998. Ribosome-binding domain of eukaryotic initiation factor-2 kinase GCN2 facilitates translation control. J. Biol. Chem. 273 1808–1814. [DOI] [PubMed] [Google Scholar]
- Zhu, S., Sobolev, A.Y., and Wek, R.C. 1996. Histidyl-tRNA synthetase-related sequences in GCN2 protein kinase regulate in vitro phosphorylation of eIF-2. J. Biol. Chem. 271 24989–24994. [DOI] [PubMed] [Google Scholar]