

Abstract
Amino acid residues associated with functional specificity of cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), glycogen synthase kinases (GSKs), and CDK-like kinases (CLKs), which are collectively termed the CMGC group, were identified by categorizing and quantifying the selective constraints acting upon these proteins during evolution. Many constraints specific to CMGC kinases correspond to residues between the N-terminal end of the activation segment and a CMGC-conserved insert segment associated with coprotein binding. The strongest such constraint is imposed on a “CMGC-arginine” near the substrate phosphorylation site with a side chain that plays a role both in substrate recognition and in kinase activation. Two nearby buried waters, which are also present in non-CMGC kinases, typically position the main chain of this arginine relative to the catalytic loop. These and other CMGC-specific features suggest a structural linkage between coprotein binding, substrate recognition, and kinase activation. Constraints specific to individual subfamilies point to mechanisms for CMGC kinase specialization. Within casein kinase 2α (CK2α), for example, the binding of one of the buried waters appears prohibited by the side chain of a leucine that is highly conserved within CK2α and that, along with substitution of lysine for the CMGC-arginine, may contribute to the broad substrate specificity of CK2α by relaxing characteristically conserved, precise interactions near the active site. This leucine is replaced by a conserved isoleucine or valine in other CMGC kinases, thereby illustrating the potential functional significance of subtle amino acid substitutions. Analysis of other CMGC kinases similarly suggests candidate family-specific residues for experimental follow-up.
Keywords: CHAIN analysis, proline-directed kinases, contrast hierarchical alignment, sky1p
Eukaryotic protein kinases propagate and amplify extracellular and intracellular signals (Karin and Hunter 1995; Johnson and Lapadat 2002) and thus play important regulatory roles in diverse cellular processes, such as metabolism, transcription, cell cycle progression, apoptosis, and neuronal development. They achieve this by transferring γ phosphate from ATP to a serine, threonine or tyrosine hydroxyl group on a protein substrate, often thereby influencing the conformational state of the protein and, as a result, downstream signaling events (for review, see Johnson and Lewis 2001; Huse and Kuriyan 2002; Lu et al. 2002).
Protein kinases themselves are commonly regulated by phosphorylation: either directly via phosphorylation of the kinase domain or indirectly via prephosphorylation of the substrate. Activation of cyclin-dependent kinase 2 (CDK2) and mitogen-activated protein (MAP) kinases (MAPKs), for example, occurs via phosphorylation of residues within a flexible activation loop (Johnson et al. 1996). Activation of glycogen synthase kinases (GSKs), on the other hand, occurs via prephosphorylation of a target substrate site that then stabilizes the activation loop for phosphorylation of a second nearby residue in the substrate (ter Haar et al. 2001). Interestingly, the activation loop of GSK constitutively resembles the active conformation of phosphorylation-regulated loops. Other modes of regulation, such as one involving the C-terminal extension of calcium/calmodulin-dependent protein kinase I (Goldberg et al. 1996), also occur.
Protein kinases are often very specific to the substrates they phosphorylate. One way that they achieve this is through direct interactions with substrate residues flanking the phosphorylation (P) site (Pinna and Ruzzene 1996; Songyang et al. 1996). For example, CDKs and MAPKs show a preference for proline at the P + 1 position and thus are termed proline-directed kinases (Pinna and Ruzzene 1996). This preference may be linked to downstream signaling mechanisms mediated by Pin1 proline isomerization (Zhou et al. 1999; Lu et al. 2002).
Downstream signal transmission typically involves co-proteins that form a complex with and participate in the function of a particular protein kinase (Pawson and Nash 2003). CDK2, for example, binds both to a regulatory cyclin subunit and to Cks1, which appears to modulate substrate recognition and/or phosphorylation (Bourne et al. 1996; for review, see Harper 2001). Likewise, GSK binds to Axin (Dajani et al. 2003), a scaffold protein associated with the β catenin signaling pathway (Li et al. 2002), and to the FRAT-tide peptide (Bax et al. 2001), which blocks GSK-3 from interacting with Axin. Notably, both the Cks1 binding site in CDK2 (Bourne et al. 1996) and the Axin/FRATtide binding site in GSK correspond to a insert (see Results and Discussion) present within the C-terminal domain of CMGC kinases, which include CDK, MAPK, GSK, and Cdc-like kinases (CLKs; Hanks and Hunter 1995; Manning et al. 2002).
Protein families within the CMGC group are often highly conserved across organisms that diverged over a billion years ago, implying that important structural features or mechanisms are associated with these conserved residues. Furthermore, such residues fall into functional categories inasmuch as certain residues are conserved in nearly all protein kinases, whereas others are largely conserved only within certain kinase groups (such as the CMGC group) or within specific families or subfamilies. Although some of these residues have known functions, the mere existence of so many conserved residues of unknown function implies that our understanding is skewed toward those kinase structural features most amenable to current experimental approaches. Can we learn anything about the possible roles and relative importance of these uncharacterized, yet important residues based on their patterns of conservation and on their structural locations and mutual interactions?
Here we address this question for CMGC kinases by using an approach called contrast hierarchical alignment and interaction network (CHAIN) analysis (Neuwald et al. 2003) that categorizes and measures the selective constraints imposed on protein sequences and maps these constraints to structural features. Other computational approaches—such as hierarchical analysis of residue conservation (Livingstone and Barton 1993), principal component analysis (Casari et al. 1995), evolutionary trace (Lichtarge et al. 1996), positional entropy (Hannenhalli and Russell 2000), site-specific rate shifts (for review, see Gaucher et al. 2002), and specificity determining residues (Mirny and Gelfand 2002; Li et al. 2003)—likewise seek to obtain evolutionarily insights into protein function from multiply aligned sequences. CHAIN analysis differs from these in that it uses (1) routines that can accurately align thousands of related sequences (using a combination of structurally and Gibbs sampling–based procedures), (2) an automated rigorous statistical procedure to optimally detect aligned sequence subgroups (each of which is characterized by strikingly conserved residues that are strikingly nonconserved outside of that subgroup), and (3) routines to identify corresponding specific structural interaction networks (including classical and weak hydrogen bonds, CH–π interactions, van der Waals contacts, and aromatic-aromatic interactions). This identifies residues within each category that are subject to the strongest selective constraints and thus are most distinctive of that category. As applied here, this reveals canonical CMGC structural features associated with kinase activation, substrate recognition, and the CMGC-insert region. Both deviations from and additions to these canonical features appear to contribute to functional specialization within individual CMGC families and subfamilies.
Results and Discussion
CHAIN analysis of CMGC kinases
CHAIN analysis involves construction of “contrast hierarchical alignments” as well as structural displays of the corresponding molecular interactions. Figure 1 ▶ shows two contrast hierarchical alignments of CMGC kinases. (Note that these alignments span only those sequence regions pertinent to our analysis here.)
Figure 1.
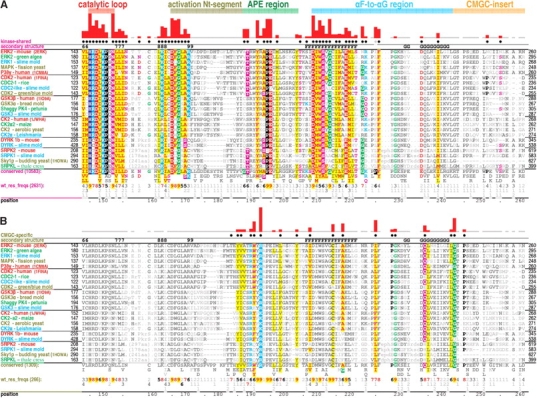
Contrast hierarchical alignments of various CMGC kinase families with representative sequences from distinct eukaryotic kingdoms for each family. The structural regions shown in Figures 3 ▶–6 ▶ and discussed in the text are indicated at the top. The histograms above the alignments plot the strength of the category-specific selective constraints imposed at each position (essentially using logarithmic scaling); these constraints also are indicated qualitatively through residue highlighting (with biochemically similar residues colored similarly). Dots below the histograms indicate residues assigned to that alignment’s category. Secondary structure is indicated directly above the aligned sequences, with β strands indicated by their number designations (i.e., 6–9 correspond to the β6–β9 strands, respectively) and helices by their letter designations (i.e., F and G correspond to the F-helix and the G-helix, respectively). The leftmost column of each alignment gives protein descriptions using the following color code to specify major eukaryotic taxa: metazoans, red; plants, green; fungi, dark yellow; and protozoans, cyan. See Materials and Methods for sequence identifiers. The foreground sequences (see text) are shown indirectly via the consensus patterns and corresponding weighted residue frequencies (wt_res_freqs) below the aligned sequences actually displayed. (Such sequence weighting adjusts for overrepresented families in the alignment.) Foreground alignment residue frequencies are indicated in integer tenths where, for example, a 5 indicates that the corresponding residue directly above it occurs in 50% to 60% of the weighted sequences. (A) Contrast hierarchical alignment of kinase-shared constraints. All protein kinases (10,583 sequences) comprise the foreground and all proteins, the background (see text for details). (B) Contrast hierarchical alignment of CMGC-specific constraints. CMGC kinases (1309 sequences) comprise the foreground and all protein kinases (i.e., the foreground set in A), the background sequences. Note that the alignment of SRPKs residues against P227 and G228 of the query (ERK2) is uncertain due to SRPK-specific insertions in this region; their structural locations are likewise uncertain due to disordering of this region in the Sky1p crystal structure. This may be related to SRPK-specific functions (see text).
In constructing a contrast hierarchical alignment, three sets of related sequences are multiply aligned: (1) a “foreground set,” which corresponds to the category of sequences with selective constraints that are being measured; (2) a “display set,” which is a subset of the foreground set; and (3) a “background set,” which is a superset of the foreground set. Only sequences in the display set are explicitly shown in the alignment. The foreground sequences are represented merely as conserved patterns and residue frequencies below the alignment (as in Fig. 1A,B ▶). The display set in Figure 1 ▶ is composed of representative CMGC kinases from distinct eukaryotic “kingdoms” for each subfamily examined here. Likewise, each of the display sets in Figure 2 ▶, A through F, corresponds to representative sequences from distinct phyla for a specific kinase subfamily.
Figure 2.
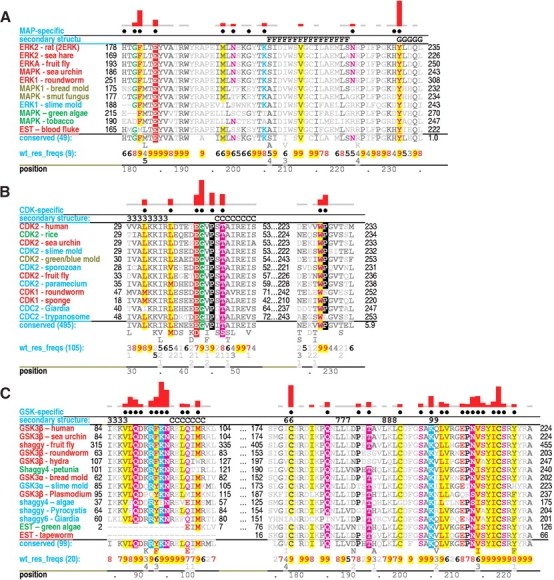
Critical regions of contrast hierarchical alignments showing various family-specific constraints within CMGC kinases. CMGC kinases (operationally defined as protein kinases with either an arginine or a lysine at the CMGC-arginine position) constitute the background sequence set for each alignment. See legend to Figure 1 ▶ for a further description of alignment highlighting and notation. See Materials and Methods for sequence identifiers. For each alignment, the sequences displayed are from distinct phyla. See text for details. (A) MAP-specific constraints. (B) CDK-specific constraints. (C) GSK-specific constraints. (D) SRPK-specific constraints. (E) DYRK-specific constraints. (F) CK2-specific constraints.
Category-specific selective constraints are measured by the degree to which residues in the foreground contrast with residues observed at corresponding positions in the background, which consists of a broader category of sequences than the foreground. More specifically, constraints are measured in terms of the difficulty of randomly drawing the amino acids observed at a particular position in the foreground from the background distribution at that position. Foreground positions with compositions that closely resemble the background thus will have little or no constraints, whereas positions with compositions incompatible (or that contrast) with the background will have strong constraints. This procedure thus emphasizes those strongly conserved subcategory-specific features that most diverge from the higher category-specific features and that, therefore, are more likely to play important roles in the distinct biological function of that subcategory.
Here we examine kinase-shared, CMGC-specific, and family- or subfamily-specific constraints. For kinase-shared constraints (i.e., those acting on all or nearly all protein kinases) sequences corresponding to all protein kinases constitute the foreground set, whereas the overall frequency of amino acids generally observed in all proteins serves as an implicit background at each position (Fig. 1A ▶). For CMGC-specific constraints, which distinguish the CMGC kinases from other protein kinases, CMGC kinases constitute the foreground, whereas all available protein kinases constitute the background (Fig. 1B ▶). For family or subfamily-specific constraints, which distinguish specific kinases from other CMGC kinases, the sequences corresponding to a specific family or subfamily constitute the foreground, whereas CMGC kinases constitute the background (Fig. 2 ▶). Our analysis also allows for another category of “intermediate” constraints, which here corresponds to residue positions that are highly conserved either within CMGC kinases as a whole or within a particular CMGC family or subfamily, but that are inconsistently conserved across the categories specifically examined here. These category-specific constraints are interpreted in light of available structural data below (a summary of which is given in Table 1). Note also that sequence fragments and other sequences that, for any reason, failed to align over the entire region of interest were eliminated from alignments.
Table 1.
Structural and functional observations and potential roles of CMGC-kinase residues
| Residue | Location | Comments | Reference |
| Residues in ERK2 characteristic of CMGC kinases | |||
| Y185 | Activation loop | Second phosphorylation site | Canagarajah et al. 1997 |
| V186 | Activation loop | Upon activation packs up against the HRD arginine | See text |
| R189 | APE region | P-2i-arginine: hydrogen bonds to substrate binding loop and to second activation loop phosphate moiety | Canagarajah et al. 1997; Brown et al. 1999; Dajani et al. 2001 |
| W190 | APE region | P-3i-aromatic: in Cdk2 interacts with substrate P-3 position | Brown et al. 1999 |
| R192 | APE region; P+1 binding pocket | The CMGC-arginine: hydrogen bonds to the carbonyl oxygen of the conformationally strained residue (A187) and interacts with phosphorylated tyrosine (Y186) | Brown et al. 1999; Canagarajah et al. 1997 |
| L198 | APE region | Mutation of this residue to alanine in ERK2 disrupt substrate binding | Lee et al. 2004 |
| Y203 | APE region | Stabilizes the backbone of the phosphorylated threonine (T160) | Johnson et al. 1996 |
| 1207 | αF helix | Packs up against Y203 | See text |
| C214 | αF helix | Potentially critical interaction with kinase-shared W210 | |
| A217 | αF helix | Packing interaction with L225 | |
| L225 | αF-to-αG loop | Mediates a CH-pi interaction with kinase conserved F226 | |
| P227, G228 | αF-to-αG region | Involved in hydrogen bonding interactions with the CMGC-glutamine and with peptide substrate | See text |
| Q234 | αF-to-αG region | CMGC-glutamine: hydrogen bonds to the backbone atoms linked to substrate binding | See text |
| L242, G243 | CMGC-insert | Possible structural roles related to the CMGC-insert. | See text |
| Resident characteristic of individual families | |||
| F181Erk2 | Activation loop | May mediate interaction with CMGC-insert upon kinase deactivation | See text |
| Y231Erk2 | N terminus of αG | Possible second phosphorylated tyrosine that could interact with CMGC-arginine; mutation of this residue disrupts substrate binding | See text; Lee et al. 2004 |
| G43Cdk2 | Loop connecting to αC | Provides flexibility in the PSTAIRE helix; hydrogen bonds to a cyclin A lysine that is subject to a strong cyclin A-specific constraint. | Russo et al. 1996 |
| Helix (PSTAIRE) | See text | ||
| P45Cdk2 | N terminus of αC | Cyclin binding and C-helix conformation | Russo et al. 1996 |
| T47Cdk2 | Helix (PSTAIRE) | ||
| W227Cdk2 | CMGC-insert | Packing of the CMGC-insert against the kinase C-terminal domain | See text |
| P228Cdk2 | |||
| Q89-N95Gsk2 | Loop before αC helix | Immediately before an arginine (R96) that hydrogen bonds with the activation loop first phosphorylation site | See text |
| C178Gsk2 | Catalytic loop | The side-chain sulfur packs against conserved hydrogen bonds between the HRD-backbone and a kinase-shared aspartate | See text |
| K205Gsk | Activation loop | Potential interaction with preprimed substrate phosphorylation site | Dajani et al. 2001 |
| C218Gsk | P+1 binding pocket | Corresponds to strained position interacting with CMGC-arginine | |
| N553Sky1p | Activation loop | Could potentially interact with the P+1 residue in the substrate | (Fig. 5e ▶) |
| Q566Sky1p | P+1 binding pocket | Corresponds to strained position interacting with CMGC-arginine | See text |
| P606Sky1p | αF-to-αG region | Occurs just before or within the Sky1p-specific insert | |
| H618Sky1p | αF-to-αG region | Replaces the CMGC-glutamine (Q234ERK2) | See text |
| C289Dyrk | Catalytic loop | Potential disulphide bond with C312 in the activation loop | See text |
| C312Dyrk | Activation loop | Potential disulphide bond with C289 | |
| Y319Dyrk | Activation loop | First phosphorylation site (normally threonine) | Becker and Joost 1999 |
| Q323Dyrk | P+1 binding pocket | Corresponds to strained position interacting with CMGC-arginine | See text |
| W176Ck2 | Activation loop | Packs against the C-helix | |
| E180Ck2 | Activation loop | Hydrogen bonds to an N-terminal CK2-specific tyrosine | Niefind et al. 2001 |
| K198Ck2 | P+1 binding pocket | Replaces the CMGC-arginine | See text |
| G199Ck2 | APE region | Replaces the APE alanine | |
| L213Ck2 | AF helix | Displaces the buried water typically found in protein kinases | See text |
Protein kinase-shared constraints
Because CMGC-conserved features are built upon the common structural framework that these proteins share with other kinases, we first briefly consider residues that are both relevant to our analysis here and also generally conserved in all protein kinases (Hanks and Hunter 1995).
We focus here on regions of the alignment within which most CMGC-conserved residues occur (Fig. 1 ▶). Eukaryotic protein kinases contain an N-terminal ATP binding domain and a C-terminal substrate-binding domain (Knighton et al. 1991; Engh and Bossemeyer 2002); nearly all of these CMGC-specific regions occur within the C-terminal domain. For conceptual and representational clarity, we sometimes define these regions based on the clusters of conserved interactions observed in our analysis (Fig. 3A ▶), rather than on conventional kinase terminology. Listed by their order in the sequence, these are as follows: (1) the αC region, which essentially corresponds to the protein kinase C-helix; (2) the catalytic loop, which contains an HRD motif; (3) the activation Nt-segment, which corresponds to the N-terminal part of the activation segment (Johnson et al. 1996) up to a conformationally strained residue (see below); (4) the APE region, which corresponds to conserved residues on either side of an APE motif and which thus includes the C-terminal part of the activation segment; (5) the αF-to-αG region, which stretches from the F-helix to the G-helix and which structurally surrounds the APE region; and (6) the CMGC insert. The CMGC insert, which is discussed at length below, is absent from other protein kinases and thus is a distinctive characteristic of the CMGC group.
Figure 3.

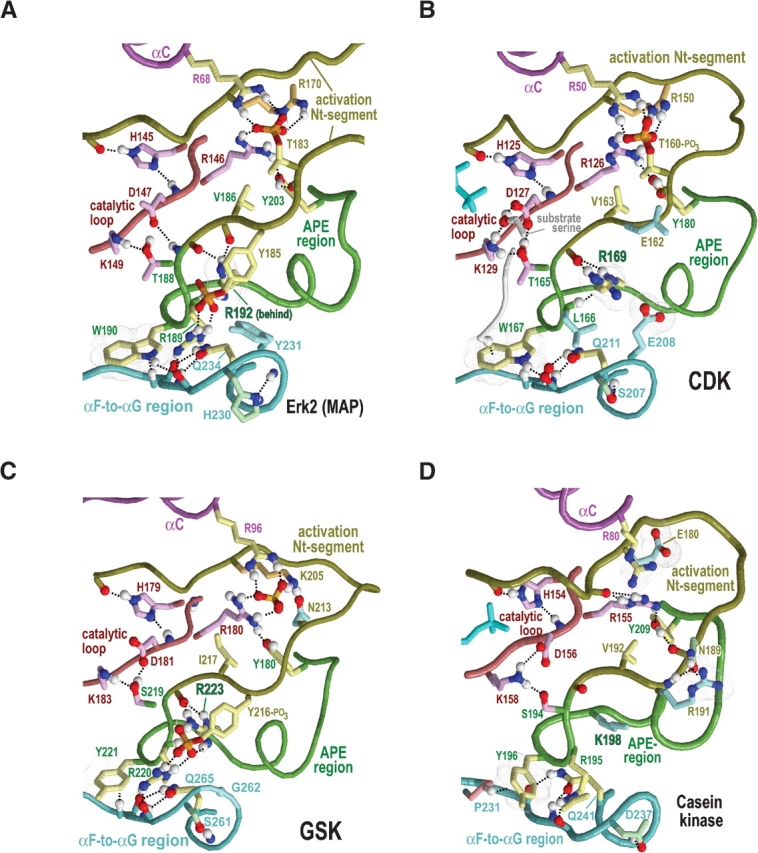
Structural features of CMGC protein kinases discussed in the text. The structure of p38 MAP kinase (PDB 1cm8) is shown as a prototype of the CMGC group. Color scheme is as follows: Main-chain traces of key regions, colored as indicated at the top of Figure 1 ▶; main-chain traces of other regions are light gray; ATPs are cyan; phosphate moieties use the standard CPK color scheme; oxygen, nitrogen, and hydrogen atoms establishing hydrogen bonds are red, blue, and white, respectively; side chains of kinase-shared residues are pale magenta; and CMGC-specific residues are light yellow. Hydrogen bonds are depicted as dotted lines; CH–π interactions (Weiss et al. 2001) are depicted as dotted lines into dot clouds. (A) Key regions within the protein kinase N- and C-terminal domains. The locations of the αC, αF, and αG helices are indicated. (B) Close-up of regions that stabilize and interact with the CMGC-arginine (R192). Modeled substrate with a P + 1 proline is shown.
The contrast hierarchical alignment in Figure 1A ▶ shows residue positions subject to kinase-shared constraints between the catalytic loop and the αF-to-αG region. (The side chains of such residues are shaded pale magenta in Figs. 3 ▶–6 ▶.) Certain residues in this category (Smith et al. 1999) either have clear functional roles in catalysis or directly interact with ATP, with phosphorylated residues, or with substrate. Nevertheless, many other residues are also subject to strong kinase-shared constraints, implying that they too play important, though still mysterious functions.
Figure 6.

CMGC canonical structural features near the αF-to-αG region and the CMGC insert. See the legends to Figures 3 ▶ and 4 ▶ for bond representations and coloring conventions; see text for details and discussion. (A) ERK2 MAP kinase (PDB 2erk). (B) CDK2 (PDB 1qmz). (C) GSK (PDB 1gng). (D) CK2α (PDB 1lp4). (E) Sky1p (PDB 1how). (F) Sky1p structural features near a histidine (H618) that replaces the CMGC-glutamine residue within SRPKs.
These functions are unlikely to involve merely the maintenance of the protein kinase fold, as this would require only very weak selective constraints, considering that sequences with essentially undetectable similarity may encode very similar folds. On the other hand, protein kinases cycle through distinct states associated with activation loop phosphorylation and dephosphorylation, with ATP binding, with substrate phosphorylation and ADP release, with activator or inhibitor binding, and with recognition, binding, and release of substrate proteins. It thus seems likely that many kinase-shared constraints involve mechanisms associated with the maintenance of or with choreographed transitions between these various states.
Certain interactions involving kinase-shared residues play a major role in coupling activation loop phosphorylation to the catalytic transfer mechanism (Johnson et al. 1996; Johnson and Lewis 2001). For example, an interaction between the side chain of the HRD-arginine and one of the activation loop phosphorylation sites (Fig. 3A ▶) helps reposition the activation loop for substrate binding (Johnson et al. 1996). Likewise, a kinase-shared threonine or serine in the APE region (T188 in Fig. 3B ▶) helps reposition the activation loop for catalysis by hydrogen bonding with both a lysine and a aspartate in the catalytic loop (K155 and D153 in Fig. 3B ▶; Madhusudan et al. 1994). These and other kinase-shared residues that serve as a structural foundation for group-specific features are considered in the context of CMGC-specific constraints in the following section.
CMGC-specific constraints
A contrast hierarchical alignment corresponding to CMGC-specific constraints is shown in Figure 1B ▶. Because MAPKs best conserve the CMGC canonical features, we use two MAPK subfamilies, ERK2 and p38, as prototypes of this group. Most CMGC-specific residues (the side chains of which are shaded light yellow in Figs. 3 ▶–6 ▶) are located from just before the APE region to just beyond the αF-to-αG region (Fig. 1 ▶). A few other CMGC-specific residues occur outside of the aligned regions shown in Figure 1 ▶, but only two of these are discussed here: a canonical arginine (R68ERK in Fig. 5A ▶, below) within the C-helix and a canonical phenylalanine/tyrosine (F294ERK in Fig. 6A ▶, below) ~20 residues from the end of the kinase C-terminal domain.
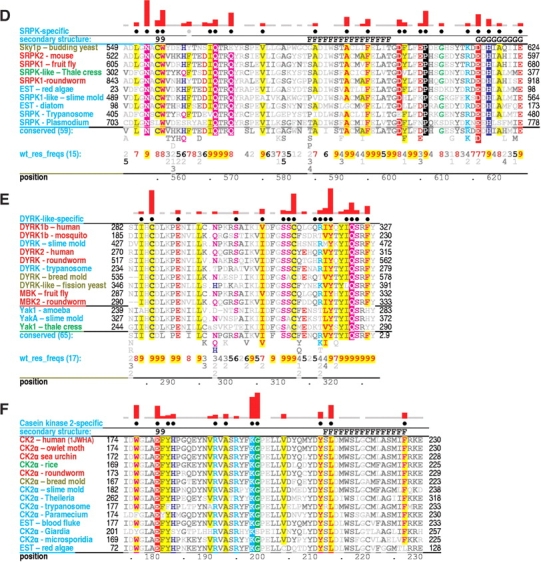
Figure 5.

Category-specific structural features within and surrounding the activation segment. Electrostatic interactions are depicted as dot clouds. See the legends to Figures 3 ▶ and 4 ▶ for other bond representations and coloring conventions; see text for details. (A) ERK2 MAP kinase (PDB 2erk). (B) CDK2 (PDB 1qmz). (C) GSK (PDB 1gng). (D) CK2α (PDB 1ds5). (E) Model of substrate-bound SRPKs. This model is based on the non–substrate-bound form of the SRPK-related Sky1p protein (PDB 1how) and on CMGC-specific interactions revealed by our analysis. Residue numbering is based on the Sky1p structure. (F) Hypothetical model of DYRK (constructed as described in the text).
The CMGC-arginine: Structural context
The most characteristic CMGC feature corresponds to what we term the CMCG arginine, which lines the base of the P + 1 substrate-binding pocket and is near the center of most of the other canonical features. The main chain of this ar-ginine is stabilized by interactions involving kinase-shared residues. For example, the conformation of a kinase-shared tyrosine directly before the CMGC-arginine (Y191ERK1 in Fig. 1A ▶) is stabilized via two canonical kinase-shared interactions: a CH–π interaction with the main chain of the catalytic loop (Y191p38 in Fig. 3B ▶) and a hydrogen bond with a kinase-shared glutamate (E218ERK1 in Fig. 1A ▶; not shown in Fig. 3B ▶). Likewise, the alanine and proline of the kinase-shared “APE” motif—two residues that directly follow the CMGC-arginine (see Fig. 1 ▶)—form canonical CH–π interactions with a kinase-shared tryptophan (W210p38 in Fig. 3B ▶) within the αF helix. Furthermore, a kinase-shared serine residue (S211p38 in Fig. 3B ▶) that is sequence adjacent to this tryptophan hydrogen bonds to a main-chain oxygen located next to the CMGC-arginine.
In nearly all CMGC kinases of known structure (apart from casein kinase 2; see below) this serine also hydrogen bonds to or contacts two buried waters, one of which, in turn, often hydrogen bonds to the main chain adjacent to the CMGC-arginine (Fig. 4 ▶). The second buried water extends this hydrogen-bonding network to the main-chain nitrogen of a catalytic aspartate and to another kinase-shared aspartate with a side chain that, in turn, hydrogen bonds to the catalytic loop (D147ERK and D208ERK, respectively, in Fig. 4A ▶). These buried waters are also found in non-CMGC kinases of known structure and thus may play important structural roles in all protein kinases. Together the buried waters and kinase-shared residues thus appear to stabilize and precisely position the conformation of the main chain of CMGC-arginine relative to the αF helix and the catalytic loop (Fig. 4 ▶).
Figure 4.


Structural features surrounding buried waters located between the APE region and the catalytic loop in CMGC and other protein kinases. In the main figures, the buried waters are shown as oxygens surrounded by turquoise dot clouds; in the inset, predicted hydrogen bonds formed by these waters are shown. Residue side chains and canonical glycine main chains are colored by functional categories as follows: kinase-shared, pale magenta; CMGC-specific, light yellow; intermediate between kinase-shared and CMGC-specific, light orange to pale red; family-specific, pale cyan; intermediate between CMGC-specific and family-specific, pale green; and subfamily-specific, light blue. Hydrogen bonding carbons are colored as their corresponding side chains, and van der Waals interactions are depicted as dot clouds. See the legend to Figure 3 ▶ for other bond representations and coloring conventions; see text for details. (A) Erk2 MAP kinase (PDB 2erk). (B) CDK2 (PDB 1qmz). (C) GSK (PDB 1gng). (D) CK2α (PDB 1lp4). In CK2α the water nearer the APE region is missing, presumably due to filling of its binding cavity by the side chain of L213, which is distinctively conserved within CK2α. (E) The SRPK-related Sky1p kinase (PDB 1how). The water nearer the APE region is replaced by the side chain of Q566, which occupies the conformationally strained position in SRPKs. Note that both the nitrogen and the oxygen of the glutamine side chain participate in hydrogen bonds that, in other protein kinases, are typically established by this water. (F) The (non-CMGC) phosphorylase kinase (PDB 2phk). As shown here, these buried waters also occur in non-CMGC protein kinases.
The CMGC-arginine: Side-chain interactions
In the active form (and in some inactive forms) of most CMGC kinases, the CMGC-arginine side-chain hydrogen bonds to the main-chain oxygen of a nonglycine residue (A187ERK in the Fig. 4A ▶ inset) that is located in the activation loop and that undergoes main-chain torsion angle strain upon activation loop phosphorylation (Wiechmann et al. 2003). This hydrogen bond lies at the base of the P + 1 substrate-binding pocket and thus may be involved in P + 1 proline recognition (Brown et al. 1999) inasmuch as it neutralizes the dipole moment of the main-chain oxygen of the strained residue and thereby allows hydrophobic interaction with proline (within non–proline-directed kinases, the corresponding main-chain oxygen hydrogen bonds to the substrate P + 1 residue main-chain nitrogen, which proline lacks; Lowe et al. 1997). Consistent with this, direct evidence for CDK, MAPK, and DYRK and anecdotal observations for other CMGC kinases, such as GSK (Pinna and Ruzzene 1996), suggest that many, if not most CMGC-arginine containing kinases are, at least to some extent, P + 1 proline-directed.
The side chain of the CMGC-arginine also electrostatically interacts with and hydrogen bonds to a second activation loop phosphate moiety present in several CMGC kinase structures (Canagarajah et al. 1997; Bax et al. 2001), implying that it also plays a key role in the activation mechanism. We will call this site the “second phosphorylation site” to distinguish it from the site sensed by the HRD-arginine, which we term the “first phosphorylation site”. Indeed, MAP kinases, which contain this second phosphorylation site, are fully activated only upon phosphorylation of both sites (Figs. 3A ▶, 5A ▶; Canagarajah et al. 1997), and each site has a distinct role: phosphorylation of the first site promotes ATP binding, whereas phosphorylation of both sites is required for substrate binding (Prowse et al. 2001). The second site corresponds to a CMGC-specific tyrosine (Y185ERK in Fig. 5A ▶) that occurs in three of the four major CMGC subgroups (i.e., MAPKs, GSKs and CLKs; Manning et al. 2002).
Cdc7 kinases, which CHAIN analysis clearly places outside of the CMGC group, also contain an arginine at the CMGC-arginine position. Notably cdc7 kinases typically possess a very large activation loop, which suggests an atypical activation mechanism, and, as do most non-CMGC kinases, conserve a glycine at the residue position corresponding to the CMGC conformationally strained position.
Other canonical features of CMGC kinases
The canonical features of CMGC kinases are clustered into several regions of the multiple sequence alignment, but by far the most striking of these extends from the activation segment to the αF-to-αG region (Fig. 1 ▶). As described in the following sections, these residues appear to link together the substrate, the activation loop phosphorylation sites, and the CMGC-insert.
CMGC canonical residues within the activation loop and APE region
There are two CMGC canonical residues within the activation loop: the second phosphorylation site tyrosine (Y185ERK), and a valine or isoleucine (V186ERK) that directly follows this tyrosine in the sequence. This valine/ isoleucine, upon phosphorylation of the first activation loop site in ERK2, comes into contact with the aliphatic region of the HRD-arginine side chain (see R146ERK in Fig. 5A ▶). At the same time, it also packs against another CMGC canonical tyrosine (Y203ERK) that is within the APE region and that hydrogen bonds to the main chain of the first phosphorylation site (T183ERK; Figs. 4 ▶, 5 ▶). Furthermore, this valine/isoleucine directly precedes the conformationally strained position (A187ERK), and is thus sandwiched between two residue positions (i.e., Y185ERK and A187ERK) linked to activation loop conformational changes. Taken together, these observations suggest that this valine/isoleucine plays a role in CMGC kinase activation.
Within the APE region there are two other CMGC canonical residues: an arginine (R189ERK) that likely interacts with the P-2 substrate position (and thus is termed the P-2i-arginine), and an aromatic residue, which is most often a tryptophan (W190ERK), that likely interacts with the P-3 substrate position (and thus termed the P-3i-aromatic; Fig. 4 ▶). We infer these substrate interactions based on homology to three distinct peptide-bound structures (Protein Data Bank [PDB] codes 1IR3, 1JBP, and 1QMZ). The P-2i-ar-ginine, which is sequence adjacent to the kinase-shared serine or threonine (T188ERK) that interacts with the catalytic aspartate (D147ERK), typically hydrogen bonds to a loop between the αF and αG helices that is involved in substrate binding (Brown et al. 1999). Similar to the CMGC-arginine, it also hydrogen bonds with the second activation loop phosphate moiety, when present (Canagarajah et al. 1997; Dajani et al. 2001). Indeed, our analysis indicates that conservation of the P-2i-arginine is most highly correlated with conservation of tyrosine at the second phosphorylation site and vice versa (data not shown), suggesting a functional coupling between this arginine and phosphorylation of this tyrosine. Incidentally, for CDKs, which lack a second activation loop phosphorylation site, the P-2–interacting residue is a conserved leucine instead of the canonical arginine; this leucine thus may play an important CDK-specific role.
CMGC structural features within the αF-to-αG region
The αF-to-αG region, which consists of a loop flanked by helices (Fig. 3 ▶), contains several CMGC canonical residues that appear to link this loop to the αG helix and to the substrate-binding pocket. For example, the side chain of a canonical glutamine (Q234ERK; termed the CMGC-glutamine) within the αG helix typically hydrogen bonds to main-chain atoms located on either side of a particular loop residue that is most often a proline (P227ERK). The Cα carbon of this loop residue often forms a CH–π interaction with the P-3i-aromatic residue (W190ERK in Fig. 6A ▶). Additional canonical interactions within this region involve both kinase-shared and CMGC-specific conserved residues (as shown in Fig. 6A ▶–E), as well as residues that are non-conserved or inconsistently conserved at the sequence level. For example, a hydrophilic residue (H230ERK) two positions beyond a CMGC-conserved glycine (G228ERK) typically serves as an N-cap for the αG helix (Fig. 6 ▶). Similarly, the main-chain nitrogen of the residue preceding this hydrophilic position forms a hydrogen bond with the side chain of a residue that is usually an aspartate (D233ERK) and that immediately precedes the CMGC-glutamine. Together, these interactions form a structural link between the substrate-binding region and the CMGC insert via the αG helix (Fig. 6 ▶).
An LG[ST]P motif associated with the CMGC-insert
The CMGC-specific consensus pattern LG[ST]P (corresponding to residues 242–245 of mouse ERK2 in Fig. 1 ▶) occurs just beyond the αG helix at the start of the CMGC insert. Curiously, the threonine or serine position of this pattern (S244ERK) has a high predictive probability of being phosphorylated (see Materials and Methods), although for some CMGC families, such as DYRK and related kinases, this serine or threonine is nonconserved. The glycine of this motif (G243ERK) is at the end of and perhaps also terminates the αG helix. Both the leucine and proline of this motif typically pack up against a CMGC canonical phenylalanine or tyrosine (F294ERK in Fig. 6A ▶; not shown in the Fig. 1 ▶ alignment) near the C-terminal end of the kinase domain. Conservation of the LG.P residues implies that they perform an important structural role, possibly linked to the adjacent CMGC insert. This insert may be involved in coprotein binding, considering that adaptor or scaffold-like proteins bind to this region in CDK2 and GSK3β (Bourne et al. 1996; Bax et al. 2001; Dajani et al. 2003) and that point mutations in the CMGC insert directly or indirectly affect binding of MEK1 to ERK2 (Robinson et al. 2002).
Links between substrate recognition, activation, and the CMGC insert
The CMGC-arginine and the nearby P-2i-arginine are predicted to interact with bound substrate and with the second activation loop phosphorylated site. Similarly, other CMGC canonical residues directly interact either with one of the activation loop phosphorylation sites or with residues that do. Still other CMGC-specific residues are located between the CMGC insert and the substrate or the activation loop (Figs. 4 ▶–6 ▶). What function or mechanism is responsible for the selective constraints acting on these residues? One possibility is that they couple coprotein binding to substrate recognition and kinase activation. Consistent with this notion, hydrogen exchange experiments (Lee et al. 2004) show that substrate docking results in conformational mobility within the P + 1 binding pocket, the αF-to-αG region, and the CMGC insert region of ERK2. Similarly, phosphorylation of the ERK2 activation loop induces conformational changes within the CMGC insert (Canagarajah et al. 1997). CMGC-specific features likewise may play a role in the ‘gated’ behavior observed for CDKs and MAPKs (Adams 2003), in which phosphorylation induces an activation loop switch from an unfavorable to a favorable substrate binding conformation. Specific aspects of such a mechanism would, of course, depend on the particular CMGC subgroup or family—evolutionary analysis of which may likewise provide useful clues.
Structural features specific to CMGC subgroups
Phylogenetic analysis (Manning et al. 2002) classifies CMGC kinases into four major families: CDKs, MAPKs, GSKs, and CLKs. CHAIN analysis of these reveals family-specific features, which we explore in the light of CMGC canonical features.
MAPK-specific constraints
The MAPKs (Johnson and Lapadat 2002), which include p38 and ERK2, best conserve CMGC canonical features and thus serve as a prototype. The p38 kinases regulate cytokine signaling, whereas ERK2 regulates mitosis, meiosis, and postmitotic functions in differentiated cells. MAPKs are highly P + 1 proline directed Ser/Thr kinases, which is consistent with a role for the CMGC-arginine in proline recognition (see above). High ERK2 activity requires phosphorylation of two activation loop residues (T183 and Y185 in Fig. 5A ▶; Robbins et al. 1993).
A phenylalanine (F181ERK) within the activation loop and a tyrosine (Y231ERK) at the N-terminal end of the αG helix most distinguish ERK2 and closely related kinases from other CMGC kinases (Fig. 2A ▶). In the inactive form, the phenylalanine packs up against the CMGC insert, whereas in the active form, it swings away from the CMGC insert and becomes substantially more solvent exposed (data not shown), which suggests a function related to the CMGC insert. A phosphorylated form of the tyrosine could interact with the CMGC arginine and with the P-2i-arginine more or less as does the tyrosine at the second activation loop phosphorylation site (Y231ERK in Fig. 5A ▶)—perhaps thereby altering substrate specificity or rate of catalysis.
CDK-specific constraints
Distinct cyclin-dependent kinases are sequential activated by cyclins and thereby regulate the ordering of events associated with DNA replication and cell division (for review, see Endicott et al. 1999). A prominent feature distinguishing CDKs from other CMGC kinases is the consensus pattern EG.P.T (residues 42–47 of CDK2 in Fig. 2B ▶). This pattern directly precedes and partially overlaps with the cyclin-interacting αC helix that corresponds to the consensus pattern PSTAIRE. The function of the PSTAIRE residues in mediating interactions with cyclin are clear from the structures of cyclin-bound CDKs (Russo et al. 1996; Brown et al. 1999). Nevertheless, CHAIN analysis assigns the strongest CDK-specific constraint to the glycine of the EG.P.T pattern (Fig. 2B ▶) and, likewise, assigns the strongest cyclin A constraint (data not shown) to a lysine residue (K266) that hydrogen bonds to main-chain oxygens on either side of this glycine. Thus the interaction between these two residues seems quite important.
Another feature distinguishing CDKs from other CMGC kinases is the consensus pattern WP within the CMGC insert (residues 227–228 of CDK2 in Fig. 2B ▶). The tryptophan of this pattern packs up against the kinase C-terminal domain proper and thus may serve as an important link to the CMGC insert. Another distinguishing feature of many CDKs is replacement of the CMGC canonical P-2i-arginine with another conserved residue, typically a leucine (L166CDK2 in Figs. 4C ▶, 5C ▶). Notably, CDKs typically lack the second phosphorylation site and thus may not require the P-2i-arginine for phosphate binding. The CDK7 subfamily, however, retains the canonical P-2i-arginine at this position, even though it also lacks a second phosphorylation site. A similar arrangement occurs within SR protein kinases (SRPKs), which may use an alternative activation mechanism involving a surrogate phosphorylation site donated by the substrate (see below).
The CDK2 subfamily manifests specific features absent in other CDKs. One such feature is a conserved arginine (R122CDK2) that is highly buried upon binding to cyclin and that, in the cyclin-bound form, forms a salt bridge with a glutamate (E57CDK2) that is also highly conserved within the CDK2 family (alignments and structures not shown). Also in place of the second phosphorylation site, CDK2 has a highly conserved glutamate (E162CDK2 in Fig. 5B ▶) that potentially could replace the phosphate electrostatic interaction with the CMGC-arginine; however, such an interaction is observed in only one of the known active form structures of CDK, namely, phospho-CDK2/cyclin A bound to a recruitment peptide (PDB 1h24; Lowe et al. 2002).
GSK-specific constraints
GSK-3 is a key regulator of glycogen metabolism, the Wnt signaling pathway, protein synthesis, and cell proliferation and differentiation (for review, see Grimes and Jope 2001; Harwood 2001; Doble and Woodgett 2003). Efficient phosphorylation of many of its substrates requires prior phosphorylation at the substrate P + 4 position by another kinase (Dajani et al. 2001; Frame et al. 2001). GSK-3 lacks the first activation loop phosphorylation site, which is located near where this preprimed substrate site is likely to occur. Moreover, in the active structure of GSK3β, both the HRD-arginine and a CMGC-specific arginine interact with a sulfate ion (Figs. 4C ▶, 5C ▶), which can mimic a phosphate and is predicted to occupy the same site as the P + 4 phosphate in the preprimed substrate (Dajani et al. 2001; ter Haar et al. 2001). This preprimed substrate thus may serve as a surrogate for the first activation loop phosphorylated site. Likewise, a GSK-specific lysine (K205GSK3β) occurs within the activation loop and, in the active form, hydrogen bonds to a sulfate ion. Because this position corresponds to a conserved arginine that interacts with the first phosphorylated site (R170ERK2 and R150CDK2 in Fig. 5 ▶), this lysine seems likely to interact with the substrate preprimed phosphate as well (Fig. 5C ▶). It also hydrogen bonds to a GSK-specific conserved asparagine (N213 in Fig. 5C ▶) that corresponds to the first phosphorylation site threonine in other CMGC kinases. GSK3β also contains a second activation loop (tyrosine) phosphorylation site (Bax et al. 2001).
Some of the strongest GSK-specific constraints are imposed on a tight cluster of conserved residues, namely, Q89, R92, F93, K94, and N95 (Fig. 2C ▶). These residues seem likely to perform a regulatory role, as this pattern occurs just before a CMGC-specific arginine (R96GSK3β) that typically hydrogen bonds to the phosphate of the first phosphorylation site. Two of the strongest GSK-specific constraints (Fig. 2C ▶) correspond to two cysteines: one (C218) at the strained position that interacts with the CMGC-arginine (Fig. 4C ▶, inset) and another (C178) directly before the HRD motif. The side-chain sulfur of this second cysteine (data not shown) packs up against two hydrogen bonds that are conserved in essentially all protein kinases and that are formed between the HRD catalytic loop and a conserved aspartate in the αF helix (D239GSK3β in Fig. 4C ▶). Thus, this cysteine may influence the chemical nature of these critical interactions.
CLK kinases
The CDK-like kinases are functionally diverse and conserve fewer of the CMGC canonical features. In addition, they generally lack the HRD-arginine and instead often harbor another highly conserved residue at that position. Instead of the hydrophobic residues usually found in other CMGC-kinases at the conformationally strained position (see above), CDK-like kinases typically contain glutamine or serine, the polar side chains of which can form hydrogen bonds. Within this group we specifically examine SRPKs and DYRKs.
SR protein kinases
SRPKs phosphorylate ‘SR’ dipeptide repeats in RNA processing factors (Colwill et al. 1996). Unlike MAP and CDK kinases, SRPKs do not have a strong requirement for proline at the P + 1 position but rather typically accommodate arginine there. They exhibit a level of constitutive activity but may require a preprimed substrate phosphate for optimum in vivo activity, considering that the structures of SRPKs contain a sulfate ion (which can mimic a phosphate; Nolen et al. 2001, 2003) near its predicted site of interaction with the substrate P + 2 position. This situation thus is analogous to that of GSK3β, the substrate of which must be prephosphorylated for recognition (see above).
To explore the structurally feasibility of an interaction between the P + 2 phosphorylated substrate serine and the CMGC- and P-2i-arginines, both of which are conserved in SRPKs, we constructed an homology model of SRPK with bound substrate (see Materials and Methods). This model indeed suggests that the previously phosphorylated P + 2 substrate position, which is likely to be in roughly the same structural location as a second activation loop phosphate, may function as a surrogate activation site phosphate (Fig. 5E ▶). This P + 2 phosphate may function to ensure processive phosphorylation of SR proteins (Aubol et al. 2003) rather than or in addition to activating the kinase. Another distinctive feature of SRPKs with a possible role in recognition of the substrate P + 2 phosphate is the insertion of six additional residues within the αF-to-αG region (these correspond to positions 231–233 of ERK2 in the hierarchical alignment of Fig. 1 ▶). Some of these inserted residues are located very near the CMGC-arginine and the P-2i-arginine (disordered region in Fig. 6E ▶), and all of these are near the proposed surrogate phosphorylation site.
SRPKs substantially diverge from canonical features relative to other CMGC kinases. In particular, the P-3i-aromatic residue is typically replaced by a conserved glutamine (position 569 in Fig. 2D ▶). The yeast Sky1p kinase, is unusual, however, inasmuch as it contains a glutamate instead of a glutamine at this position (E569Sky1p in Fig. 4D ▶). This may be due to the fact that the Saccharomyces cerevisiae genome lacks SR protein encoding genes, implying that Sky1p is a paralog rather than an ortholog of SRPKs. In any case, this glutamine (termed here the SKPK-specific glutamine) appears well situated to interact with a substrate P-2 serine, as is shown in the homology model for substrate-bound SRPKs (Fig. 5E ▶).
Another divergent, highly conserved SRPK feature is the replacement of the CMGC-glutamine by a histidine (H618Sky1p in Fig. 2D ▶), a substitution also observed for the SRPK-related Lammer and CDC-like kinases, many of which are also known to phosphorylate serine/arginine rich substrates (Nikolakaki et al. 2002). Unlike the CMGC-glutamine, the Sky1p histidine fails to interact with the main chain of the substrate interaction loop and instead interacts with a buried water (Nolen et al. 2001, 2003). This water, in turn, hydrogen bonds to the side chain of the kinase-shared tryptophan (W588Sky1p in Fig. 6F ▶) within the αF helix and to the main chain of the APE region and thus, together with H618Sky1p, forms a network of precise interactions positioning key residues within the APE loop (Fig. 6F ▶).
A feature of SRPKs possibly related to substrate specificity is a highly conserved glutamine (Q566Sky1p in Fig. 2D ▶) at the conformationally strained position with a main chain that typically hydrogen bonds to the side chain of the CMGC-arginine. A comparison of the Sky1p structure with those of other CMGC kinases reveals that the side chain of this glutamine displaces the buried water that forms hydrogen bonds to main-chain atoms on either side of the CMGC-arginine (Fig. 4 ▶, insets), resulting in a different geometric arrangement. More specifically, both the side-chain oxygen and nitrogen of this glutamine hydrogen bond to the main-chain atoms on either side of the CMGC-arginine. The side-chain nitrogen also hydrogen bonds to a main-chain oxygen directly preceding a kinase-shared aspartate (D586Sky1p in Fig. 4D ▶) that, in turn, hydrogen bonds to the main chain of the catalytic loop. Together, these interactions thus displace the hydrogen bonds typical of those CMGC kinases containing water at this position and, as a result, appear to reposition the catalytic loop and APE region relative to each other—presumably, in a manner more favorable to the specific function of SRPK.
Yet another feature possibly related to SR specificity is a SRPK-specific asparagine (N553 in Fig. 2D ▶) directly following the protein kinase DFG motif (which, in fact, is most often DLG in SRPKs). This asparagine is predicted to pack up against the substrate P + 1 position (Fig. 5E ▶), given that, in the structure of CDK2 bound to substrate, the corresponding residue, which is a leucine, extensively packs up against the proline at the substrate P + 1 position. Indeed, the area of contact of the leucine with the P + 1 proline is greater than that of any other residue in CDK2. This SRPK asparagine thus may perform an analogous role in substrate P + 1 arginine recognition.
There is anecdotal evidence, however, that SRPKs favor both arginine and proline at the substrate P + 1 position (Colwill et al. 1996). For example, Npl3p, a budding yeast shuttling protein that is the natural substrate of Sky1p, is phosphorylated by mammalian SRPK1 on a serine followed by a proline (Gilbert et al. 2001). Sky1p likewise can phosphorylate serine residues within RS domains of mammalian proteins (Nolen et al. 2001), which are substrates of mammalian SRPKs. Furthermore, a pattern-based analysis of SR repeat regions within SR proteins (see Materials and Methods) reveals a highly elevated propensity for both arginine and proline at the ambiguous position (x) within the pattern S-R-x, as follows:
Table t2.
| Pattern | Expected | Observed | E-value |
| RSR | 482 | 1277 | 10−196 |
| RSP | 219 | 531 | 10−69 |
| RSY | 63 | 108 | 0.00001 |
| RSL | 101 | 138 | 0.006 |
This implies a strong selective pressure for proline following RS patterns within RS domains. One possible explanation for this is that proline is also favored at the P + 1 substrate position by SRPKs. This hypothesis also helps explain conservation in SRPKs of the CMGC-arginine.
DYRK and DYRK-like kinases
Dual specificity tyrosine phosphorylated and regulated kinases (DYRKs) phosphorylate serine, threonine, and tyro-sine residues and—though possessing significant constitutive activity—are fully activated only after autophosphorylation on a Y-x-Y pattern corresponding to their two activation loop phosphorylation sites (Becker and Joost 1999). Upon full activation, they are specific to substrates with either proline or arginine at the P + 1 position (Himpel et al. 2000; Campbell and Proud 2002).
As for SR kinases, a distinguishing feature of DYRKs is a glutamine (Q323DYRK in Fig. 2E ▶) at the conformationally strained position within the P + 1 binding pocket. The function of this residue may thus be similar to that in SR kinases. Notably, mutation of this glutamine to asparagine within one DYRK had as great an effect on catalytic activity as mutation of the second phosphorylation site tyrosine to phenylalanine (Wiechmann et al. 2003). Another distinguishing feature is replacement of the HRD-arginine with cysteine (C286DYRK). A homology model of the active form of DYRK, based both on CMGC canonical features and known active conformation structures, suggests that this cysteine might stabilize the activation loop through disulphide bond formation with another DYRK-specific cysteine located nearby in the hypothetical structure (C312DYRK in Fig. 5F ▶).
Casein kinase 2
CK2 is the only family within the CMGC group that replaces the CMGC-arginine with a lysine. This may allow phosphorylation of substrates with either proline or nonproline at the P + 1 position due to the side-chain flexibility of lysine, which allows hydrogen bonding to the main-chain oxygen at the strained position, as does the CMGC-arginine, yet can accommodate alternative hydrogen bonds as well. A glycine residue (G199) that directly follows this lysine and that likewise is subject to strong CK2-specific constraints (Fig. 2F ▶) may contribute to the inherent conformational flexibility of the CK2 lysine by allowing a greater range of main-chain conformations.
CK2α lacks the same buried water that is absent in SR-PKs, namely, the water that hydrogen bonds to the main chain near the CMGC-arginine position. (Out of nine available CK2α structures, only the structure of a, possibly functionally deficient, C-terminal deletion mutant [Ermakova et al. 2003] contains a water molecule at this position.) In SRPK this water cavity is occupied by the side-chain atom of a glutamine located at the “strained position” (Fig. 4E ▶), but in CK2α s this water cavity typically overlaps with a side-chain methyl group of a CK2α-specific leucine (L213CK2α in Fig. 4D ▶, inset). Notably, although leucine is invariant or nearly invariant at this position in CK2α, it apparently never occurs at this position in other CMGC kinases but rather is typically replaced by a CMGC-specific isoleucine or valine, neither of which prohibits the buried water. It thus appears that even conservative replacement of this leucine by isoleucine or valine or vice versa is highly selected against in these families, implying that even very subtle amino acid differences may have profound effects on protein function. Unlike isoleucine or valine, which apparently forms a Cβ-H hydrogen bond to the oxygen of the water at this position, leucine is incapable of such a bond. This leucine thus may contribute to the broad substrate specificity of CK2α by relaxing CMGC-canonical interactions near the active site.
A well-conserved CK2-specific glutamate (E180 in Fig. 2F ▶) seems likely to play a role in N-terminal–mediated regulation of the activation loop. In many other CMGC kinases, this residue corresponds to an activation loop arginine that participates in binding to the phosphate of the first activation loop phosphorylation site (Fig. 5 ▶). This glutamate instead hydrogen bonds to an invariant tyrosine (Y23) within the N-terminal region of CK2 (data not shown) and directly precedes three CK2-specific aromatic residues that likewise interact with the N-terminal region (data not shown).
Conclusion
CMGC canonical residues appear to couple kinase activation and substrate recognition to substrate and coprotein binding, whereas both variation of and additions to these features within individual subcategories presumably contribute to CMGC functional specialization. Conserved residues generally shared by all protein kinases and buried waters located below the APE region appear to play important roles in precise geometric positioning of key CMGC-specific residues. Our analysis suggests hypothetical roles for these residues and provides guidance for mutational studies to explore these roles—including, for example, conversion of the CMGC-glutamine to glutamate to explore the role of the side-chain nitrogen or mutation of the CK2 leucine to isoleucine. Similar conservative mutations aimed at broadening our understanding of CMGC kinase function can readily be proposed.
Materials and methods
CHAIN analysis
CHAIN analysis is described in Neuwald et al. (2003); for a conceptual description, see Results and Discussion. In essence, CHAIN analysis focuses on finding sequence subgroups, each of which share a strikingly conserved pattern that is strikingly nonconserved in sequences outside of that subgroup. An important underlying assumption is that the co-conserved residues of such a pattern roughly correspond to a functional module that—given its persistence over a billion years or more of evolution—likely mediates structural mechanisms important for survival. A second assumption is that the degree to which aligned residue positions within a subgroup have shifted away from the composition observed at that position in sequences outside of that subgroup provides a measure of the subgroup-specific selective pressure acting on that residue position.
Other structural and sequence analysis procedures
Protein hydrogen atoms were added to structural coordinates by using the Reduce program (Word et al. 1999); to add hydrogens to water, we used the method of Hooft et al. (1996). Homology models based on our analysis were manually constructed and optimized by using the RAMP suite of programs (Samudrala et al. 2000) and the O program (Jones 1978). In particular, homology models for DYRK were based on CDK2 (PDB 1qmz), ERK2 (PDB 2erk), and Sky1p (PDB 1how). Structural images were created by using Rasmol (Sayle and Milner-White 1995). Ramachandran plots were examined by using the program PROCHECK (Morris et al. 1992). Secondary structure assignments were made by using the DSSP program (Kabsch and Sander 1983). Phosphorylation site predictions in kinase sequences were performed by using the NetPhos program (Blom et al. 1999). Statistically significant amino acid patterns associated with SR proteins were examined by using the ASSET program (Neuwald and Green 1994). Structural alignments were performed by using the CE program (Shindyalov and Bourne 1998), as previously described (Neuwald 2003).
Sequences displayed in alignments
National Center for Biotechnology Information (NCBI) sequence identifiers for the CMGC alignments in Figure 1 ▶ are as follows: ERK2-mouse (2ERK), 6754632; ERK2-green algae, 11275338; ERK1-slime mold, 1169550; MAPK-fission yeast, 19113755; P38γ (1CM8A)-human, 8569500; CDK2-human (1FINA), 16936528; CDC2–1-rice, 231706; CDC2-like-slime mold, 461704; CDK2-green/blue mold, 2499588; GSK3β-human (1IO9A), 20455502; GSK3α-bread mold, 32405824; Shaggy PK4-petunia, 1076649; GSK3-slime mold, 1730041; CK2-human (1JWHA), 20150571; CK2α2-maize, 11527006; CK2-aerobic yeast, 1694914; CK2α-Leishmania, 10046857; DYRK1b-mouse, 12054926; DYRK-slime mold, 28829499; SRPK2-mouse, 18043214; SRPK1-slime mold, 28829647; Sky1p-budding yeast (1HOWA), 6323872; and SRPKL-thale cress, 11259819.
NCBI sequence identifiers for the ERK2 alignment in Figure 2A ▶ are as follows: Erk2-rat, 3318705; Erk2-sea hare, 1110512; ErkA-fruit fly, 17977692; MAPK-sea urchin, 24286498; ERK1-roundworm, 32564571; MAPK1-bread mold, 32421451; MAPK-smut fungus, 6457281; ERK1-slime mold, 1362214; MAPK-green algae, 11275338; MAPK(Nrk1)-tobacco, 12718824; and EST-blood fluke, 28325407.
NCBI sequence identifiers for the CDK2 alignment in Figure 2B ▶ are as follows: Cdk2-human, 16936528; Cdc2-rice, 231706; Cdk2-sea urchin, 2956719; Cdk2-slime mold, 461704; Cdk2-green/blue mold, 2499588; Cdk2-sporozoan, 1420882; Cdk2-fruit fly, 115918; Cdk2-paramecium, 4959457; Cdk1-roundworm, 17554940; Cdk1-sponge, 21304629; Cdc2-Giardia, 29409213; and Cdc2-trypanosome, 1705673.
NCBI sequence identifiers for the GSK alignment in Figure 2C ▶ are as follows: GSK3β-human, 24987247; GSK3β-sea urchin, 2959981; shaggy-fruit fly, 103318; GSK3β-roundworm, 17509723; GSK3-hydra, 10178642; Shaggy4-petunia, 1076649; GSK3α-bread mold, 32405824; Gsk3α-slime mold, 1730041; GSK3β-Plasmodium, 23957759; shaggy4-algae, 13811965; shaggy-Pyrocystis, 27450763; shaggy6-Giardia, 29249328; EST-green algae, 15697726; and EST-tapeworm, 22789432.
NCBI sequence identifiers for the SRPK alignment in Figure 2D ▶ are as follows: Sky1p-budding yeast, 6323872; SRPK2-mouse, 18043214; SRPK1-fruit fly, 10242347; SRPK-like-thale cress, 11259819; SRPK1-roundworm, 1353067; EST-red alga, AV432962_EST; SRPK1-like-slime mold, 28829647; EST-diatom, CD381433_EST; SRPK-trypanosome, 27447393; and SRPK-Plasmodium, 14578289.
NCBI sequence identifiers for the DYRK alignment in Figure 2E ▶ are as follows: Dyrk1b-human, 18765754; Dyrk1b-mosquito, 31226065; DYRK-slime mold, 28829499; DYRK2-human, 4503427; DYRK-roundworm, 7507072; DYRK-trypanosome, 19263269; DYRK-bread mold, 32416200; DYRK-like-fission yeast, 2130387; MBK-fruit.fly, 24642876; MBK2-roundworm, 7503839; YAK1-amoeba, 17980211; YakA-slime mold, 7489897; and Yak1-thale cress, 15239248.
NCBI sequence identifiers for the CK2 alignment in Figure 2F ▶ are as follows: CK2-human, 20150571; CK2α-owlet moth, 13628721; CK2α-sea urchin, 7209841; CK2α-rice, 22831318; CK2α-roundworm, 17505290; CK2α-bread mold, 30580436; CK2α-slime mold, 28830167; CK2α-Theileria, 125272; CK2α-trypanosome, 14532298; CK2α-Paramecium, 13940371; CK2α-blood fluke, 28354096; CK2α-Giardia parasite, 29245163; CK2α-microsporidia, 19173691; and EST-red algae, 8588611.
Crystal structures used in our analysis
The crystal structure coordinate files used for the figures were obtained from PDB (Berman et al. 2000) and have the following identifiers: 1FIN (Jeffrey et al. 1995), 1JWH (Niefind et al. 2001), 1CM8 (Wang et al. 1997), 2ERK (Canagarajah et al. 1997), 1QMZ (Brown et al. 1999), 1GNG (Bax et al. 2001), 1DS5 (Battistutta et al. 2000), 1HOW (Nolen et al. 2001), and 1LP4 (Niefind et al. 1998).
Acknowledgments
This work was supported by NIH (NLM) grant LM06747 to A.F.N. and by a Cold Spring Harbor Association postdoctoral fellowship to N.K.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04637904.
References
- Adams, J.A. 2003. Activation loop phosphorylation and catalysis in protein kinases: Is there functional evidence for the autoinhibitor model? Biochemistry 42 601–607. [DOI] [PubMed] [Google Scholar]
- Aubol, B.E., Chakrabarti, S., Ngo, J., Shaffer, J., Nolen, B., Fu, X.D., Ghosh, G., and Adams, J.A. 2003. Processive phosphorylation of alternative splicing factor/splicing factor 2. Proc. Natl. Acad. Sci. 100 12601–12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistutta, R., Sarno, S., De Moliner, E., Marin, O., Issinger, O.G., Zanotti, G., and Pinna, L.A. 2000. The crystal structure of the complex of Zea mays α subunit with a fragment of human β subunit provides the clue to the architecture of protein kinase CK2 holoenzyme. Eur. J. Biochem. 267 5184–5190. [DOI] [PubMed] [Google Scholar]
- Bax, B., Carter, P.S., Lewis, C., Guy, A.R., Bridges, A., Tanner, R., Pettman, G., Mannix, C., Culbert, A.A., Brown, M.J., et al. 2001. The structure of phosphorylated GSK-3β complexed with a peptide, FRATtide, that inhibits β-catenin phosphorylation. Structure 9 1143–1152. [DOI] [PubMed] [Google Scholar]
- Becker, W. and Joost, H.G. 1999. Structural and functional characteristics of DYRK, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acid Res. Mol. Biol. 62 1–17. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom, N., Gammeltoft, S., and Brunak, S. 1999. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294 1351–1362. [DOI] [PubMed] [Google Scholar]
- Bourne, Y., Watson, M.H., Hickey, M.J., Holmes, W., Rocque, W., Reed, S.I., and Tainer, J.A. 1996. Crystal structure and mutational analysis of the human CDK2 kinase complex with cell cycle–regulatory protein CksHs1. Cell 84 863–874. [DOI] [PubMed] [Google Scholar]
- Brown, N.R., Noble, M.E., Endicott, J.A., and Johnson, L.N. 1999. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell. Biol. 1 438–443. [DOI] [PubMed] [Google Scholar]
- Campbell, L.E. and Proud, C.G. 2002. Differing substrate specificities of members of the DYRK family of arginine-directed protein kinases. FEBS Lett. 510 31–36. [DOI] [PubMed] [Google Scholar]
- Canagarajah, B.J., Khokhlatchev, A., Cobb, M.H., and Goldsmith, E.J. 1997. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 90 859–869. [DOI] [PubMed] [Google Scholar]
- Casari, G., Sander, C., and Valencia, A. 1995. A method to predict functional residues in proteins. Nat. Struct. Biol. 2 171–178. [DOI] [PubMed] [Google Scholar]
- Colwill, K., Feng, L.L., Yeakley, J.M., Gish, G.D., Caceres, J.F., Pawson, T., and Fu, X.D. 1996. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J. Biol. Chem. 271 24569–24575. [DOI] [PubMed] [Google Scholar]
- Dajani, R., Fraser, E., Roe, S.M., Young, N., Good, V., Dale, T.C., and Pearl, L.H. 2001. Crystal structure of glycogen synthase kinase 3 β: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105 721–732. [DOI] [PubMed] [Google Scholar]
- Dajani, R., Fraser, E., Roe, S.M., Yeo, M., Good, V.M., Thompson, V., Dale, T.C., and Pearl, L.H. 2003. Structural basis for recruitment of glycogen synthase kinase 3β to the axin-APC scaffold complex. EMBO J. 22 494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble, B.W. and Woodgett, J.R. 2003. GSK-3: Tricks of the trade for a multitasking kinase. J. Cell. Sci. 116 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott, J.A., Noble, M.E., and Tucker, J.A. 1999. Cyclin-dependent kinases: Inhibition and substrate recognition. Curr. Opin. Struct. Biol. 9 738–744. [DOI] [PubMed] [Google Scholar]
- Engh, R. and Bossemeyer, D. 2002. Structural aspects of protein kinase control—role of conformational flexibility. Pharmacol. Ther. 93 99. [DOI] [PubMed] [Google Scholar]
- Ermakova, I., Boldyreff, B., Issinger, O.G., and Niefind, K. 2003. Crystal structure of a C-terminal deletion mutant of human protein kinase CK2 catalytic subunit. J. Mol. Biol. 330 925–934. [DOI] [PubMed] [Google Scholar]
- Frame, S., Cohen, P., and Biondi, R.M. 2001. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 7 1321–1327. [DOI] [PubMed] [Google Scholar]
- Gaucher, E.A., Gu, X., Miyamoto, M.M., and Benner, S.A. 2002. Predicting functional divergence in protein evolution by site-specific rate shifts. Trends Biochem. Sci. 27 315–321. [DOI] [PubMed] [Google Scholar]
- Gilbert, W., Siebel, C.W., and Guthrie, C. 2001. Phosphorylation by Sky1p promotes Npl3p shuttling and mRNA dissociation. RNA 7 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, J., Nairn, A.C., and Kuriyan, J. 1996. Structural basis for the auto-inhibition of calcium/calmodulin-dependent protein kinase I. Cell 84 875–887. [DOI] [PubMed] [Google Scholar]
- Grimes, C.A. and Jope, R.S. 2001. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 65 391–426. [DOI] [PubMed] [Google Scholar]
- Hanks, S.K. and Hunter, T. 1995. Protein kinases 6: The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 9 576–596. [PubMed] [Google Scholar]
- Hannenhalli, S.S. and Russell, R.B. 2000. Analysis and prediction of functional sub-types from protein sequence alignments. J. Mol. Biol. 303 61–76. [DOI] [PubMed] [Google Scholar]
- Harper, J.W. 2001. Protein destruction: Adapting roles for Cks proteins. Curr. Biol. 11 R431–R435. [DOI] [PubMed] [Google Scholar]
- Harwood, A.J. 2001. Regulation of GSK-3: A cellular multiprocessor. Cell 105 821–824. [DOI] [PubMed] [Google Scholar]
- Himpel, S., Tegge, W., Frank, R., Leder, S., Joost, H.G., and Becker, W. 2000. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem. 275 2431–2438. [DOI] [PubMed] [Google Scholar]
- Hooft, R.W., Sander, C., and Vriend, G. 1996. Positioning hydrogen atoms by optimizing hydrogen-bond networks in protein structures. Proteins 26 363–376. [DOI] [PubMed] [Google Scholar]
- Huse, M. and Kuriyan, J. 2002. The conformational plasticity of protein kinases. Cell 109 275–282. [DOI] [PubMed] [Google Scholar]
- Jeffrey, P.D., Russo, A.A., Polyak, K., Gibbs, E., Hurwitz, J., Massague, J., and Pavletich, N.P. 1995. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376 313–320. [DOI] [PubMed] [Google Scholar]
- Johnson, G.L. and Lapadat, R. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298 1911–1912. [DOI] [PubMed] [Google Scholar]
- Johnson, L.N. and Lewis, R.J. 2001. Structural basis for control by phosphorylation. Chem. Rev. 101 2209–2242. [DOI] [PubMed] [Google Scholar]
- Johnson, L.N., Noble, M.E., and Owen, D.J. 1996. Active and inactive protein kinases: Structural basis for regulation. Cell 85 149–158. [DOI] [PubMed] [Google Scholar]
- Jones, T.A. 1978. A graphics model building and refinement system for macromolecules. J. Appl. Crystallogr. 11 268–272. [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Karin, M. and Hunter, T. 1995. Transcriptional control by protein phosphorylation: Signal transmission from the cell surface to the nucleus. Curr. Biol. 5 747–757. [DOI] [PubMed] [Google Scholar]
- Knighton, D.R., Zheng, J.H., Ten Eyck, L.F., Ashford, V.A., Xuong, N.H., Taylor, S.S., and Sowadski, J.M. 1991. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253 407–414. [DOI] [PubMed] [Google Scholar]
- Lee, T., Hoofnagle, A.N., Kabuyama, Y., Stroud, J., Min, X., Goldsmith, E.J., Chen, L., Resing, K.A., and Ahn, N.G. 2004. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14 43–55. [DOI] [PubMed] [Google Scholar]
- Li, H., Pamukcu, R., and Thompson, W.J. 2002. β-Catenin signaling: Therapeutic strategies in oncology. Cancer Biol. Ther. 1 621–625. [DOI] [PubMed] [Google Scholar]
- Li, L., Shakhnovich, E.I., and Mirny, L.A. 2003. Amino acids determining enzyme-substrate specificity in prokaryotic and eukaryotic protein kinases. Proc. Natl. Acad. Sci. 100 4463–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge, O., Bourne, H.R., and Cohen, F.E. 1996. Evolutionarily conserved Gαβγ binding surfaces support a model of the G protein–receptor complex. Proc. Natl. Acad. Sci. 93 7507–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone, C.D. and Barton, G.J. 1993. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 9 745–756. [DOI] [PubMed] [Google Scholar]
- Lowe, E.D., Noble, M.E., Skamnaki, V.T., Oikonomakos, N.G., Owen, D.J., and Johnson, L.N. 1997. The crystal structure of a phosphorylase kinase peptide substrate complex: Kinase substrate recognition. EMBO J. 16 6646–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, E.D., Tews, I., Cheng, K.Y., Brown, N.R., Gul, S., Noble, M.E., Gamblin, S.J., and Johnson, L.N. 2002. Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry 41 15625–15634. [DOI] [PubMed] [Google Scholar]
- Lu, K.P., Liou, Y.C., and Zhou, X.Z. 2002. Pinning down proline-directed phosphorylation signaling. Trends Cell. Biol. 12 164–172. [DOI] [PubMed] [Google Scholar]
- Madhusudan, Trafny, E.A., Xuong, N.H., Adams, J.A., Ten Eyck, L.F., Taylor, S.S., and Sowadski, J.M. 1994. cAMP-dependent protein kinase: Crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 3 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning, G., Whyte, D.B., Martinez, R., Hunter, T., and Sudarsanam, S. 2002. The protein kinase complement of the human genome. Science 298 1912–1934. [DOI] [PubMed] [Google Scholar]
- Mirny, L.A. and Gelfand, M.S. 2002. Using orthologous and paralogous proteins to identify specificity determining residues. Genome Biol. 3 PREPRINT0002. [DOI] [PubMed] [Google Scholar]
- Morris, A.L., MacArthur, M.W., Hutchinson, E.G., and Thornton, J.M. 1992. Stereochemical quality of protein structure coordinates. Proteins 12 345–364. [DOI] [PubMed] [Google Scholar]
- Neuwald, A.F. 2003. Evolutionary clues to DNA polymerase III β clamp structural mechanisms. Nucleic Acids Res. 31 4503–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald, A.F. and Green, P. 1994. Detecting patterns in protein sequences. J. Mol. Biol. 239 698–712. [DOI] [PubMed] [Google Scholar]
- Neuwald, A.F., Kannan, N., Poleksic, A., Hata, N., and Liu, J.S. 2003. Ran’s C-terminal, basic patch and nucleotide exchange mechanisms in light of a canonical structure for Rab, Rho, Ras and Ran GTPases. Genome Res. 13 673–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niefind, K., Guerra, B., Pinna, L.A., Issinger, O.G., and Schomburg, D. 1998. Crystal structure of the catalytic subunit of protein kinase CK2 from Zea mays at 2.1 Å resolution. EMBO J. 17 2451–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niefind, K., Guerra, B., Ermakowa, I., and Issinger, O.G. 2001. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. EMBO J. 20 5320–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakaki, E., Du, C., Lai, J., Giannakouros, T., Cantley, L., and Rabinow, L. 2002. Phosphorylation by LAMMER protein kinases: Determination of a consensus site, identification of in vitro substrates, and implications for substrate preferences. Biochemistry 41 2055–2066. [DOI] [PubMed] [Google Scholar]
- Nolen, B., Yun, C.Y., Wong, C.F., McCammon, J.A., Fu, X.D., and Ghosh, G. 2001. The structure of Sky1p reveals a novel mechanism for constitutive activity. Nat. Struct. Biol. 8 176–183. [DOI] [PubMed] [Google Scholar]
- Nolen, B., Ngo, J., Chakrabarti, S., Vu, D., Adams, J.A., and Ghosh, G. 2003. Nucleotide-induced conformational changes in the Saccharomyces cerevisiae SR protein kinase, Sky1p, revealed by X-ray crystallography. Biochemistry 42 9575–9585. [DOI] [PubMed] [Google Scholar]
- Pawson, T. and Nash, P. 2003. Assembly of cell regulatory systems through protein interaction domains. Science 300 445–452. [DOI] [PubMed] [Google Scholar]
- Pinna, L.A. and Ruzzene, M. 1996. How do protein kinases recognize their substrates? Biochim. Biophys. Acta 1314 191–225. [DOI] [PubMed] [Google Scholar]
- Prowse, C.N., Deal, M.S., and Lew, J. 2001. The complete pathway for catalytic activation of the mitogen-activated protein kinase, ERK2. J. Biol. Chem. 276 40817–40823. [DOI] [PubMed] [Google Scholar]
- Robbins, D.J., Zhen, E., Owaki, H., Vanderbilt, C.A., Ebert, D., Geppert, T.D., and Cobb, M.H. 1993. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 268 5097–5106. [PubMed] [Google Scholar]
- Robinson, F.L., Whitehurst, A.W., Raman, M., and Cobb, M.H. 2002. Identification of novel point mutations in ERK2 that selectively disrupt binding to MEK1. J. Biol. Chem. 277 14844–14852. [DOI] [PubMed] [Google Scholar]
- Russo, A.A., Jeffrey, P.D., and Pavletich, N.P. 1996. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 3 696–700. [DOI] [PubMed] [Google Scholar]
- Samudrala, R., Huang, E.S., Koehl, P., and Levitt, M. 2000. Constructing side chains on near-native main chains for ab initio protein structure prediction. Protein Eng. 13 453–457. [DOI] [PubMed] [Google Scholar]
- Sayle, R.A. and Milner-White, E.J. 1995. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 20 374. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Smith, C.M., Radzio-Andzelm, E., Madhusudan, Akamine, P., and Taylor, S.S. 1999. The catalytic subunit of cAMP-dependent protein kinase: Prototype for an extended network of communication. Prog. Biophys. Mol. Biol. 71 313–341. [DOI] [PubMed] [Google Scholar]
- Songyang, Z., Lu, K.P., Kwon, Y.T., Tsai, L.H., Filhol, O., Cochet, C., Brickey, D.A., Soderling, T.R., Bartleson, C., Graves, D.J., et al. 1996. A structural basis for substrate specificities of protein Ser/Thr kinases: Primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 16 6486–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Haar, E., Coll, J.T., Austen, D.A., Hsiao, H.M., Swenson, L., and Jain, J. 2001. Structure of GSK3β reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 8 593–596. [DOI] [PubMed] [Google Scholar]
- Wang, Z., Harkins, P.C., Ulevitch, R.J., Han, J., Cobb, M.H., and Goldsmith, E.J. 1997. The structure of mitogen-activated protein kinase p38 at 2.1-Å resolution. Proc. Natl. Acad. Sci. 94 2327–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, M.S., Brandl, M., Suhnel, J., Pal, D., and Hilgenfeld, R. 2001. More hydrogen bonds for the (structural) biologist. Trends Biochem. Sci. 26 521–523. [DOI] [PubMed] [Google Scholar]
- Wiechmann, S., Czajkowska, H., de Graaf, K., Grotzinger, J., Joost, H.G., and Becker, W. 2003. Unusual function of the activation loop in the protein kinase DYRK1A. Biochem. Biophys. Res. Commun. 302 403–408. [DOI] [PubMed] [Google Scholar]
- Word, J.M., Lovell, S.C., Richardson, J.S., and Richardson, D.C. 1999. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285 1735–1747. [DOI] [PubMed] [Google Scholar]
- Zhou, X.Z., Lu, P.J., Wulf, G., and Lu, K.P. 1999. Phosphorylation-dependent prolyl isomerization: A novel signaling regulatory mechanism. Cell. Mol. Life Sci. 56 788–806. [DOI] [PMC free article] [PubMed] [Google Scholar]