

Abstract
Double-stranded DNA bacteriophages and herpesviruses assemble their heads in a similar fashion; a pre-formed precursor called a prohead or procapsid undergoes a conformational transition to give rise to a mature head or capsid. A virus-encoded prohead or procapsid protease is often required in this maturation process. Through computational analysis, we infer homology between bacteriophage prohead proteases (MEROPS families U9 and U35) and herpesvirus protease (MEROPS family S21), and unify them into a procapsid protease superfamily. We also extend this superfamily to include an uncharacterized cluster of orthologs (COG3566) and many other phage or bacteria-encoded hypothetical proteins. On the basis of this homology and the herpesvirus protease structure and catalytic mechanism, we predict that bacteriophage prohead proteases adopt the herpesvirus protease fold and exploit a conserved Ser and His residue pair in catalysis. Our study provides further support for the proposed evolutionary link between dsDNA bacteriophages and herpesviruses.
Keywords: dsDNA bacteriophage, prohead protease, MEROPS, homology detection, structure prediction, gene organization, evolution
Homology, or evolutionary relationship, is frequently used to predict protein structure and function. With the development of profile-based sequence similarity search tools such as PSI-BLAST (Altschul et al. 1997) and HMMer (Krogh et al. 1994; Bateman et al. 1999), increasingly remote homologs can be detected, which enables automated and large-scale functional annotation for sequences generated by ge-nome projects. Additional improvements in fold-recognition methods, in particular, the development of Meta Servers (Lundstrom et al. 2001; Fischer 2003; Ginalski et al. 2003), greatly facilitate structure prediction and add power to sequence-based searching tools. However, in detailed studies of a protein family, human inspection based on biological knowledge of that particular family is often indispensable in guiding the process of automatic searches, as well as in ensuring the reliability and improving the sensitivity of these searches (Pei and Grishin 2001, 2003; Kinch and Grishin 2002b). Using such an approach, we were able to detect the remote homologous relationship between several families of bacteriophage prohead proteases and herpesvirus protease.
Double-stranded DNA bacteriophages assemble their heads in two steps; first, capsid proteins gather around a scaffold to form a prohead (or procapsid); then, the prohead undergoes a maturation process to expand into the final, mature head (Dokland 1999). Interestingly, this two-stage assembly strategy is also exploited by herpesviruses (Lata et al. 2000). Prohead maturation in some viruses requires the activity of a virus-encoded prohead protease, whose major responsibilities are to cleave the scaffold proteins and/or process the capsid proteins (Dokland 1999; Lata et al. 2000). The peptidase classification database MEROPS (Barrett et al. 2001; Rawlings et al. 2002) defines two prohead protease families, U9 and U35. MEROPS names each peptidase family by its catalytic type, aspartic (A), cysteine (C), metallo (M), serine (S), threonine (T), and unknown (U). Thus, the catalytic mechanisms of U9 and U35 are still unknown. Family U9 is represented by bacteriophage T4 gp21 protease. Family U35 consists of two subgroups, U35.001 represented by bacteriophage HK97 gp4 protease and U35.002 represented by phage Mu gpI protein. In addition, the herpesvirus procapsid maturation protease, which is also called assemblin or UL26 protein (numbered according to herpes simplex virus type 1 or HSV-1; Homa and Brown 1997), is classified as family S21 in MEROPS.
Using computational methods, we gather evidence that dsDNA bacteriophage prohead protease families U35.001, U35.002, and U9 are homologs. Furthermore, we link these phage prohead protease families with herpesvirus protease and unify them into a procapsid protease superfamily. Because herpesvirus protease has a known 3D structure, the homology inferred here provides important clues to further our understanding of the structure and mechanism of the bacteriophage prohead proteases.
Results and Discussion
Sequence similarity searches
U35.001 family
Starting with the U35.001 representative bacteriophage HK97 gp4 protease sequence (gi|9634157, residues 1–225), we carried out extensive transitive PSI-BLAST searches (e-value cutoff 0.001) against the nonredundant database (nr) to detect distant members in this family. These transitive searches converged after the third round, extending the U35.001 family to include a total of 78 sequences (a group of lipoproteins were removed from the BLAST hits as false positives). The sequence numbers reported here and below are all counted after removal of redundant and incomplete sequences.
Most of these 78 sequences in the U35.001 family belong to either bacteriophages or bacteria. When used as queries in searching CDD (Conserved Domain Database; Marchler-Bauer et al. 2003), many of them (including bacterial sequences) readily found COG3740 (Tatusov et al. 2001), phage head maturation protease, with good e-values (<0.01). This close relationship suggests that the bacterial sequences in this family probably resulted from integrated phage genomes. The words prophage or phage frequently appear in the annotations of these bacterial sequences.
S21 family
Most interestingly, the above-mentioned transitive searches to expand the U35.001 family found a group of herpesvirus proteases (44 sequences in total) in addition to the 78 U35.001 family members from phages or bacteria, suggesting a homologous relationship between phage prohead protease and herpesvirus protease (MEROPS family S21). Regular PSI-BLAST searches provided further evidence for this putative homologous relationship. For example, we used the full-length HK97 gp4 protein (gi|9634157) as a query to run PSI-BLAST (Altschul et al. 1997) on the NCBI nonredundant (nr) database (December 29, 2003: 1,585,607 sequences; 519,349,222 total letters; e-value cutoff 0.01). The UL26 capsid maturation protease of Meleagrid herpesvirus 1 (gi|12084854) was found in the second iteration with a significant e-value 0.009.
U9 family
The U9 family in MEROPS is represented by bacteriophage T4 gp21 protease. Starting with this protein (gi|75965, residues 1–212), we performed transitive PSI-BLAST (e-value cutoff 0.001) to detect possible U9 family members. These searches converged after the first round, yielding only eight sequences. All of these sequences are phage proteins, often annotated as gp21 prohead core scaffold protein and protease. The small number of sequences in the U9 family suggests that it is a singleton family in the current nr database, and that its sequence profile is not good enough for detecting its similarity to distant homologs. However, in the course of expanding the U35.001 family, we found statistical evidence that the U9 family and the U35.001 family are remote homologs. For example, starting with the U35.001 representative HK97 gp4 protease (gi|9634157, residues 1–225), PSI-BLAST search found a close homolog gi|26988298 with e-value 2e-18 in the first iteration. Using this sequence as query to run PSI-BLAST (default parameters in NCBI Web site), we found a U9 family member (gi|30044105) in the third iteration with a significant e-value 0.003.
U35.002 family and COG3566
In MEROPS, the U35.002 family contains only the bacteriophage Mu gpI protein. Using this protein as a query (gi|9633523, residues 1–361), we performed extensive transitive PSI-BLAST searches (e-value cutoff 0.001) to expand the U35.002 family. These searches converged after the third round, yielding a total of 69 sequences. (We manually inspected the hits in each round to remove false positives.) Much like the U35.001 family, most of these 69 sequences also come from either bacteriophages or bacteria. Many of the bacterial sequences probably resulted from integrated phages, as suggested by their annotations.
We clustered these 69 sequences into four groups on the basis of sequence conservation and the results of Euclidian distance mapping (see “Sequence clustering”): three U35.002 subfamilies (U35.002.a, U35.002.b, and U35.002.c) and a fourth group corresponding to an uncharacterized cluster of orthologs (COG3566;Tatusov et al. 2001). U35.002.a contains enterobacteria phage Mu gpI protein and its close homologs (10 sequences in total); U35.002.b contains enterobacteria phage P2 gpO protein and its close homologs (32 sequences in total); U35.002.c includes 10 hypothetical proteins (nine from bacteria, one from archaea); and COG3566 consists of Burkholderia cepacia phage Bcep1 gp15 protein and its close homologs (17 sequences in total).
Because U35.001 and U35.002 are in the same U35 family in MEROPS, and MEROPS families consist of sequences with significant statistical similarity (http://merops.sanger.ac.uk/), we expected U35.001 and U35.002 family members to be close homologs. Surprisingly, during the above-mentioned extensive transitive PSI-BLAST searches, the U35.001/S21/U9 family members could not find any of the U35.002/COG3566 family members and vice versa. To explore the possible link between U35.001/S21/U9 and U35.002/COG3566, we made a global multiple sequence alignment of U35.001 combined with S21 (122 sequences, only protease domains) to seed BLAST searches. A U35.002 family member (gi|34496934) was found with a significant e-value 3e-04 by the query gi|9634157. BLAST searches seeded with a multiple sequence alignment of U35.002/ COG3566 families (69 sequences, full length) found a S21 member, the human herpesvirus 8 protease (gi|2246545), with a higher e-value 0.038.
Sequence clustering: Euclidian distance mapping and average pairwise sequence identities
The transitive and alignment-seeded PSI-BLAST searches have expanded and linked several MEROPS families, including phage prohead protease families U35.001, U35.002, U9, and herpesvirus protease family S21. To better understand the relationships between these families, we used a manually adjusted PCMA (Pei et al. 2003) multiple sequence alignment of all the detected sequences (199 in total) for Euclidian distance mapping (Grishin and Grishin 2002) and sequence identity calculations.
In distance mapping, each sequence is represented by a point in a multidimensional Euclidian space and the distances between these points reflect the evolutionary distances between the sequences. A two-dimensional projection of this space is shown in Figure 2A ▶ (below). This plot offers a visualization of the sequence clustering; each family or subfamily appears as a rather distinct group, except for U35.002.a and U35.002.c. These two subfamilies are divided on the basis of sequence conservation and insertion/ deletion patterns, for example, U35.002.c sequences have a two-residue insertion before the putative oxyanion-binding site highlighted in red in Figure 1 ▶.
Figure 2.

Euclidian distance mapping and sequence identities. (A) Euclidian distance mapping. This diagram is a two-dimensional projection of a multidimensional Euclidian space with X and Y-axes selected for best visualization effect. Different families or subfamilies are represented by various symbols. (B) Average pairwise sequence identities (percent) within and between families. Shown in bold are within-group identities of a family or a subfamily. Shown in italic are between-group identities >17%. The number in parentheses under each family name is the number of sequences in that family.
Figure 1.
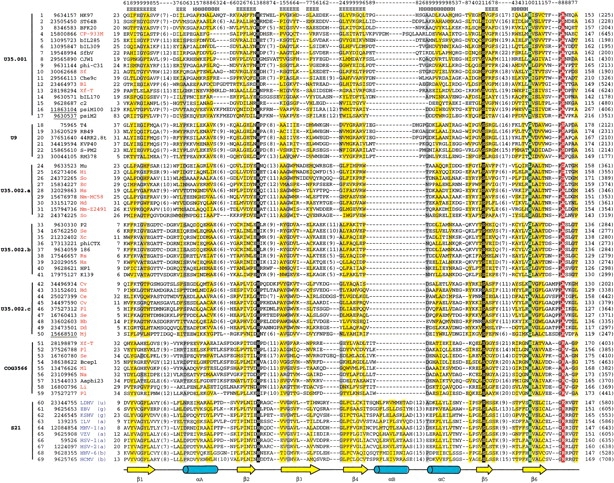
Multiple sequence alignment of the procapsid protease superfamily. Except for COG3566, each group is named according to a MEROPS family—phage prohead protease families U35.001, U35.002, U9, and herpesvirus protease family S21. U35.002 is divided into three subfamilies on the basis of the results of Euclidian distance mapping and sequence conservation. For each sequence in this multiple alignment, the NCBI gene identification (gi) number and the species name abbreviation are shown after the serial number. The abbreviations for bacteria or archaea names are colored in red, whereas those for phages and herpesviruses are in black and in blue, respectively. The gi numbers for archaeal phage or archaeal sequences are underlined. In S21 group, the lowercase letters in parentheses after the species name abbreviations indicate the subfamily to which that herpesvirus belongs according to NCBI taxonomy browser (http://www.ncbi.nlm.nih.gov/Taxonomy/): αherpesvirus (a), βherpesvirus (b), γherpesvirus (g), and unclassified herpesvirus (u). The first and the last residue numbers are shown before and after each sequence, respectively, and the total lengths are shown in parentheses at the end. Long insertions in loop regions are omitted for clarity, and the number of omitted residues is indicated in parentheses. Uncharged residues (any residue except K, R, E, D) at mainly hydrophobic positions are highlighted in yellow and small residues (G, A, C, P, T, S, V, D, N) at positions containing mainly small residues are highlighted in gray. The conserved catalytic H and S are boxed in black and the position corresponding to the His third member of herpesvirus protease catalytic triad is boxed in green. The position corresponding to the oxyanion-binding site of herpesvirus protease is highlighted in red. Shown at the bottom of this alignment is the secondary structure elements diagram of human cytomegalovirus protease (PDB: 1wpo, chain A). α-helices and β-strands are represented as blue cylinders and yellow arrows, respectively. Psipred secondary structure prediction results (strand, E; helix, H) and reliability (highest, 9; lowest, 0) for HK97 prohead protease (gi|9634157) are shown on the top of this alignment. Species name abbreviations are as follows: 186, Enterobacteria phage 186; 44RR2.8t, Bacteriophage 44RR2.8t; Aaphi23, Bacteriophage Aaphi23; Bb, Bordetella bronchiseptica RB50; Bcep1, Burkholderia cepacia phage Bcep1; BFK20, Bacteriophage BFK20; bIL170, Bacteriophage bIL170; bIL285, Bacteriophage bIL285; bIL309, Bacteriophage bIL309; Bl, Bifidobacterium longum NCC2705; Ce, Corynebacterium efficiens YS-314; Che9c, Mycobacteriophage Che9c; CJW1, Mycobacterio-phage CJW1; CP-933M, Escherichia coli O157:H7 EDL933 cryptic prophage CP-933M; Cv, Chromobacterium violaceum ATCC 12472; Dd, Desulfo-vibrio desulfuricans G20; EBV, Epstein-Barr virus; Ec, Escherichia coli O157:H7; HCMV, Human cytomegalovirus; Hd, Haemophilus ducreyi 35000HP; HHV-6, Human herpesvirus 6; HSV-1, Herpes simplex virus type 1; HSV-2, Herpes simplex virus type 2; Hi, Haemophilus influenzae Rd KW20; HK97, Bacteriophage HK97; HP1, Haemophilus phage HP1; Hs, Haemophilus somnus 2336; ILV, Infectious laryngotracheitis virus; K139, Bacteriophage K139; KSHV, Kaposi’s sarcoma-associated herpesvirus; KVP40, Bacteriophage KVP40; LDHV, lung-eye-trachea disease-associated herpesvirus; Li, Listeria innocua; MHV-1, Meleagrid herpesvirus 1; Mj, Methanococcus jannaschii; Ml, Mesorhizobium loti; Mu, Enterobacteria phage Mu; Na, Novosphingobium aromaticivorans; Nm-MC58, Neisseria meningitidis MC58; Nm-Z2491, Neisseria meningitidis Z2491; P2, Enterobacteria phage P2; phi-C31, Bacteriophage phi-C31; phiCTX, Bacteriophage phi CTX; Pl, Photorhabdus luminescens subsp. laumondii TTO1; psiM100, Methanothermobacter wolfeii prophage psiM100; psiM2, Methanobacterium phage psiM2; RB49, Enterobacteria phage RB49; RM378, Bacteriophage RM 378; Rs, Ralstonia solanacearum; Se, Salmonella enterica subsp. enterica serovar Typhi; Sf, Shigella flexneri 2a str. 2457T; SfbV, Shigella flexneri bacteriophage V; So, Shewanella oneidensis MR-1; S-PM2, cyanophage S-PM2; ST64B, Salmonella typhimurium phage ST64B; T4, phage T4; VZV, Varicellazoster virus; Xc, Xanthomonas campestris pv. campestris str. ATCC 33913; Xf-T, Xylella fastidiosa Temecula1.
The average pairwise sequence identities within and between each family or subfamily are shown in Figure 2B ▶. The within-group identities are usually above 35%, with the only exception of U35.001, which is the most diverse family with the largest number of members and the lowest within-group identity of 24.3%. In contrast to the rather high within-group identities, the between-group identities are much lower, with most falling between 10% and 15%, reflecting the extreme sequence diversity of the procapsid protease superfamily. Interestingly, COG3566 and herpes-virus protease family S21 share a comparatively high sequence identity of 17.9%, indicating that these two groups are more similar to each other than to other families. In the multiple sequence alignment (Fig. 1 ▶) discussed below, their similarities around the active site are apparent.
Fold and active site prediction for the phage prohead proteases
The inferred homology between phage prohead proteases and herpesvirus protease offers a tentative prediction that these two kinds of proteases may adopt a similar fold. To explore this possibility further, we submitted representative sequences from each of the phage prohead protease families to the fold-recognition Meta server (Ginalski et al. 2003). Some of the queries found herpesvirus protease fold among their top hits. For example, when HK97 gp4 (gi|9634157, residues 1–225) was used as a query, all of the 3D-Jury hits are herpesvirus proteases with significant consensus scores ranging from 70 to 93 (scores above 50 are considered to be significant; Ginalski et al. 2003). The representative sequences for U35.002.c (gi|34496934, residues 1–202) and COG3566 (gi|38638622, residues 1–200) also found the herpesvirus protease fold with the best consensus scores of 50 and 24, respectively.
To validate the homologous relationship between phage prohead proteases and herpesvirus protease, we constructed a PCMA (Pei et al. 2003) multiple-sequence alignment of all of the detected sequences (199 in total) in the procapsid protease superfamily. This alignment was manually adjusted according to sequence conservation, secondary structure predictions by PSI-PRED (Jones 1999), and comparisons with PSI-BLAST local alignments. Figure 1 ▶ shows the representative sequences in each family. The secondary structure predictions of phage prohead protease families exhibit an overall agreement with the experimental structure of herpesvirus protease, which is also reflected by the conservation of hydrophobicity pattern and small residue positions (yellow and gray shadings in Fig. 1 ▶). The structural core of herpesvirus protease fold is a seven-stranded β-barrel composed of two orthogonally packed, four-stranded β-sheets (β3β4β1β7 and β3β2β6β5) (Fig. 3 ▶). A sharp bend near a conserved Gly enables β3 to participate in both sheets (Chen et al. 1996). This central, mainly antiparallel β-barrel is capped by αA at one end and αBαC at the other, whereas the remaining four C-terminal helices (αDαEαFαG) and the loops between them form a circle surrounding the barrel (Tong et al. 1996; Tong 2002). The active form of herpesvirus protease is a homodimer, in which the monomer–monomer interaction is mediated mainly by αF and partially by αB and αC. The connection between the secondary structure elements is as follows: β1–αA-β2–β3–β4–αB-αC-β5–β6–β7–αD-αE-αF-αG (Fig. 3 ▶; Tong et al. 1996). The multiple sequence alignment in Figure 1 ▶ covers only β1–β6, as the rest of the sequences are too divergent to ensure reliable alignment. However, the covered region constitutes the major part of the herpesvirus protease fold and includes all of the active site residues (catalytic residues and oxyanion-binding loop). Each of the aligned secondary structure elements (β1–β6) finds its counterpart in phage prohead protease sequences except αB, which corresponds to a long gap in the phage families. The secondary structure prediction results indicate that there is only one helix between β4 and β5 in phage families. We aligned this predicted helix with αC in herpesvirus protease instead of αB, as this alignment gives a better sequence conservation pattern. Because αB is involved in homodimer formation in herpesvirus protease, the absence of this helix might affect the manner or even the capability of phage prohead protease dimerization.
Figure 3.
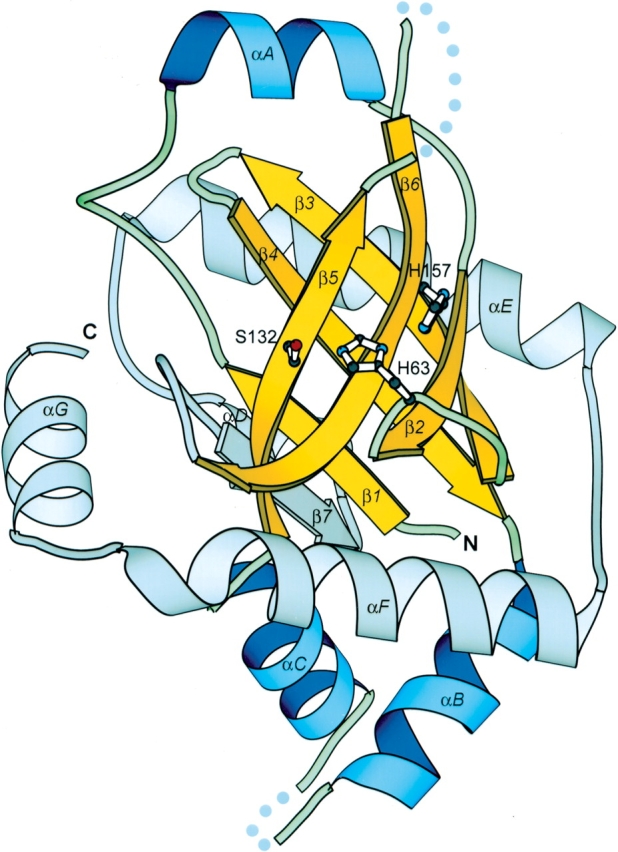
The structure of herpes simplex virus type 2 protease (PDB code 1at3, chain A). β-strands and α-helices included in the multiple sequence alignment in Figure 1 ▶ are shown in yellow and in blue, respectively. The remaining secondary structure elements are shown in gray. β-strands are labeled numerically and α-helices alphabetically. The catalytic triad (Ser 132, His 63, and His 157 according to HCMV protease) is shown in ball-and-stick representation. N and C termini are labeled. Dotted lines represent disordered regions in the structure. This diagram is drawn by MOLSCRIPT (Kraulis 1991).
Including the consistency of hydrophobicity patterns and conservation of small residue positions, the multiple sequence alignment also reveals two invariant residues across all of the families (boxed in black in Fig. 1 ▶). These two residues (His63 in the loop between β2 and β3 and Ser 132 in the middle of β5 in human cytomegalovirus or HCMV protease; Tong et al. 1996) constitute the essential catalytic elements for serine proteases; Ser 132 is the nucleophile and His 63 is the general base (Chen et al. 1996). The invariance of these two residues in families U35.001, U35.002, U9, and COG3566 strongly suggests that these phage prohead proteases are serine proteases and share the same overall catalytic mechanism with herpesvirus protease. However, the third member of the herpesvirus protease catalytic triad (His 157 in HCMV protease; Chen et al. 1996) is not conserved in phage prohead proteases (shaded in green in Fig. 1 ▶). This histidine third member is a distinctive feature of the herpesvirus protease catalytic triad, as classical serine proteases usually use an Asp or Glu in this position. Detailed biochemical and structural studies have shown that this His contributes very little to catalysis, making herpesvirus protease a rather slow enzyme compared with classical serine proteases (Khayat et al. 2001; Tong 2002). In phage prohead protease families, the position corresponding to this His is occupied by various residues: in U35.001, mostly Glu and Asp, like in classical serine proteases; in COG3566 and U35.002.c, mainly His, like in herpesvirus proteases; and in U9, U35.002.a, and U35.002.b, mainly Gly and Ala. Because Gly and Ala are unlikely to participate in catalysis, members in U9, U35.002.a, and U35.002.b might just rely on a catalytic diad composed of the conserved Ser and His residue pair to perform chemistry. Mutagenesis studies on classical serine proteases and herpesvirus protease have shown that this kind of Ser/His catalytic diad is able to sustain the enzymatic activity at a lower efficiency (Khayat et al. 2001; Tong 2002). Alternatively, the U9, U35.002.a, and U35.002.b enzymes might still use a catalytic triad, except that the third member in this triad has migrated to some other position. Such active-site residue migrations were observed in many enzyme superfamilies (Todd et al. 2001; Kinch and Grishin 2002a). In family U9, for instance, a possible candidate for the third member is the conserved Asp just two positions C-terminal from the Gly/Ala highlighted in green (Fig. 1 ▶). In any case, members in this procapsid protease superfamily have different residue types in the position corresponding to the third member in the catalytic triad. This variability may imply differences in detailed catalytic mechanisms and enzymatic efficiency, and reflect varied requirements on procapsid protease activity to match the head maturation process in different viruses.
The oxyanion-binding loop in herpesvirus protease consists of residues Gly 164 to Thr 169 (residue number according to HCMV protease), with the backbone amide of Arg 165 (boxed in red in Fig. 1 ▶) directly contributing to the oxyanion hole (Reiling et al. 2000). In herpesvirus protease family S21, this oxyanion-binding loop is highly conserved. However, in phage prohead protease families U35.001, U35.002, and U9, a different conservation pattern is observed in this region. For example, the position corresponding to Gly 164 is occupied instead by a highly conserved Pro. More importantly, the oxyanion hole residue Arg 165 in S21 family is substituted by a conserved small residue (mainly A, sometimes S or G). Because the backbone amide, but not the side chain, forms an H-bond with the oxyanion, we expect that these small residues in this position could also play the oxyanion-binding role.
Although the oxyanion loop region in family S21 differs from its corresponding regions in families U35.001, U35.002, and U9, it is surprisingly similar to that found in COG3566. The shared features include the oxyanion hole residue R165 and the conserved G164 and G168. Furthermore, COG3566 also has a conserved His in the position occupied by the His third member of herpesvirus protease catalytic triad, whereas the residues in other families vary in this position. These similarities make COG3566 the most similar group to herpesvirus proteases in this procapsid protease superfamily. This similarity is also reflected in the sequence identity analysis; S21 family shows the highest between-group identity with COG3566, and vice versa.
Gene organization of the phage head-assembly module
In phage genomes, the genes functioning in head assembly usually cluster together to form a module (Hendrix 2003). Important members in this module include a portal protein, a prohead protease, a scaffolding protein, and capsid protein(s) (Duda et al. 1995). During this study, we observed three different situations concerning the position of the prohead protease gene in this module relative to those of the scaffolding and capsid protein genes (Fig. 4 ▶).
Figure 4.

Organizations of head assembly genes in different viruses (virus name abbreviations according to Fig. 1 ▶). Each gene is represented by a rectangular bar with the gene name on it. Genes are shaded according to their functions. The different shadings of HK97 gene 5 and c2 l5 N-terminal parts indicate their putative roles as scaffolding protein or protease, respectively. The dotted lines in Mu and HSV-1 diagrams indicate omitted genes. The diagrams for T4, λ, HK97, and T7 are adapted with modifications from Duda et al. (1995) with permission from Elsevier © 1995. Functional assignments for Mu and HSV-1 genes are according to Grimaud (1996), Morgan et al. (2002), Sheaffer et al. (2000), Newcomb et al. (2001), and Perelygina et al. (2003). The assignment of Mu gpH as portal protein is speculative.
In the first case, the prohead protease is followed by a scaffolding protein and then by a capsid protein (Fig. 4 ▶; T4 and HK97). For instance, phage T4’s prohead protease (gp21) is followed by its scaffolding protein (gp22) and major capsid protein (gp23; Duda et al. 1995; Dokland 1999). Similar gene arrangement is also observed in phage HK97. This phage does not have a specific scaffolding protein. However, the N-terminal part of its capsid protein (gp5) has been suggested to fulfill this role (Duda et al. 1995; Hendrix and Duda 1998). Thus, HK97 prohead protease gp4 is still followed by the scaffolding (N-terminal part of gp5) and capsid protein (the remaining of gp5), although the scaffolding and capsid proteins are fused together in this case.
In the second scenario, the prohead protease and the scaffolding protein share the same gene (Fig. 4 ▶; Mu). In phage Mu, the putative protease gpI has 361 residues. However, only the first half can be aligned with other prohead proteases (Fig. 1 ▶; U35.002.a). The C-terminal 183 residues of gpI correspond to another protein, gpZ, which is experimentally confirmed to be the Mu scaffolding protein (Grimaud 1996; Morgan et al. 2002). Thus, the scaffolding protein gpZ uses the same reading frame and the same stop codon as the protease gpI; only the start codon is different (Morgan et al. 2002). This gene arrangement is strikingly similar to that observed in gpC and gpNu3 proteins of bacteriophage λ (Fig. 4 ▶; λ; Morgan et al. 2002). However, λ gpC is not a member in this procapsid protease superfamily. Instead, it is homologous to Escherichia coli protease IV (discussed below). Interestingly, herpesvirus protease and scaffolding protein are also arranged in a nested way similar to phage Mu (Fig. 4 ▶; HSV-1; Sheaffer et al. 2000). The protease is encoded by the full-length version of UL26 gene, whereas the scaffolding protein is encoded by a truncated version (UL26.5). Thus, the scaffolding protein has exactly the same sequence as the C-terminal part of the protease.
In the last scenario, the prohead protease is fused to the capsid protein (Fig. 4 ▶; c2). For example, the predicted lactococcal bacteriophage c2 prohead protease covers the N-terminal part of gene l5 (Fig. 1 ▶; U35.001, gi|9628687), whereas the C-terminal part (residues 206–480) is indicated by biochemical studies to be the c2 major capsid protein (Lubbers et al. 1995). The fusion of protease and capsid protein is also observed in Rhodobacter prophage φRcM1 (Smith et al. 1999).
The fusion of prohead protease to scaffolding protein or capsid protein suggests the possibility of autoproteolysis. In fact, autocleavage has been observed for herpesvirus protease (Homa and Brown 1997; Sheaffer et al. 2000).
It is also worth mentioning that many phages do not encode a protease in their head assembly gene module. For instance, phage T7 (Fig. 4 ▶; T7) head assembly does not involve proteolysis (Duda et al. 1995).
Distribution of procapsid protease superfamily members
Altogether, we detected 199 sequences in this procapsid protease superfamily: 89 from bacteria, one from archaea, 65 from bacterial or archaeal phages, and 44 from herpes-viruses. No homologs were detected from other kinds of viruses or eukaryotes. As discussed above, the bacterial and archaeal sequences probably originate from integrated prophages.
The phage sequences come from 65 different phages, of which 48 are in the order Caudovirales (Maniloff and Ackermann 1998) and 17 are unclassified bacteriophages. Among the Caudovirales phages, 20 are from the family Myoviridae (e.g., Mu, T4, P2), 26 from Siphoviridae (e.g., HK97, c2), and two from Podoviridae (e.g., ST64B). All of these are bacteriophages, except for psiM2 and psiM100, which are closely related archaeophages. (In fact, psiM100 is a defective prophage found in archaeon Methanothermobacter wolfeii [Luo et al. 2001].)
Interestingly, some phages known to carry out head-maturation proteolysis do not possess a procapsid protease. Instead, another kind of protease is found in their head-assembly gene module at roughly the same position as the prohead protease, suggesting that this second type of protease mediates the maturation proteolysis. Bacteriophage λ putative protease gpC is an example of this second type (Baird et al. 1991). In MEROPS, gpC is classified in the protease IV family S49. However, it also shows significant sequence similarity to the ClpP proteases (MEROPS S14). For example, CDD search using λ gpC (gi|9626248, residues 1–439) as query revealed COG0740 or ClpP with evalue 3e-06. Thus, we will refer to this second type of phage protease as protease IV/ClpP-type. In addition to λ, we found a protease IV/ClpP-type protease in the head-assembly module in coliphage 21 (Smith and Feiss 1993), Streptococcus thermophilus phage Sfi21 (Desiere et al. 1999), Staphylococcus aureus phage φ12 (Iandolo et al. 2002), and a few other phages. ClpP has a known 3D structure (Wang et al. 1997), which has a fold different from the herpesvirus protease (Tong et al. 1996).
Surprisingly, certain phages possess both types of proteases (procapsid proteases and protease IV/ClpP-type). For example, mycobacteriophage CJW1 gp11 is in the prohead protease family U35.001 (Fig. 1 ▶, gi|29565890). However, its gp103 is homologous to ClpP protease (Pedulla et al. 2003). Because gp11 is located in the head-assembly module, but gp103 is far away downstream, we expect that the maturation proteolysis be carried out by gp11. Further experiments are needed to elucidate the function of gp103.
Evolutionary implications
In this study, we argue that herpesvirus protease (UL26 protein) is homologous to dsDNA phage prohead proteases. This relationship is consistent with the well-established morphological similarities shared between herpesviruses and dsDNA phages in their head-assembly pathways. These similarities mainly include the requirement and subsequent removal of scaffolding proteins; and the preassembly and subsequent maturation of prohead or procapsid (Homa and Brown 1997; Dokland 1999). Our results provide evidence at the molecular level that herpesvirus and dsDNA phage head-assembly pathways have a common origin. Actually, the head-assembly pathway also includes DNA packaging, a process to encapsulate the viral genome. A similar DNA packaging mechanism is also shared by herpesviruses and dsDNA phages, in which a concatemer DNA is cleaved to unit genomes and translocated into the head or capsid by a virus-encoded terminase (Catalano 2000; Newcomb et al. 2001). Recently, the large subunit of this terminase was found to be homologous in herpesviruses and dsDNA phages (Mitchell et al. 2002; Przech et al. 2003). In addition to procapsid protease and terminase large subunit, the herpesvirus alkaline exonuclease, which functions in DNA recombination and replication, also shares homology with its counterpart in dsDNA phages (Bujnicki and Rychlewski 2001; Reuven et al. 2003). Importantly, these three herpes-virus enzymes (protease, terminase, and exonuclease) do not have counterparts detected from eukaryotic cells to date (Bujnicki and Rychlewski 2001; Mitchell et al. 2002). Thus, it is unlikely that herpesviruses acquired these enzymes from their eukaryotic hosts. A parsimonious explanation is that herpesviruses and dsDNA phages are evolutionarily related (Dokland 1999; Newcomb et al. 2001; Mitchell et al. 2002), and these shared enzymes are inherited from an ancestral virus.
Conclusions
Through computational sequence and structure analysis, we infer homology between phage prohead proteases (MEROPS families U35.001, U35.002, and U9) and herpesvirus protease (MEROPS family S21), and unify them to form a procapsid protease superfamily. This homology offers a fold prediction for phage prohead proteases and implies that they are serine proteases. Members in this procapsid protease superfamily are found in bacteriophages, archaeophages, herpesviruses, bacteria, and archaea. Our study presents evidence that herpesvirus and dsDNA phages are evolutionarily related.
Materials and methods
Sequence similarity searches, multiple sequence alignment, secondary structure prediction, and fold recognition
Sequence similarity searches were carried out against the nonredundant database (nr, from Dec. 11, 2003 with 1,551,548 sequences to Jan. 1, 2004 with 1,584,649 sequences, filtered for low complexity regions). The SEALS package (Walker and Koonin 1997) was used to script transitive PSI-BLAST searches; starting with a single query sequence, PSI-BLAST (Altschul et al. 1997) with specified parameters (BLOSUM62 scoring matrix, e-value cutoff 0.001) was iterated until convergence; found homologs were grouped using single-linkage clustering (1 bit per site threshold, ~50% identity), and representative sequences from these groups were used as new queries in subsequent PSI-BLAST rounds. These searches were repeated until no more new hits were found.
Multiple sequence alignments were constructed using the PCMA program (Pei et al. 2003) followed by manual adjustment. PCMA first aligns similar sequences in a fast way by ClustalW (Thompson et al. 1994) to form prealigned groups. These pre-aligned sequence groups are then aligned by a consistency objective function (Notredame et al. 2000) to improve alignment accuracy for divergent sequences. The similarity threshold of PCMA is set to 50% in this study (Pei et al. 2003).
In seeded BLAST searches, a position-specific scoring matrix was generated according to the input PCMA alignment. Then, each sequence in the input alignment was used as a query to run one round of PSI-BLAST using the alignment-generated matrix.
Secondary structure prediction was performed using PSI-PRED (Jones 1999). Representative sequences from each family were submitted to the 3D-Jury system on the fold recognition Meta server (Ginalski et al. 2003; http://bioinfo.pl/Meta).
Euclidian distance mapping and average pairwise sequence identity
The global multiple alignment of all the detected sequences (199 in total) was used for distance mapping and pairwise identity calculations. Euclidian distance mapping was performed by the EESG program (Grishin and Grishin 2002). Columns with more than 40% gaps were removed from the input multiple sequence alignment. In calculating the pairwise percent identity between two sequences, the numerator was the number of identical residue pairs and the denominator was the number of aligned positions in which both sequences had an amino acid. The pairwise percent identities were calculated for all sequence pairs within a family and averaged to give the within-group sequence identity for that family. The between-group identity for two families was calculated in a similar way, only that the two sequences in a sequence pair came from different families.
Electronic supplemental material
A multiple sequence alignment of all the detected sequences in this procapsid protease superfamily (199 in total) can be downloaded from ftp://iole.swmed.edu/pub/cheng/prohead. Also available on this site are a table of the taxonomy information for all the 199 sequences, a list of all the gene annotations, and a figure summarizing the PSI-BLAST scheme of the procapsid protease superfamily delineation.
Acknowledgments
We thank Lisa Kinch for critical reading of the manuscript and helpful suggestions. H.C. thanks Mei Qi for assistance in programming. This work was supported by NIH grant GM67165 to N.V.G.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
PDB, Protein Data Bank
PSI-BLAST, position-specific iterated basic local alignment search tool
COG, clusters of orthologous groups
3D, three-dimensional
dsDNA, double-stranded DNA
PCMA, profile consistency multiple sequence alignment.
Supplemental material: see ftp://iole.swmed.edu/pub/cheng/prohead
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04726004.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, L., Lipinska, B., Raina, S., and Georgopoulos, C. 1991. Identification of the Escherichia coli sohB gene, a multicopy suppressor of the HtrA (DegP) null phenotype. J. Bacteriol. 173 5763–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, A.J., Rawlings, N.D., and O’Brien, E.A. 2001. The MEROPS database as a protease information system. J. Struct. Biol. 134 95–102. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Finn, R.D., and Sonnhammer, E.L. 1999. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27 260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujnicki, J.M. and Rychlewski, L. 2001. The herpesvirus alkaline exonuclease belongs to the restriction endonuclease PD-(D/E)XK superfamily: Insight from molecular modeling and phylogenetic analysis. Virus Genes 22 219–230. [DOI] [PubMed] [Google Scholar]
- Catalano, C.E. 2000. The terminase enzyme from bacteriophage λ: A DNA-packaging machine. Cell. Mol. Life Sci. 57 128–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P., Tsuge, H., Almassy, R.J., Gribskov, C.L., Katoh, S., Vanderpool, D.L., Margosiak, S.A., Pinko, C., Matthews, D.A., and Kan, C.C. 1996. Structure of the human cytomegalovirus protease catalytic domain reveals a novel serine protease fold and catalytic triad. Cell 86 835–843. [DOI] [PubMed] [Google Scholar]
- Desiere, F., Lucchini, S., and Brussow, H. 1999. Comparative sequence analysis of the DNA packaging, head, and tail morphogenesis modules in the temperate cossite Streptococcus thermophilus bacteriophage Sfi21. Virology 260 244–253. [DOI] [PubMed] [Google Scholar]
- Dokland, T. 1999. Scaffolding proteins and their role in viral assembly. Cell. Mol. Life Sci. 56 580–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda, R.L., Martincic, K., Xie, Z., and Hendrix, R.W. 1995. Bacteriophage HK97 head assembly. FEMS Microbiol. Rev. 17 41–46. [DOI] [PubMed] [Google Scholar]
- Fischer, D. 2003. 3D-SHOTGUN: A novel, cooperative, fold-recognition meta-predictor. Proteins 51 434–441. [DOI] [PubMed] [Google Scholar]
- Ginalski, K., Elofsson, A., Fischer, D., and Rychlewski, L. 2003. 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics 19 1015–1018. [DOI] [PubMed] [Google Scholar]
- Grimaud, R. 1996. Bacteriophage Mu head assembly. Virology 217 200–210. [DOI] [PubMed] [Google Scholar]
- Grishin, V.N. and Grishin, N.V. 2002. Euclidian space and grouping of biological objects. Bioinformatics 18 1523–1534. [DOI] [PubMed] [Google Scholar]
- Hendrix, R.W. 2003. Bacteriophage genomics. Curr. Opin. Microbiol. 6 506–511. [DOI] [PubMed] [Google Scholar]
- Hendrix, R.W. and Duda, R.L. 1998. Bacteriophage HK97 head assembly: A protein ballet. Adv. Virus Res. 50 235–288. [DOI] [PubMed] [Google Scholar]
- Homa, F.L. and Brown, J.C. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 7 107–122. [DOI] [PubMed] [Google Scholar]
- Iandolo, J.J., Worrell, V., Groicher, K.H., Qian, Y., Tian, R., Kenton, S., Dorman, A., Ji, H., Lin, S., Loh, P., et al. 2002. Comparative analysis of the genomes of the temperate bacteriophages φ 11, φ 12 and φ 13 of Staphylococcus aureus 8325. Gene 289 109–118. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Khayat, R., Batra, R., Massariol, M.J., Lagace, L., and Tong, L. 2001. Investigating the role of histidine 157 in the catalytic activity of human cytomegalovirus protease. Biochemistry 40 6344–6351. [DOI] [PubMed] [Google Scholar]
- Kinch, L.N. and Grishin, N.V. 2002a. Evolution of protein structures and functions. Curr. Opin. Struct. Biol. 12 400–408. [DOI] [PubMed] [Google Scholar]
- ———. 2002b. Expanding the nitrogen regulatory protein superfamily: Homology detection at below random sequence identity. Proteins 48 75–84. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Krogh, A., Brown, M., Mian, I.S., Sjolander, K., and Haussler, D. 1994. Hidden Markov models in computational biology. Applications to protein modeling. J. Mol. Biol. 235 1501–1531. [DOI] [PubMed] [Google Scholar]
- Lata, R., Conway, J.F., Cheng, N., Duda, R.L., Hendrix, R.W., Wikoff, W.R., Johnson, J.E., Tsuruta, H., and Steven, A.C. 2000. Maturation dynamics of a viral capsid: Visualization of transitional intermediate states. Cell 100 253–263. [DOI] [PubMed] [Google Scholar]
- Lubbers, M.W., Waterfield, N.R., Beresford, T.P., Le Page, R.W., and Jarvis, A.W. 1995. Sequencing and analysis of the prolate-headed lactococcal bacteriophage c2 genome and identification of the structural genes. Appl. Environ. Microbiol. 61 4348–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundstrom, J., Rychlewski, L., Bujnicki, J., and Elofsson, A. 2001. Pcons: A neural-network-based consensus predictor that improves fold recognition. Protein Sci. 10 2354–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y., Pfister, P., Leisinger, T., and Wasserfallen, A. 2001. The genome of archaeal prophage PsiM100 encodes the lytic enzyme responsible for autolysis of Methanothermobacter wolfeii. J. Bacteriol. 183 5788–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniloff, J. and Ackermann, H.W. 1998. Taxonomy of bacterial viruses: Establishment of tailed virus genera and the order Caudovirales. Arch. Virol. 143 2051–2063. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer, A., Anderson, J.B., DeWeese-Scott, C., Fedorova, N.D., Geer, L.Y., He, S., Hurwitz, D.I., Jackson, J.D., Jacobs, A.R., Lanczycki, C.J., et al. 2003. CDD: A curated Entrez database of conserved domain alignments. Nucleic Acids Res. 31 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, M.S., Matsuzaki, S., Imai, S., and Rao, V.B. 2002. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 30 4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, G.J., Hatfull, G.F., Casjens, S., and Hendrix, R.W. 2002. Bacteriophage Mu genome sequence: Analysis and comparison with Mulike prophages in Haemophilus, Neisseria and Deinococcus. J. Mol. Biol. 317 337–359. [DOI] [PubMed] [Google Scholar]
- Newcomb, W.W., Juhas, R.M., Thomsen, D.R., Homa, F.L., Burch, A.D., Weller, S.K., and Brown, J.C. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75 10923–10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302 205–217. [DOI] [PubMed] [Google Scholar]
- Pedulla, M.L., Ford, M.E., Houtz, J.M., Karthikeyan, T., Wadsworth, C., Lewis, J.A., Jacobs-Sera, D., Falbo, J., Gross, J., Pannunzio, N.R., et al. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113 171–182. [DOI] [PubMed] [Google Scholar]
- Pei, J. and Grishin, N.V. 2001. GGDEF domain is homologous to adenylyl cyclase. Proteins 42 210–216. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Peptidase family U34 belongs to the superfamily of N-terminal nucleophile hydrolases. Protein Sci. 12 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, J., Sadreyev, R., and Grishin, N.V. 2003. PCMA: Fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics 19 427–428. [DOI] [PubMed] [Google Scholar]
- Perelygina, L., Zhu, L., Zurkuhlen, H., Mills, R., Borodovsky, M., and Hilliard, J.K. 2003. Complete sequence and comparative analysis of the genome of herpes B virus (Cercopithecine herpesvirus 1) from a rhesus monkey. J. Virol. 77 6167–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przech, A.J., Yu, D., and Weller, S.K. 2003. Point mutations in exon I of the herpes simplex virus putative terminase subunit, UL15, indicate that the most conserved residues are essential for cleavage and packaging. J. Virol. 77 9613–9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings, N.D., O’Brien, E., and Barrett, A.J. 2002. MEROPS: The protease database. Nucleic Acids Res. 30 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling, K.K., Pray, T.R., Craik, C.S., and Stroud, R.M. 2000. Functional consequences of the Kaposi’s sarcoma-associated herpesvirus protease structure: Regulation of activity and dimerization by conserved structural elements. Biochemistry 39 12796–12803. [DOI] [PubMed] [Google Scholar]
- Reuven, N.B., Staire, A.E., Myers, R.S., and Weller, S.K. 2003. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 77 7425–7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaffer, A.K., Newcomb, W.W., Brown, J.C., Gao, M., Weller, S.K., and Tenney, D.J. 2000. Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J. Virol. 74 6838–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M.P. and Feiss, M. 1993. Sequence analysis of the phage 21 genes for prohead assembly and head completion. Gene 126 1–7. [DOI] [PubMed] [Google Scholar]
- Smith, M.C., Burns, R.N., Wilson, S.E., and Gregory, M.A. 1999. The complete genome sequence of the Streptomyces temperate phage straight phiC31: Evolutionary relationships to other viruses. Nucleic Acids Res. 27 2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Natale, D.A., Garkavtsev, I.V., Tatusova, T.A., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D., and Koonin, E.V. 2001. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, A.E., Orengo, C.A., and Thornton, J.M. 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307 1113–1143. [DOI] [PubMed] [Google Scholar]
- Tong, L. 2002. Viral proteases. Chem. Rev. 102 4609–4626. [DOI] [PubMed] [Google Scholar]
- Tong, L., Qian, C., Massariol, M.J., Bonneau, P.R., Cordingley, M.G., and Lagace, L. 1996. A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature 383 272–275. [DOI] [PubMed] [Google Scholar]
- Walker, D.R. and Koonin, E.V. 1997. SEALS: A system for easy analysis of lots of sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5 333–339. [PubMed] [Google Scholar]
- Wang, J., Hartling, J.A., and Flanagan, J.M. 1997. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell 91 447–456. [DOI] [PubMed] [Google Scholar]