

Abstract
Dehydroquinate synthase (DHQS) is the N-terminal domain of the pentafunctional AROM protein that catalyses steps 2 to 7 in the shikimate pathway in microbial eukaryotes. DHQS converts 3-deoxy-D-arabino-heptulosonate-7-phosphate (DAHP) to dehydroquinate in a reaction that includes alcohol oxidation, phosphate β-elimination, carbonyl reduction, ring opening, and intramolecular aldol condensation. Kinetic analysis of the isolated DHQS domains with the AROM protein showed that for the substrate DAHP the difference in Km is less than a factor of 3, that the turnover numbers differed by 24%, and that the Km for NAD+ differs by a factor of 3. Isothermal titration calorimetry revealed that a second (inhibitory) site for divalent metal binding has an approximately 4000-fold increase in KD compared to the catalytic binding site. Inhibitor studies have suggested the enzyme could act as a simple oxidoreductase with several of the reactions occurring spontaneously, whereas structural studies have implied that DHQS participates in all steps of the reaction. Analysis of site-directed mutants experimentally test and support this latter hypothesis. Differential scanning calorimetry, circular dichroism spectroscopy, and molecular exclusion chromatography demonstrate that the mutant DHQS retain their secondary and quaternary structures and their ligand binding capacity. R130K has a 135-fold reduction in specific activity with DAHP and a greater than 1100-fold decrease in the kcat/Km ratio, whereas R130A is inactive.
Keywords: dehydroquinate synthase, AROM, biocalorimetry, site-directed mutagenesis
The AROM protein is a pentafunctional enzyme that catalyzes the steps 2 to 7 (the conversion of 3-deoxy-D-arabino-heptulosonate-7-phosphate [DAHP] to chorismate) in the shikimate pathway of microbial eukaryotes; however, in many prokaryotes the equivalent five enzymes are mono-functional (Bentley 1990). The shikimate pathway is absent in mammals, and the pathogenic bacteria mutant in this pathway is attenuated for virulence (Gunel-Ozcan et al. 1997). The pathway therefore provides an attractive target for the development of antimicrobial drugs, and has already been exploited to generate an effective postemergence herbicide (Kishore and Shah 1988).
In many microbial eukaryotes the AROM protein shares a common pool of the substrates dehydroquinate and dehydroshikimate with the type II dehydroquinase and the dehydroshikimate dehydratase of the quinic acid utilization pathway. Metabolic control analysis has shown that the AROM protein is very leaky, and that the quinic acid pathway dehydroshikimate dehydratase has a flux control coefficient of -1 in the shikimate pathway. These observations prompted the proposal that the AROM protein may have a low level channelling function to protect against substrate depletion by dehydroshikimate dehydratase under conditions where the exogenous supply of quinic acid is rapidly diminished (Lamb et al. 1992; Wheeler et al. 1996). This putative channelling function could simply be due to the close juxtaposition of the five active sites within AROM. However, it is also possible that each of the enzyme domains has evolved to function with optimal catalytic activity only as part of the AROM protein.
The N-terminal domain of the AROM protein is the enzyme dehydroquinate synthase (DHQS), and this catalyzes the conversion of DAHP to dehydroquinate (DHQ; Giles et al. 1967; Lumsden and Coggins 1977; Charles et al. 1986; Bentley 1990; Hawkins et al. 1993a; Moore et al. 1994). The isolated DHQS domain of the AROM protein from Aspergillus nidulans has been crystallized and its structure determined (Carpenter et al. 1998; Nichols et al. 2003). The enzyme DHQS has generated interest because it apparently catalyzes five individual reactions (alcohol oxidation, phosphate β-elimination, carbonyl reduction, ring opening, and intramolecular aldol condensation) in a single active site (Srinivasan et al. 1963; Widlanski et al. 1989). The proposed reaction mechanism is summarized in Figure 1 ▶. The molecular mechanism of the overall reaction has been studied extensively by the use of substrate analogs (Bartlett and Satake 1988; Bender et al. 1989b; Knowles 1989; Widlanski et al. 1989; Bartlett et al. 1994), leading to the suggestion that the enzyme may be acting as a simple oxido-reductase with several of the reactions occurring spontaneously.
Figure 1.

The proposed reaction mechanism of dehydroquinate synthase. The proposed five-step reaction mechanism catalyzed by dehydroquinate synthase is shown with the possible side-chain interactions necessary for the completion of the reaction. (Reprinted with permission from Carpenter et al. 1998 © 1998, Nature Publishing Group [http://www.nature.com/].)
The structure of the ternary complex of A. nidulans DHQS with NAD+ and the nonhydrolyzable substrate analog carbaphosphonate (CBP) reveals that the active DHQS enzyme is homodimeric. Each subunit (see Fig. 2A ▶) is further divided into an N-terminal NAD+ binding α/β domain containing a Rossmann fold (“N” domain) and a C-terminal α-helical domain containing most of the residues involved in catalysis, substrate and Zn2+ binding (“C” domain). Formation of each of the two active sites within the dimer requires the interaction of amino acids from the “N” and “C” domains of one monomer together with the side chain R130 from the other monomer’s “N” domain. DHQS, in the absence of the substrate analog CBP (i.e., with NAD+, ADP, or unliganded), is in an open form where a relative rotation of 11°–14° between N- and C-terminal domains occurs. Overlapping 21 different copies of the individual N- and C-terminal DHQS domains revealed a series of pivot points about which the domain closure occurs. This analysis suggested that the structural mechanism for domain closure involved an ordered sequence of substrate binding, local rearrangement within the active site, and a propagation of torque inducing closure of the active-site cleft (Nichols et al. 2001, 2003). A preliminary report of a further open form structure of DHQS in a binary complex with NAD+ has been published (Brown et al. 2003). The crystal structures of DHQS suggested that the enzyme is actively involved in all of the five steps of the reaction and suggested that several highly conserved residues including R130, K152, R264, and H275 played an essential role in catalysis and domain closure (Carpenter et al. 1998; Nichols et al. 2003).
Figure 2.

(A) The structure of a DHQS monomer shown in cartoon format to indicate the positions of the proximal elements PE1–PE3, the distal element DE3, and helix α8. (B) A closeup cut-away view of a DHQS monomer showing the positions of the site-directed mutants. Blue, open form with cyan showing the mutated residues; red, closed form with pink showing the mutated residues; green, B-chain of the open form; purple, N-chain of the closed form. R105 is not shown, as there is no single viewpoint that allows all the mutated residues to be shown. The section of the closed form used PDB model 1dqs and the open form PDB 1nrx.
Here we report the first kinetic comparison of the DHQS activity of the complete AROM protein (hereafter referred to as the native DHQS activity) and isolated forms (hereafter referred to as the wild-type DHQS) of eukaryotic DHQS. The analysis reveals only modest differences, showing that isolation from the penta-domain AROM protein does not compromise DHQS catalytic efficiency. We also report the first biochemical and biophysical characterization of site-directed mutants of DHQS. We show that single-site mutations at residues R130, R264, H275, or K152 appear to retain their ability to form quaternary structures, and to bind the substrate and cofactor. However, all mutants have their enzyme activity abolished or sharply reduced, supporting the hypothesis that these conserved residues play key roles in the catalytic mechanism of DHQS including domain closure.
Results
Comparison of the kinetic properties of the isolated A. nidulans DHQS domain with the native AROM protein
The isolation and kinetic characterization of the equivalent AROM protein from Neurospora crassa and Saccharomyces cerevisiae has been reported (Coggins et al. 1987a; Duncan et al. 1987), but there has been no direct comparison of the kinetic properties of an isolated N-terminal DHQS domain compared to native AROM from the same species. We wished to carry out this comparison to test the hypothesis for DHQS that the enzyme domains of AROM had evolved for maximum catalytic activity only as part of the complete AROM protein. Recombinant A. nidulans AROM protein was purified by the novel protocol described in Materials and Methods. Table 1 summarizes the purification protocol, and Figure 3 ▶ shows the extent of purification at each stage. AROM was isolated to greater than 98% purity, with a yield of 2.7 mg per liter of cell culture. Purified AROM and the separately expressed DHQS domain were kinetically characterized with respect to their substrate DAHP and the cofactor NAD+, and analyzed using GRAFIT; the results are summarized in Table 2. The comparison (see Table 2) shows that for the substrate DAHP the difference in Km is less than a factor of 3, that the turnover numbers differ by 24%, and that the Km for NAD+ differs by a factor of 3. These values demonstrate that isolation of DHQS domain from the rest of the AROM protein has overall modest effects, and does not drastically compromise enzyme efficiency.
Table 1.
AROM purification from E. coli cell lysates
| Purification step | Protein (mg) | 3-DHQase activity (units) | DHQS activity (units) | Ratio DHQS:DHQase | Yield (%) | 3-DHQase specific activity (units mg−1) | DHQS specific activity (units mg−1) | Purification (fold) |
| 1. Cell free extract | 2176 | 558.3 | 352.5 | 0.6:1 | 100 | 0.26 | 0.16 | 1 |
| 2. 30% (NH4)2SO4 supernatant | 1841 | 599.1 | 393.4 | 0.7:1 | 107 | 0.33 | 0.21 | 1.3 |
| 3. 55% (NH4)2SO4 pellet | 1007 | 483.7 | 311.5 | 0.6:1 | 87 | 0.48 | 0.31 | 1.8 |
| 4. Phenyl sepharose | 290 | 360.6 | 237.7 | 0.7:1 | 65 | 1.24 | 0.82 | 4.8 |
| 5. Sephacryl | 74 | 262.3 | 196.7 | 0.7:1 | 47 | 3.54 | 2.66 | 13.6 |
| 6. Hydroxyapatite | 54 | 204.9 | 123 | 0.6:1 | 37 | 3.79 | 2.28 | 14.6 |
| 7. FPLC | 17.4 | 140.2 | 109.8 | 0.7:1 | 25.1 | 8.16 | 6.31 | 31.4 |
The specific activities of the type I dehydroquinase and the dehydroquinate synthase activities of the AROM protein at each stage in the purification protocol are shown with the overall yield and purification factor.
Figure 3.

SDS PAGE analysis of the purification of the A. nidulans AROM protein from E. coli cell lysates. The proteins present at each stage in the purification protocol are shown. Lane 1, boiled codon plus cells containing pEKA15; lane 2, clarified supernatant postsonication; lane 3, pellet from the 55% (NH4)2SO4 precipitation; lane 4, sample from the phenyl-Sepharose pool; lane 5, sample from the sephacryl pool; lane 6, sample from the hydroxyapatite pool; lane 7, sample from the DEAE FPLC pool; lane 7, molecular mass markers of 45 kDa (ovalbumin), 66 kDa (bovine serum albumin), and 97 kDa (Phosphorylase B).
Table 2.
Comparative kinetic analysis of the AROM protein with the wild-type and mutant isolated AROM dehydroquinate synthase domains
| Enzyme | Substrate | Km μM | Specific activity U mg−1 | Turnover number |
| Wild-type and mutant DHQS | ||||
| Wild-type DHQS | DAHP | 21 (2) | 9.50 | 6.8 |
| NAD+ | 1.9 (0.1) | |||
| R130K mutant DHQS | DAHP | 228 (19) | 0.07 | |
| NAD+ | 14.1 (1.5) | |||
| R130A mutant DHQS | NA | — | — | |
| K152A mutant DHQS | NA | — | — | |
| H275L mutant DHQS | NA | — | — | |
| R264A mutant DHQS | NA | — | — | |
| Pentafunctional AROM | ||||
| DHQS domain | DAHP | 9.3 (0.9) | 1.78 | 5.1 (19.0) |
| NAD+ | 0.7 (0.04) | |||
| Type 1 dehydroquinase domain | DHQ | 40 (3) | 3.63 | 10.5 (19.4) |
| Shikimate dehydrogenase domain | DHS | 311 (18) | 0.52 | 1.5 (51.2) |
| NADPH | 13.5 (1.5) | |||
| Shikimate kinase domain | ATP | 4800 (342) | 5.35 | 15.4 (18.3) |
| Shikimate | 22.3 (1.2) | |||
NA = not applicable.
The Km and the turnover number for the native AROM protein, the wild-type DHQS domain and the mutant R130K DHQS domain are shown. All values were calculated using GRAFIT, standard errors for each Km measurement are given in brackets. The values given in brackets following the turnover number are the published values for the equivalent AROM protein of N. crassa (Lambert et al. 1985; Coggins et al. 1987a).
Additionally, we measured the Km and turnover number with respect to substrate and any cofactors of the type 1 dehydroquinase, shikimate kinase, and shikimate dehydrogenase activities of the AROM protein. We were unable to measure the EPSP synthase activity of the AROM protein due to the lack of the availability of substrate. The Km values and the turnover numbers are shown in Table 2, and the values in parentheses in the turnover number column are those reported for the equivalent AROM protein from N. crassa (Lambert et al. 1985; Coggins et al. 1987a). With the exception of shikimate dehydrogenase, which has an apparent 35-fold difference, the turnover numbers differ by less than a factor of 4. The shikimate dehydrogenase activity in the A. nidulans AROM is sensitive to substrate inhibition, making accurate measurement of Vmax problematic, and possibly accounts for the difference from turn over number reported for the N. crassa enzyme. Interpretation of the Km values and turnover number for shikimate dehydrogenase shown in Table 2 are therefore subject to this caveat.
Measuring the affinity of an inhibitory metal binding site
We have previously shown that in addition to Zn2+, other metals such as Co2+, Fe2+, Ni2+, Eu3+, and Sm3+ can all reactivate metal-depleted A. nidulans DHQS to differing extents (Moore et al. 1998). In common with DHQS from prokaryotic sources, the A. nidulans DHQS appears to have two metal binding sites, one of which is of low affinity, and acts in an inhibitory manner when occupied. To determine the relative affinities of the two metal binding sites we generated metal-depleted wild-type DHQS and attempted to measure the binding affinities of the two putative Zn2+ sites by ITC. However, the affinity was higher than can be measured by this technique. Therefore, we repeated the experiments using the much weaker binding Ni2+ metal, and found that we were able to measure the affinity of both sites by ITC. Figure 4A ▶ shows the isotherm generated, and Figure 4B ▶ shows the data after peak integration, subtraction of blank titration data (not shown), concentration normalization, and analysis by the Origin (version 5) suite of programs. A sequential two-site binding model adequately described the data. Analysis of these data showed that the primary metal binding site has a KD of around 245 nM for Ni2+ binding, and the secondary site has KD of 1 mM, an approximately 4000-fold difference. It must be borne in mind that derivation of the KD values for the secondary site from the fit to the model shown is only approximate, and that the possibility of multiple weaker binding sites cannot be excluded. However, the single secondary site model is consistent with the known kinetic characteristics of the wild-type enzyme from A. nidulans and Escherichia coli in the presence of increasing concentrations of metal ions (Bender et al. 1989a; Moore et al. 1998).
Figure 4.

ITC analysis of Ni2+ binding by the wild-type DHQS domain. (A) Raw data showing the heat pulses resulting from a titration of metal free wild-type DHQS domain (93 μM) in the calorimetric cell with a 2-μL injection of 2 mM Ni2+ followed by 24 subsequent 10-μL injections. (B) Integrated heat pulses, normalized per mole of injectant, giving a differential binding curve that is adequately described by a two-site sequential binding model.
Analysis of site-directed mutants of A. nidulans DHQS
Structural studies of A. nidulans DHQS as a ternary complex with NAD+ and a nonhydrolyzable analog of the DAHP substrate CBP (Bender et al. 1989b; Widlanski et al. 1989) have allowed the determination of the precise geometry of the catalytically competent closed form active site. The structure also leads to the proposal for an ordered multistep reaction mechanism highlighting the critical role of residues R130, K152, R264, and H275 (Carpenter et al. 1998). Analysis of the rigid body changes has identified eight structural elements in DHQS that show ligand-dependent changes (Nichols et al. 2003). These can be further divided into three proximal elements containing residues that contact the substrate in the closed form (PE1–3) and five distal elements (DE1–5) whose conformation is also ligand dependent but that make no direct contact with the substrate (Nichols et al. 2003). The interaction of these elements leading to the domain closure is complex. but the essential process is one of local rearrangement induced by substrate/CBP binding followed by propagation of changes through to distal locations. As CBP binds it is likely that the close interaction of the inhibitor (and also DAHP) with R130 and N268 induces tightening of the local conformation, these contacts leading to the crosslinking of the N and C faces. Single-domain overlays of open and closed form structures suggest that the formation of two pair of “pincers” (K152/N162 and R264/N268) is critical for correct domain closure. We wished to test the hypothesis that amino acid residues R130, K152, R264, and H275 are all essential for correct domain closure and completion of the DHQS reaction mechanism. To do this we constructed the mutants R130K, R130A, K152A, R264A, and H275L and determined steady-state kinetic parameters of the DHQS substrate DAHP and cofactor NAD+ (see Table 2). Table 3 shows the oligonucleotides used to generate these mutants.
Table 3.
The sequences of the oligonucleotides used in the site-directed mutagenesis of dehydroquinate synthase
| Mutation | Mutagenic oligonucleotide | Nonmutagenic oligonucleotide |
| H275L | GAC TCT ATT GGC CTG GCC ATT GAA GCC | GAC CCC AGT TCA AAA GGT TCC |
| K152A | TCG ATC GGC GGG GCA ACT GCC ATC GAC | TGA ATC TAC CAT GGC CAG AAG |
| R130K | TCC ACC TAC ATG AAA GGT GTC CGT TAT | AGC GAC GAA TCC TGT CAG ATC |
| R130A | TCC ACC TAC ATG GCT GGT GTC CGT TAT | AGC GAC GAA TCC TGT CAG ATC |
| R264A | GAG GGG GGT CTC GCA AAC CTT TTG AAC | ACG TTC GTC TGC TGA GAC AAC |
The sequences of the oligonucleotides used in the site-directed mutagenesis are shown written 5′ to 3′.
Substitution of R130 with lysine provides an equivalent positive charge to that associated with the R130 guanidium group, but potentially with nonoptimal interactions due to differences in side-chain length and the stereochemistry of the functional groups. Although the typical pK values for the ionization of the lysine side chain NH3+ group is 10 compared to 12 for the guanidium group of arginine, at physiological pH both groups will be essentially fully ionized. Therefore, in the R130K mutant enzyme the electrostatic attraction is predicted to be high enough to pull Lys130 and Arg105 into the active site so that domain closure occurs but with a likely reduction in efficiency, resulting in poorer substrate binding reflected in an increased value of Km. The R130A mutation is predicted to disrupt the necessary inward movement of both itself as well as R105 required for stabilization of the closed form, and consequently this mutant DHQS, should be catalytically inactive. The R130 side chain stabilizes the base of PE1 by crosslinking to the rigid core domain so an additional predicted negative effect of the R130A mutation is to cause loss of structural rigidity within the cleft. Consistent with these predictions, the R130A mutant was inactive and the R130K mutant had a kcat of 2.6 × 10-3 μmoles min−1 (standard error = 0.9 × 10−4) compared to 0.28 μmoles min−1 (standard error = 0.01) for the wild-type domain. This approximately 100-fold reduction in Vmax and approximately 11-fold increase in the Km (see Table 2) equate to a greater than an 1100-fold decrease in the kcat/Km ratio.
The mutation R264A is predicted to disrupt the PE2 “pincer-pair,” preventing strain propagation through the PE2 loop and α8 transfer helix movement. The effect of this disruption should be to prevent correct domain closure and abolish enzyme activity. Confirming this prediction, the R264A mutant enzyme lacked detectable activity (see Table 2).
Steps 1 and 3 of the complex reaction mechanism are a reversible oxidation of the substrate C5 by NAD+, and H275 is proposed to be part of a proton-shuttling system. Substitution of H275 with leucine is therefore predicted to disrupt the putative proton shuttling system necessary for steps 1 and 3 of the DHQS reaction mechanism. Step 2 is β-elimination of the substrate phosphate group, and H275 and K152 are proposed to be part of a phosphate-binding pocket completed by N162, N268, and K356. Steps 4 and 5 consist of ring opening and intramolecular aldol condensation. K152A forms half of the PE1 pincer-pair involved in the initiation of domain closure; thus, this mutation should stop the fold-in of PE1 and strain propagation, so preventing the hinge closure and hence reducing binding of the substrate and potentially abolishing enzyme activity. The side chains of K152 (and possibly R264) are also implicated in preventing epimerization at C2 by interacting with the carboxylic acid group on C2 of the proposed intermediate 4 shown in Figure 1 ▶. The side chain of K152 may be involved in stabilizing the negative charge generated during the intramolecular aldol condensation shown in intermediate 5 in Figure 1 ▶. Consistent with these predictions, the mutant enzymes H275L and K152A lacked detectable activity.
Biophysical characterization of wild-type and mutant DHQS domains
The disruption of enzymatic activity seen in the mutant DHQS domains shown in Table 2 is consistent with the view that the wild-type residues are all involved in the reaction mechanism described above. However, the data are also consistent with the view that the lack of activity is because the mutations cause some change in the secondary, tertiary, or quaternary structure, or alternatively, that the changes compromise substrate, cofactor, or Zn2+ binding. To address these different possibilities we determined the native molecular weights, near- and far-UV circular dichroism (CD) spectra of the proteins, and characterized their substrate, metal, and cofactor binding ability by differential scanning calorimetry (DSC) and isothermal titration calorimetry (ITC).
CD spectroscopy and native molecular weight determinations
The near- and far-UV CD spectra of the wild-type and mutant DHQS enzymes were compared to look for any evidence that the mutations had caused disruption to the secondary or tertiary structure of the proteins. The near-UV spectra of the wild-type and mutant enzymes were featureless in the region 270 nm to 310 nm, and examples of far-UV spectra are shown in Figure 5 ▶. Comparison of the spectra in Figure 5 ▶ reveals minimal differences between them, implying that the mutations were not disrupting the DHQS secondary structure.
Figure 5.
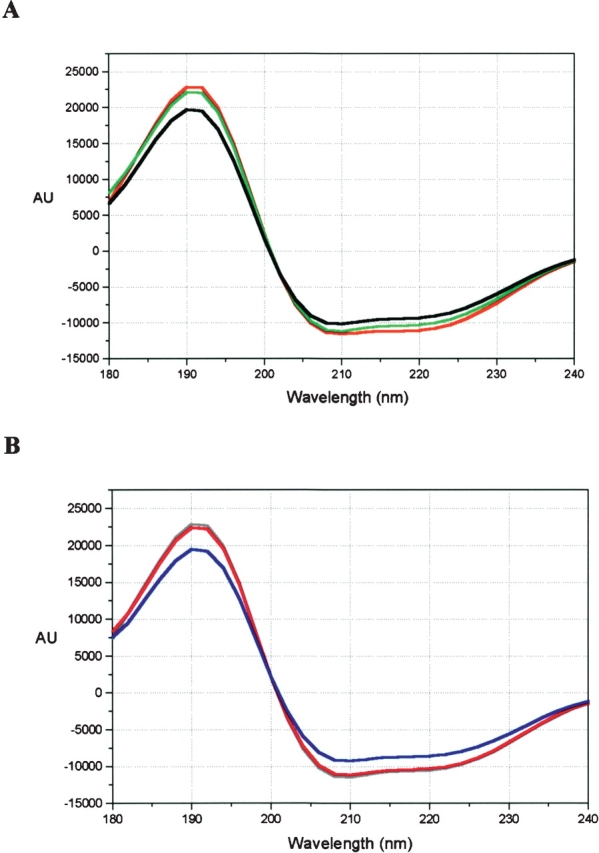
Far-UV spectra of wild-type and mutant DHQS. Far-UV CD spectra comparing the secondary structure content of wild-type and mutant DHQS. (A) Wild-type (red), R130K (green), R130A (black). (B) H275L (gray), K152A (pink), and R264A (blue), wild-type (not shown) exactly overlaid the H275L spectrum.
Gel filtration on calibrated columns was used to determine the molecular weight of wild-type mutant DHQS. Wild-type A. nidulans DHQS has a calculated molecular weight of 86 kDa and eluted in a single peak at a position in the elution profile equivalent to a molecular weight of 77 kDa. All the mutant enzymes eluted as a single peak at exactly the same position in the elution profile as the wild-type enzyme, implying that the mutant enzymes were also dimeric under these conditions. DSC data also show that the mutant enzymes have a very similar thermal stability to the wild-type enzyme in solution (see Table 4 and Fig. 6 ▶). Taken together, these data indicate than the single-site mutations do not disrupt the folding of the enzyme, and that the reduction in enzyme activity observed in these mutants more likely arises from disruption of specific protein–substrate and/or protein–protein interactions.
Table 4.
Thermal stability and ligand binding analysis of the wild-type and mutant dehydroquinate synthase enzymes
| Sample | Ligand | Tm (°C) | ΔH × 10−5 (kcal mole−1) | Error +/−×10−3 | Δ-5HVH × 10 (kcal mole−1) | KD (μM) |
| WT | Nonea | 42.77 (0.07) | 1.30 (0.03) | 3.2 | 1.27 (0.04) | |
| Zinca | 50.76 (0.09) | 1.39 (0.04) | 4.4 | 1.34 (0.05) | 0.15 | |
| Zincb | 50.55 (0.12) | 1.29 (0.05) | 5.1 | 1.27 (0.06) | ||
| DAHP | 52.50 (0.08) | 1.36 (0.04) | 3.6 | 1.33 (0.04) | 440 | |
| NAD+ | 54.62 (0.05) | 1.55 (0.03) | 3.0 | 1.51 (0.04) | 180 | |
| H275L | Nonea | 41.49 (0.09) | 1.21 (0.04) | 3.8 | 1.20 (0.05) | |
| Zinca | 52.19 (0.13) | 1.21 (0.05) | 4.6 | 1.10 (0.05) | 0.45 | |
| Zincb | 51.99 (0.22) | 1.19 (0.09) | 8.7 | 1.20 (0.11) | ||
| DAHP | 53.79 (0.17) | 1.23 (0.07) | 6.6 | 1.20 (0.08) | 320 | |
| NAD+ | 55.60 (0.07) | 1.30 (0.03) | 2.9 | 1.26 (0.04) | 97 | |
| R264A | Nonea | 43.40 (0.08) | 1.35 (0.04) | 4.4 | 1.37 (0.05) | |
| Zinca | 51.59 (0.11) | 1.45 (0.06) | 5.9 | 1.45 (0.07) | 0.11 | |
| Zincb | 50.79 (0.07) | 1.51 (0.04) | 4.4 | 1.48 (0.05) | ||
| DAHP | 52.11 (0.10) | 1.52 (0.06) | 5.9 | 1.47 (0.08) | 630 | |
| NAD+ | 55.72 (0.10) | 1.69 (0.08) | 7.5 | 1.68 (0.09) | 30 | |
| K152A | Nonea | 41.90 (0.40) | 1.19 (0.04) | 3.7 | 1.23 (0.05) | |
| Zinca | 49.57 (0.11) | 1.34 (0.05) | 5.1 | 1.28 (0.06) | 0.28 | |
| Zincb | 49.28 (0.11) | 1.43 (0.05) | 5.2 | 1.34 (0.07) | ||
| DAHP | 50.58 (0.18) | 1.45 (0.10) | 9.6 | 1.42 (0.12) | 690 | |
| NAD+ | 53.20 (0.13) | 1.73 (0.10) | 11.0 | 1.80 (0.15) | 73 | |
| R130A | Nonea | 41.20 (0.09) | 1.07 (0.03) | 3.2 | 1.09 (0.04) | |
| Zinca | 50.78 (0.09) | 1.37 (0.03) | 2.8 | 1.32 (0.03) | 0.16 | |
| Zincb | 50.82 (0.09) | 1.22 (0.04) | 4.0 | 1.27 (0.05) | ||
| DAHP | 51.48 (0.17) | 1.30 (0.03) | 2.8 | 1.25 (0.09) | 2300 | |
| NAD+ | 53.71 (0.13) | 1.42 (0.07) | 6.6 | 1.34 (0.08) | 238 | |
| R130K | Nonea | 41.60 (0.09) | 1.05 (0.03) | 2.7 | 1.01 (0.03) | |
| Zinca | 50.73 (0.09) | 1.31 (0.04) | 4.0 | 1.23 (0.05) | 0.14 | |
| Zincb | 50.52 (0.05) | 1.27 (0.02) | 2.2 | 1.32 (0.03) | ||
| DAHP | 51.86 (0.06) | 1.33 (0.03) | 2.8 | 1.35 (0.04) | 820 | |
| NAD+ | 54.14 (0.05) | 1.43 (0.02) | 2.34 | 1.41 (0.03) | 130 |
a For zinc binding, samples were treated to remove all zinc then a specified amount of zinc was added to the apoenzyme.
b For substrate binding, samples were prepared in the presence of zinc and dialyzed against 25 μM zinc prior to use.
Midpoint transition temperatures (Tm), calorimetric (ΔH) and van’t Hoff (ΔHVH) enthalpies for thermal unfolding determined by DSC in the presence and absence of ligands. The KD values for the binding of Zn2+, NAD+, and DAHP to the wild-type and mutant DHQS enzymes are derived from Tm shifts. The measured enthalpy changes, ΔH, and the calculated ΔHVH values for each experiment are shown. For each measurement the standard error is given in parentheses. Although the unfolding transitions are irreversible under the conditions we report here, the ratio of the calorimetric to van’t Hoff enthalpies, ΔHcal/3HVH are close to unity consistent with cooperative unfolding of a monomeric protein unit.
Figure 6.

DSC analysis of the ligand binding ability of wild-type and mutant DHQS domains. DSC data comparing the thermal unfolding transitions of the wild-type (A) and R130K (B) mutant DHQS domains in the presence of the ligands Zn2+, DAHP, and NAD+. Heat capacity data are corrected for instrumental (buffer) baseline and concentration normalized (1 cal = 4.184 J). In each case the order of the thermal transitions from left to right is unliganded DHQS, DHQS plus Zn2+ (40 μM), DHQS plus DAHP (1 mM), and DHQS plus NAD+(1 mM). The Tm for each transition is shown at the apex of each peak.
Zinc binding
Metal depleted wild-type and mutant DHQS enzymes were used to determine the KD for Zn2+ by DSC using the protocol described in Materials and Methods. The data are summarized in Table 4, and typical DSC thermograms are shown in Figure 6 ▶. As a control, we also compared the KD for Ni2+ binding to wild-type enzyme as determined by ITC to the value determined by DSC. It should be borne in mind that KD values determined by DSC are likely to be higher than those measured by ITC, as the former measurements are carried out at a higher temperature. The KD value of 435 nM determined by DSC compares favorably with the value of 245 nM determined by ITC for the primary Ni2+ binding site. This reasonable match between the two techniques gives confidence that the use of DSC for a comparison of the relative ligand binding properties of wild-type and mutant DHQS enzymes is valid. The data in Table 4 also show that all the mutant DHQS enzymes tested were able to bind Zn2+ under these conditions, which suggests that the loss of catalytic activity does not arise from perturbation of other residues involved in zinc binding.
Substrate and cofactor binding
Given the success of DSC for the comparative analysis of metal binding to DHQS we chose to use this technique for an analysis of substrate and cofactor binding. The use of DSC has the advantage that much smaller amounts of protein are required for the analysis compared to ITC. Table 4 summarizes the results of the comparative DSC analysis of wild-type and mutant DHQS enzymes with respect to substrate DAHP and the cofactor NAD+. Figure 6 ▶ shows typical data for the wild-type and R130K mutant DHQS enzymes. The data in Table 4 show that the KD values for DAHP and NAD+ binding are comparable, with the greatest difference between the wild-type and mutant enzymes being a factor of 5 for DAHP with respect to wild-type and R130A and 6 for NAD+ with respect to wild-type and R264A. This compares sharply with the undetectable enzyme activity of the H275L, K152A, R130A, and R264A mutants and the approximately 135-fold reduction in the activity of the R130K mutant. These data show that the lack of enzymatic activity cannot be ascribed to an inability to bind DAHP or NAD+.
Discussion
The data we present here demonstrate that the isolated DHQS domain of AROM shows only modest changes in catalytic efficiency having an approximately 41% diminution in the kcat/Km ratio when compared to the same activity measured as part of the complete pentafunctional AROM protein. The data imply that at least for this domain there is no advantage in terms of catalytic efficiency in being part of the pentafunctional AROM protein. A less detailed characterization of the isolated A. nidulans AROM type 1 dehydroquinase domain compared to published data on the N. crassa native AROM dehydroquinase suggested that the isolated domain was much less catalytically efficient (Hawkins et al. 1993b). A comparison with the A. nidulans AROM protein data reported here suggests that the kcat/Km ratio is approximately 10-fold lower for the isolated dehydroquinase domain compared to the native AROM. However, a direct comparison is problematic for two reasons. First, the activities of the isolated dehydroquinase domain and AROM were measured at different temperatures (37°C and 25°C, respectively), and second, the boundaries of the isolated domain were estimated from amino acid sequence alignments and may not correspond exactly to the optimal sequence for maximal enzymatic activity.
Dehydroshikimate dehydratase from the quinic acid utilization pathway has flux control coefficient of -1 in the shikimate pathway, and experiments with A. nidulans have shown that dehydroshikimate dehydratase is present in mycelium at approximately half the quinic acid induced level 2 h after removal of quinic acid (Lamb et al. 1992; Wheeler et al. 1996). Consequently, under these conditions the enzyme can divert flux from the shikimate pathway into the quinic acid utilization pathway and it may be advantageous to the organism to have a low level channelling function associated with the AROM protein (Lamb et al. 1992). However, it could be that the organism derives a selective advantage from having the five enzymatic activities associated with AROM produced in equimolar amounts (Coggins et al. 1987b), and that any low level channelling is simply a consequence of having the five active sites closely juxtaposed.
The site-directed mutagenesis studies we present here show that when the side chains of any of the four amino acids R130, K152, H275, and R264 are changed, DHQS activity is sharply reduced or abolished. On the basis of structural studies these four residues have been identified as important for correct domain closure and catalysis in DHQS. Spectroscopic studies found no changes in the secondary, tertiary, or quaternary structures of the mutant DHQS proteins, and microcalorimetry showed that all the mutant proteins retained their full ligand binding properties. As the mutant enzymes are all able to bind Zn2+, their substrate DAHP, and the cofactor NAD+, the lack of enzyme activity implies that the mutations have disrupted the molecular mechanism of the reaction. Our experiments represent the first rational mutational studies of this enzyme, and show the value of a combined structural and thermodynamic approach in probing complex reaction mechanisms (Carpenter et al. 1998; Nichols et al. 2003).
Materials and methods
All chemicals were from BDH, unless otherwise stated, and were of AnalaR grade or greater purity. Chloramphenicol, Coomassie brilliant blue, lysozyme, ADP, ATP, NAD, NADH, NADPH, phenol/chloroform/isoamyl alcohol mix, pyruvate kinase/lactate dehydrogenase enzymes, protease inhibitor cocktail, shikimic acid, quinic acid, and triethanolamine were from Sigma. Ampicillin, dithiothreitol (DTT), and isopropyl β-D-thiogalactoside (IPTG) were from Melford Labs UK. Growth media that included Bactotryptone yeast extract and agar were from Beckton Dickinson UK Ltd. Dehydroquinic acid was made as previously described (Grewe and Haendler 1968).
SDS PAGE was as previously described (Laemmli 1970).
Kinetic measurements
Assay of the individual activities of the AROM protein were as follows: Dehydroquinate synthase was measured by linking the product of the reaction dehydroquinate to the type 1 dehydroquinase from Salmonella typhi (Moore et al. 1993; Gourley et al. 1999) and measuring the appearance of dehydroshikimate at 240 nm. The standard assay was 1 mL containing 12.5 mM Bis-Tris propane/acetate (pH 7.0), 40 μM ZnSO4, 50 μM NAD+, 340 μM DAHP, and 2 units of Salmonella typhi type 1 dehydroquinase. The type 1 dehydroquinase activity was measured at 240 nm in 1 mL of 12.5 mM Bis-Tris propane/acetate (pH 7.0), containing 300 μM dehydroquinate. Shikimate dehydrogenase was assayed by measuring the NADPH-dependant oxidation of dehydroshikimate at 340 nm in 1 mL containing 50 mM potassium phosphate (pH 7.0), 50 μM NADPH, and 600 μM dehydroshikimate. Shikimate kinase was assayed by measuring the shikimate-dependant oxidation of NADH at 340 nm in 1 mL containing 50 mM triethanolamine/KOH (pH 7.0), 50 mM KCl, 5 mM MgSO4, 0.1 mM shikimic acid, 2.5 mM ATP, 0.5 mM PEP, 50 μM NADH, 6.6 units of pyruvate kinase mL−1, and 13.5 units of lactate dehydrogenase mL−1. All assays were carried out at 25°C, and enzyme kinetic data were analyzed using nonlinear regression with the program GRAFIT (Erithacus software).
Circular dichroism
Circular dichroism spectra were recorded from 10 accumulative scans at 20°C between 180 nm to 250 nm (far-UV) and 250 nm to 320 nm (near-UV) using a Jasco J-810 spectropolarimeter. Scans were recorded at standard sensitivity with a band width of 2 nm, scanning speed of 50 nm min−1and a protein concentration of 0.5 mg mL−1. Specific ellipticity values were calculated using concentrations derived from the absorption measurements at 280 nm on a Shimadzu UV-1601 (ɛ280 = 36,160). Calculations and data analysis were carried out using Jasco Spectra analysis software. Each spectrum was corrected by subtraction of a comparative blank.
Differential scanning calorimetry
Protein thermal transition characteristics in solution were determined by differential scanning calorimetry (DSC) using a Micro-Cal VP-DSC (cell volume 0.52 mL). Samples were scanned over a range from 25°C–65°C, with a scan rate of 90°C h−1, and a filtering period of 16 sec with protein at 20 μM–23 μM and an appropriate buffer in the reference cell. Solutions were gently degassed prior to loading. Thermograms were corrected for instrumental baseline, using control buffer:buffer scans under identical conditions, and analyzed using standard MicroCal ORIGIN software. Protein concentrations were measured spectrophotometrically using the molar absorption coefficient calculated from the amino acid sequence by the vector NTI suite of programs as previously described (Gill and von Hippel 1989). Ligand-induced shifts in thermal transition midpoint temperatures (Tm) were used to estimate approximate ligand affinities using standard thermodynamic methods as previously described (Cooper 1999; Cooper et al. 2000).
Isothermal titration calorimetry
Protein/ligand binding constants were also determined by ITC using a MicroCal VP-ITC microcalorimeter. ITC measurements were carried out at 25°C, and the titrations were carried out using a 250-μL syringe, with stirring at 300 rpm. Each titration consisted of a preliminary injection of 2 μL, followed by 24 injections of 10 μL into a cell containing approximately 1.4 mL of protein sample at 50 μM–100 μM. To correct for dilution and mixture effects a series of baselines were carried out, where injections of substrate/ ligand were carried out into buffer alone. Data were analyzed using MicroCal ORIGIN software and baselines subtracted from data to obtain accurate heat exchanges. Protein solutions for calorimetry were extensively dialyzed against buffer, and the equilibrated dialysis buffer used for ligand solutions and controls to minimize heats of dilution effects.
Site-directed mutagenesis
Site-directed mutagenesis was carried out as previously described (Hemsley et al. 1989).
Native molecular weight determinations
A Superdex 200 FPLC column in conjunction with an AKTA explorer protein purifier was used to determine the molecular weight/oligomeric state of wild-type and mutant DHQS. The void volume of the column was determined with blue dextran and cytochrome c (12 kDa), carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa), and alcohol dehydrogenase (150 kDa) were used to compile a standard curve from which the sample molecular weights were extrapolated.
Purification of the AROM protein
Overproduction of the A. nidulans AROM enzyme was carried out in E. coli expression strain Codon Plus containing the plasmid pEKA15 (Moore 1992). A 200 mL broth starter culture containing 100 μg mL−1ampicillin and 35 μg mL−1chloramphenicol was grown through the day at 27°C and then used to seed 18 × 2-liter flasks containing 500 mL of drug supplemented broth. These cultures were grown at 27° C to an attenuance (D550) of 0.75, and then induced by the addition of IPTG to a final concentration of 0.2 mg mL−1 followed by a further 6-h growth at 27°C. Cells were harvested by centrifugation and stored frozen at −18°C until needed. Cell paste (50 g) was thawed at room temperature, and cells disrupted by sonication at 15 μ in 450 mL of buffer containing 0.1 M Tris HCl, 1 mM DTT, 1 mM benzamidine (pH 7.5), 40 μM ZnSO4 (buffer 1). A cell free extract was obtained by removing cellular debris by centrifugation at 10,000g for 42 min at 4°C. The supernatant was made 30% saturated with ammonium sulphate, and the precipitated proteins removed by centrifugation at 10,000g for 45 min at 4°C. The supernatant was then made 55% saturated with ammonium sulphate and the precipitated protein recovered by centrifugation centrifugation at 10,000g for 45 min at 4°C. The recovered protein pellet was dissolved in a 50 mM potassium phosphate (pH 7.2), 1.0 M ammonium sulphate, 150 mM NaCl, 1 mM DTT (buffer 2) to a final volume of 400 mL. The supernatant was applied to a phenyl Sepharose column pre equilibrated with buffer 2. Following a 500-mL wash with buffer 2, the column was eluted with a 1-L linear reverse ammonium sulphate gradient made by using 500 mL of buffer 2 and 500 mL of 50 mM potassium phosphate (pH 7.2), 150 mM NaCl, 1 mM DTT (buffer 3), and collecting 10 mL fractions. A further 300 mL of buffer 3 was applied and fractions monitored for AROM protein by assaying the DHQS and type 1 dehydroquinase activities. Sample loading and elution of the phenyl Sepharose column were carried out at the maximum flow rate without the use of a pump. Fractions were also monitored by SDS PAGE (7.5% separating gel) and appropriate fractions were pooled and made 60% saturated with ammonium sulphate. The precipitated AROM protein was harvested by centrifugation at 10,000g for 45 min, then resuspended in the minimum required volume of 50 buffer 4. The sample was then applied to a Sephacryl S300 column (125 × 2.5 cm) at a flow rate of 0.5 mL min−1while collecting 10-mL fractions. Fractions containing AROM were identified by enzyme assay and analyzed by SDS PAGE. Appropriate fractions were pooled, dialyzed against 2 × 5l of 25 mM potassium phosphate (pH 7.2), 1 mM DTT (buffer 5) and, following filtration through a 0.45-μm filter, applied in halves to two ceramic hydroxyapatite columns (9 × 5 cm). The columns were washed with 500 mL each of buffer 5 and then eluted with a 1-L gradient of 25–400 mM potassium phosphate (pH 7.2), 1 mM DTT collecting 10-mL fractions. AROM was located by enzyme assay and SDS PAGE. For kinetic analysis the AROM protein was dialyzed into 50 mM potassium phosphate (pH 7.2), 1 mM DTT, 40 μM ZnSO4, and further purified by FPLC using a 5-mL DEAE FF Hi-trap column. AROM protein was eluted with a 160-mL 0 to 0.5-M NaCl gradient collecting 2-mL fractions. AROM was located by enzyme assay and SDS PAGE with a typical yield of 2.7 mg L−1at greater than 98% purity. The wild-type and mutant DHQS domains of the AROM protein were purified as previously described (Moore et al. 1994).
Metal depletion of buffers and enzymes
A 100-mL chelating resin column was prepared by sequentially washing with two volumes of 1 N HCl, 5 volumes of deionized water, 2 volumes of 1 N NaOH, followed by deionized water until the column effluent was at neutral pH. Once neutrality was reached, buffers previously prepared in deionized water were passed through the column to remove divalent cations. All glass and plasticware used was rinsed in buffer containing 10 mM EDTA and then rinsed five times in metal-free water. Metal-free enzyme was prepared by incubation with 1 mM EDTA for 1 h at 4°C while gently mixing the solution. EDTA was removed from the samples by dialysis once into buffer containing 50 μM EDTA and then into three changes of metal-free buffer (typically, a 20-mL sample in 5 L of dialysis buffer) at 4°C, with at least 12 h between each change of buffer.
Competing interests
I.G.C., A.R.H., and D.K.S. are founders of Arrow Therapeutics, and A.R.H. and D.K.S. are consultants to Arrow Therapeutics.
Acknowledgments
This work was funded by Arrow Therapeutics, the Biotechnology and Biological Sciences Research Council, and the Wellcome Trust. We thank Elisabeth P. Carpenter for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04705404.
References
- Bartlett, P.A. and Satake, K. 1988. Does dehydoquinate synthase synthesise dehydoquinate? J. Am. Chem. Soc. 110 1628–1630. [Google Scholar]
- Bartlett, P.A., McLaren, K.L., and Marx, M.A. 1994. Divergence between the enzyme-catalysed and non-catalysed synthesis of 3-dehydroquinate. J. Org. Chem. 59 2082–2085. [Google Scholar]
- Bender, S.L., Mehdi, S., and Knowles, J.R. 1989a. Dehydroquinate synthase: The role of divalent metal cations and of nicotinamide adenine dinucleotide in catalysis. Biochemistry 28 7555–7560. [DOI] [PubMed] [Google Scholar]
- Bender, S.L., Widlanski, T., and Knowles, J.R. 1989b. Dehydroquinate synthase: The use of substrate analogues to probe the early steps of the catalyzed reaction. Biochemistry 28 7560–7572. [DOI] [PubMed] [Google Scholar]
- Bentley, R. 1990. The shikimate pathway—A metabolic tree with many branches. Crit. Rev. Biochem. Mol. Biol. 25 307–384. [DOI] [PubMed] [Google Scholar]
- Brown, K.A., Carpenter, E.P., Watson, K.A., Coggins, J.R., Hawkins, A.R., Koch, M.H., and Svergun, D.I. 2003. Twists and turns: A tale of two shikimate-pathway enzymes. Biochem. Soc. Trans. 31 543–547. [DOI] [PubMed] [Google Scholar]
- Carpenter, E., Hawkins, A., Frost, J., and Brown, K. 1998. Structure of dehydroquinate synthase reveals an active site capable of multistep catalysis. Nature 394 299–302. [DOI] [PubMed] [Google Scholar]
- Charles, I.G., Keyte, J.W., Brammar, W.J., Smith, M., and Hawkins, A.R. 1986. The isolation and nucleotide sequence of the complex AROM locus of Aspergillus nidulans. Nucleic Acids Res. 14 2201–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins, J.R., Boocock, M.R., Chaudhuri, S., Lambert, J.M., Lumsden, J., Nimmo, G.A., and Smith, D.D. 1987a. The arom multifunctional enzyme from Neurospora crassa. Methods Enzymol. 142 325–341. [DOI] [PubMed] [Google Scholar]
- Coggins, J.R., Duncan, K., Anton, I.A., Boocock, M.R., Chaudhuri, S., Lambert, J.M., Lewendon, A., Millar, G., Mousdale, D.M., and Smith, D.D. 1987b. The anatomy of a multifunctional enzyme. Biochem. Soc. Trans. 15 754–759. [DOI] [PubMed] [Google Scholar]
- Cooper, A. 1999. Thermodynamics of protein folding and stability. In Protein: A comprehensive treatise (ed. G. Allen), pp. 217–270. JAI Press Inc, Stamford, CT.
- Cooper, A., Nutley, M.A., and Wadood, A. 2000. Differential scanning micro-calorimetry. In Protein–ligand interactions: Hydrodynamics and calorimetry (eds. S.E. Harding and B.Z. Chowdry), pp. 287–318. Oxford University Press, Oxford.
- Duncan, K., Edwards, R.M., and Coggins, J.R. 1987. The pentafunctional arom enzyme of Saccharomyces cerevisiae is a mosaic of monofunctional domains. Biochem. J. 246 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles, N.H., Case, M.E., Partridge, C.W., and Ahmed, S.I. 1967. A gene cluster in Neurospora crassa coding for an aggregate of five aromatic synthetic enzymes. Proc. Natl. Acad. Sci. 58 1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Gourley, D.G., Shrive, A.K., Polikarpov, I., Krell, T., Coggins, J.R., Hawkins, A.R., Isaacs, N.W., and Sawyer, L. 1999. The two types of 3-dehydroquinase have distinct structures but catalyze the same overall reaction. Nat. Struct. Biol. 6 521–525. [DOI] [PubMed] [Google Scholar]
- Grewe, R. and Haendler, H. 1968. 5-Dehydroquinic acid. Biochem. Prep. 11 21–26. [Google Scholar]
- Gunel-Ozcan, A., Brown, K.A., Allen, A.G., and Maskell, D.J. 1997. Salmonella typhimurium aroB mutants are attentuated in BALB/c mice. Microb. Pathog. 23 311–316. [DOI] [PubMed] [Google Scholar]
- Hawkins, A.R., Lamb, H.K., Moore, J.D., Charles, I.G., and Roberts, C.F. 1993a. The pre-chorismate (shikimate) and quinate pathways in filamentous fungi: Theoretical and practical aspects. J. Gen. Microbiol. 139 (Pt. 12) 2891–2899. [DOI] [PubMed] [Google Scholar]
- Hawkins, A.R., Moore, J.D., and Adeokun, A.M. 1993b. Characterization of the 3-dehydroquinase domain of the pentafunctional AROM protein, and the quinate dehydrogenase from Aspergillus nidulans, and the overproduction of the type II 3-dehydroquinase from Neurospora crassa. Biochem. J. 296 (Pt. 2) 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemsley, A., Arnheim, N., Toney, M.D., Cortopassi, G., and Galas, D.J. 1989. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 17 6545–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore, G.M. and Shah, D.M. 1988. Amino acid biosynthesis inhibitors as herbicides. Annu. Rev. Biochem. 57 627–663. [DOI] [PubMed] [Google Scholar]
- Knowles, J.R. 1989. Mechanistic ingenuity in enzyme catalysis: Dehydroquinate synthase. Aldrichim. Acta 22 59–67. [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lamb, H.K., van den Hombergh, J.P.T.W., Newton, G.H., Moore, J.D., Roberts, C.F., and Hawkins, A.R. 1992. Differential flux through the quinate and shikimate pathways. Implications for the channelling hypothesis. Biochem. J. 284 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, J.M., Boocock, M.R., and Coggins, J.R. 1985. The 3-dehydroquinate synthase activity of the pentafunctional arom enzyme complex of Neurospora crassa is Zn2+-dependent. Biochem. J. 226 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden, J. and Coggins, J.R. 1977. The subunit structure of the arom multienzyme complex of Neurospora crassa. A possible pentafunctional poly-peptide chain. Biochem. J. 161 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J.D. 1992. “Molecular organisation and interactions of shikimate pathway enzymes.” Ph.D. thesis, University of Newcastle upon Tyne, Newcastle upon Tyne, UK.
- Moore, J.D., Hawkins, A.R., Charles, I.G., Deka, R., Coggins, J.R., Cooper, A., Kelly, S.M., and Price, N.C. 1993. Characterization of the type I dehydroquinase from Salmonella typhi. Biochem. J. 295 (Pt. 1) 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J.D., Coggins, J.R., Virden, R., and Hawkins, A.R. 1994. Efficient independent activity of a monomeric, monofunctional dehydroquinate synthase derived from the N-terminus of the pentafunctional AROM protein of Aspergillus nidulans. Biochem. J. 301 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J.D., Skinner, M.A., Swatman, D.R., Hawkins, A.R., and Brown, K.A. 1998. Reactivation of 3-dehydroquinate synthase by lanthanide cations. J. Am. Chem. Soc. 28 7105–7106. [Google Scholar]
- Nichols, C.E., Ren, J., Lamb, H., Haldane, F., Hawkins, A.R., and Stammers, D.K. 2001. Identification of many crystal forms of Aspergillus nidulans dehydroquinate synthase. Acta Crystallogr. D Biol. Crystallogr. 57 306–309. [DOI] [PubMed] [Google Scholar]
- Nichols, C.E., Ren, J., Lamb, H.K., Hawkins, A.R., and Stammers, D.K. 2003. Ligand-induced conformational changes and a mechanism for domain closure in Aspergillus nidulans dehydroquinate synthase. J. Mol. Biol. 327 129–144. [DOI] [PubMed] [Google Scholar]
- Srinivasan, P.R., Rothchild, J., and Sprinson, D.B. 1963. The enzymic conversion of 3-deoxy-D-arabino-heptulosonic acid 7-phosphate to 5-dehydroquinate. J. Biol. Chem. 238 3176–3182. [PubMed] [Google Scholar]
- Wheeler, K.W., Lamb, H.K., and Hawkins, A.R. 1996. Control of metabolic flux through the quinate pathway in Aspergillus nidulans. Biochem. J. 1996 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlanski, T., Bender, S.L., and Knowles, J.R. 1989. Dehydroquinate synthase: The use of substrate analogues to probe the late steps of the catalyzed reaction. Biochemistry 28 7572–7582. [DOI] [PubMed] [Google Scholar]