

Abstract
In contrast to water-soluble proteins, membrane proteins reside in a heterogeneous environment, and their surfaces must interact with both polar and apolar membrane regions. As a consequence, the composition of membrane proteins’ residues varies substantially between the membrane core and the interfacial regions. The amino acid compositions of helical membrane proteins are also known to be different on the cytoplasmic and extracellular sides of the membrane. Here we report that in the 16 transmembrane β-barrel structures, the amino acid compositions of lipid-facing residues are different near the N and C termini of the individual strands. Polar amino acids are more prevalent near the C termini than near the N termini, and hydrophobic amino acids show the opposite trend. We suggest that this difference arises because it is easier for polar atoms to escape from the apolar regions of the bilayer at the C terminus of a β-strand. This new characteristic of β-barrel membrane proteins enhances our understanding of how a sequence encodes a membrane protein structure and should prove useful in identifying and predicting the structures of trans-membrane β-barrels.
Keywords: β barrel, membrane protein, snorkeling, membrane polarity, protein structure, genome
Transmembrane (TM) β-barrel proteins comprise 2%–3% of the Gram-negative bacterial genomes and belong to a variety of protein functional groups, such as nonspecific and specific pores, transporters, lipases, and proteases (Wimley 2003). As is exemplified by Omp A, the polypeptides are secreted into the periplasmic space, where they bind a chaperone (Mori and Ito 2001; Kleinschmidt and Tamm 2002). Their insertion into the outer membrane is then facilitated by binding to a periplasmic lipopolysaccharide. In eukaryotes, TM β-barrels are found in the outer membrane of mitochondria and chloroplasts (Benz 1994; Fischer et al. 1994). The known structures include β-barrels containing 8–22 β-strands (Schulz 2002). Frequently, the strands are contained within the same polypeptide chain, although there are exceptions. For example, in the heptamer, α-hemolysin, each subunit contributes two β-strands to a 14-stranded β-barrel (Song et al. 1996).
The membrane can influence the protein’s amino acid composition. Some amino acids in TM helical proteins have a bias toward residing on the cytoplasmic or extracellular side of the membrane (Sipos and von Heijne 1993). In β-barrel proteins, however, there are apparently no strong periplasmic/extracellular biases, with the exception of Lys (Ulmschneider and Sansom 2001). Recently, we uncovered another amino acid bias in α-helical membrane proteins: The amino acid composition differs between the N- and C-terminal ends of the helices (Chamberlain et al. 2004). In general, the N-terminal end contains more hydrophilic amino acids, whereas the C-terminal end contains more hydrophobic amino acids. This preference appears to arise from the geometry of the α-helix and from the preference of hydrophilic amino acids to “snorkel” their polar atoms out of the membrane. Side chains in a helix extend back toward the N terminus, making it easier for polar atoms to escape the bilayer when they reside at this terminus.
Here we report an analogous trend in the amino acid pattern of β-barrels; namely, that the amino acid distribution varies between the N- and C-terminal ends of the strands within the membrane. In TM strands, the N-terminal half contains an abundance of hydrophobic amino acids when compared to the C-terminal half. The hydrophilic amino acids have the opposite preference. These trends define a new constraint on β-barrel membrane protein structures that may also arise from the limitations imposed on the side chains by the membrane polarity gradient.
Results
We identified the transmembrane β-strands of 16 β-barrel membrane proteins and divided the transmembrane residues into groups based on their location in the structure. The 220 β-strands contained a total of 2042 residues. We categorized each residue based on its burial or exposure to the membrane and on its location in one half of the membrane. We considered the membrane to be 30.0 Å thick and divided it into either two 15.0 Å sections or six 5.0 Å sections. Here we refer to the half of the membrane toward the N- or C-terminal end of the strands as the N- or C-terminal side of the membrane.
The frequencies of the amino acids facing the lipids differ in the N- and C-terminal regions of the membrane (Fig. 1A ▶). Several hydrophobic amino acids are more frequent in the N-terminal half of the membrane than in the C-terminal half. For example, Ile is twice as frequent in the N-terminal half compared with the C-terminal half. It comprises 9.1% (48 of 528) of the amino acids in the N terminus and only 4.7% (24 of 515) in the C terminus. Leu and Val also show a similar bias for the N-terminal side. In contrast, the C terminus has a higher frequency of many polar amino acids compared with the N terminus, and particularly Arg, Gln, and Tyr. Tyr is the most common amino acid in the C-terminal half, with a frequency of 16.8%. Of the Tyr residues facing the lipids, 89 of 117 (76%) are in the C-terminal half, whereas 28 of 117 (24%) are in the N-terminal half.
Figure 1.

Amino acid frequencies in the β-strands of 16 β-barrel membrane proteins. (A) The frequency of amino acids facing the membrane lipids in the N-terminal (black) and C-terminal (gray) halves of the membrane. Glu and Arg are not found in the N-terminal half. (B) The statistical significance of the N/C bias of lipid-facing amino acids. For each amino acid, we show the random probability of having the observed distribution, or a more biased distribution, for one particular membrane half. A small probability indicates that the likelihood of having the observed bias is improbable. Amino acids with a gray bar above the drawn line have distributions with less than a 5% chance of occurring at random. (C) The frequency of amino acids facing into the β-barrel in the N-terminal (black) and C-terminal (gray) halves of the strands. (D) The statistical significance of the N/C bias of amino acids facing into the β-barrel center. The amino acids are listed in order of decreasing hydrophobicity (Engelman et al. 1986).
Because of the limited data in the current database, not all of the differences seen in Figure 1A ▶ are statistically significant. In Figure 1B ▶, we show the statistical significance of these results based on a random, binomial model of placing residues in either the N- or C-terminal half. A low probability indicates that the difference observed between the two halves is unlikely to be a chance occurrence. The significance of tyrosine’s preference for the C-terminal half is the greatest because of its strong bias and high overall abundance. The likelihood that the observed C-terminal bias for Tyr residues would be seen by chance is only 9 × 10−9. Other amino acids are also significantly biased. For Ile, Leu, Val, Gly, Gln, and Arg, there is less than a 0.05 chance (Fig. 1B ▶, horizontal bar) of a random occurrence of the observed distribution. Of the 19 lipid-facing Gln residues, five are located in the N-terminal half and 14 are in the C-terminal half, which has a modestly significant likelihood of 0.033. Of those residues with biases that are statistically significant, polar residues favor the C terminus, and apolar residues favor the N terminus.
In contrast, the amino acids facing into the core of the β-barrel do not differ appreciably in composition between the two halves of the β-strands (Fig. 1C,D ▶). Overall, the hydrophobic amino acids are less frequent and the hydrophilic amino acids are more frequent than in the residues facing the lipids. The N- and C-terminal frequencies, however, are essentially equal. Phe is the only exception, having a bias for the C-terminal half and a statistical likelihood of 0.014. Of the interior-facing residues, none of the hydrophilic residues shows a statistically significant composition bias, even though the interior of the β-barrel is generally more hydrophilic than the exterior. These results suggest that the residue composition differences arise from interactions with the bilayer, rather than from constraints intrinsic to β-barrel geometry.
The composition differences in the residues facing the membrane lipids are also seen by dividing the membrane into six 5.0 Å sections from the N- to C-terminal sides of the membrane. As shown in Figure 2A ▶, the hydrophilic amino acids Glu, Gln, Asp, Asn, Lys, and Arg are more populated in the membrane edges and, in particular, in the C-terminal edge. We found all three Glu residues and seven of eight Arg residues in the most C-terminal membrane section. Tyr makes up nearly one-fourth (23.3%) of the most C-terminal section and only 11.3% of the most N-terminal section (Fig. 2B ▶). In contrast, the hydrophobic amino acids are most frequent in the N-terminal side of the membrane core (Fig. 2C ▶). The combined total of Phe, Ile, Leu, and Val is most populated in the second membrane section, making up 64.5% of the total amino acids. This section includes residues between 5.0 and 10.0 Å from the N-terminal edge of our 30.0 Å-thick membrane. The preference of the hydrophobic amino acids for this section arises from their high frequency in the membrane core and from their bias to be more populated in the N-terminal halves of the strands.
Figure 2.
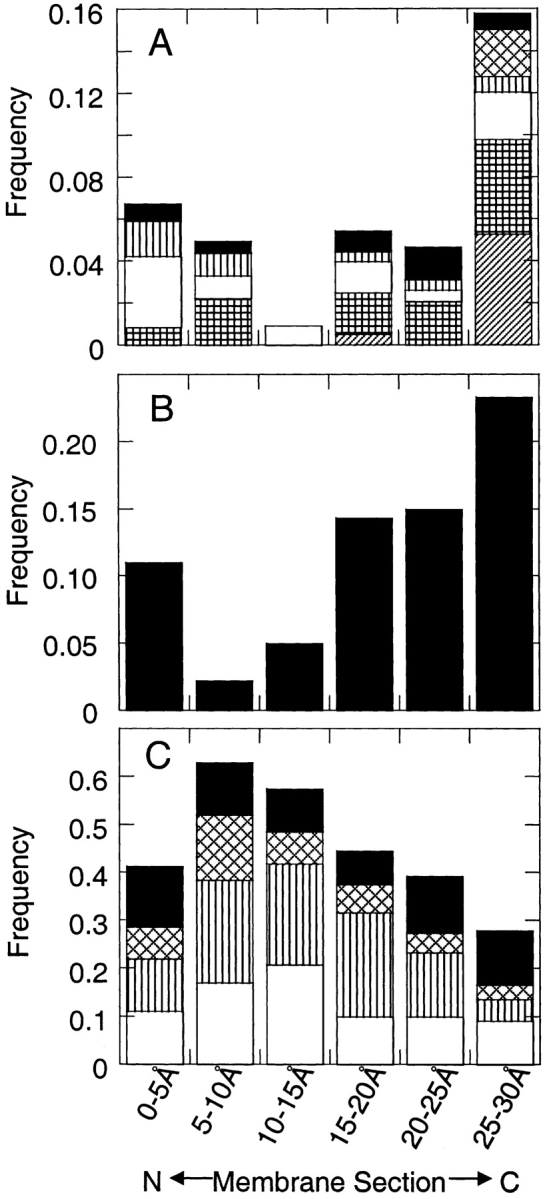
Amino acid frequencies in six 5.0 Å sections of the membrane from the N-terminal side (abscissa, left) to the C-terminal side (abscissa, right) of the membrane. (A) The frequencies of some of the polar amino acids, Asp (solid bar), Glu (diagonally hatched bar), Lys (vertical stripes), Asn (open bar), Gln (vertically hatched bar), and Arg (diagonal stripes). (B) The frequency of Tyr alone. (C) The frequencies of the hydrophobic amino acids, Phe (solid bar), Ile (diagonally hatched bar), Leu (vertically striped bar), and Val (open bar).
Discussion
We suggest that the N-terminal/C-terminal bias in amino acid composition is the result of the interactions of the strands with the membrane lipids. Each side chain should have a preferred orientation that matches its polarity gradient to that of the membrane. Polar residues in a membrane environment prefer to extend their side chains out of the membrane core and toward the aqueous regions. Side chains have three favored choices of the χ1 dihedral angle: −60°, +60° , or +180°. On the outside of a TM β-barrel, extension of the side chain out of the lipid core is better accomplished with a χ1 dihedral angle of 180° than with χ1 angles of −60° or +60°. For example, in Figure 3 ▶, a Tyr side chain with χ1 = 180° extends its Oη atom 5.0 Å toward the C-terminal side of the membrane, but the side chains with χ1 = +60° and −60° extend the Oη atom only 3.2 Å and 2.0 Å toward the N terminus, respectively. Because the largest extension occurs toward the C terminus, Tyr residues are easier to accommodate at the C terminus.
Figure 3.

Extension of the Tyr side chain out of the membrane in different rotamers. The distance the Tyr Oη atom extends out of the membrane core depends on the Tyr χ1 angle. The Oη atom extends 5.0 Å toward the C-terminal side of the membrane with χ1 = 180°, but it only extends 3.2 Å and 2.0 Å toward the N-terminal side with χ1 = +60° and −60°, respectively. These distances are the extension of the Oη atom from the Cβ along the membrane normal. For each χ1 angle, we averaged the distances of Tyr placed in eight different positions in β-strands. These values are specific to lipid-facing Tyr residues. Residues facing inside the β-barrel have a different angle between the N-Cα bond and the normal membrane, and therefore the χ1 angles affect the Oη extension differently.
This explanation is analogous to our explanation of amino acid composition differences in TM helices (Chamberlain et al. 2004). In TM helices, however, polar amino acids generally are more populated in the N terminus. The helix geometry favors side chain extension toward the N terminus because of the direction of the Cα-Cβ bond. In β-barrels, the Cα-Cβ bond extends essentially parallel to the membrane, but the tilt of the strands with respect to the membrane normal favors the χ1 = 180° extension toward the C terminus over the other χ1 angles extending toward the N terminus.
These results aid our understanding of transmembrane β-barrels and could be directly applied to the prediction of β-barrels from genomic sequences (Martelli et al. 2002; Wimley 2002; Zhai and Saier Jr. 2002). In particular, the identification of TM β-strands in a sequence should be improved by knowing the amino acid composition in each β-strand position. A more specific description of the β-strands will become feasible as more structures become available. Thus, understanding how the amino acid composition varies in different regions of β-barrels aids our interpretation of genomic information and illuminates the interactions between the membrane and transmembrane proteins.
Materials and methods
Choice of β-barrel membrane proteins
The β-barrel membrane proteins were selected from a list at the Max Planck Institute (http://www.mpibp-frankfurt.mpg.de/michel/public/memprotstruct.html). Each pair of proteins has less than 30% sequence identity (http://www.ncbi.nlm.nih.gov/BLAST/), and all structures were determined by X-ray crystallography to a resolution of 3.0 Å or better. The 16 PDB codes used were 1A0S, 1E54, 1EK9, 1FEP, 1I78, 1K24, 1KMO, 1PHO, 1PRN, 1QD6, 1QJ8, 1QJP, 2FCP, 2MPR, 2POR, and 7AHL.
Identification of transmembrane residues
The strands making up each β-barrel were identified by eye, and the secondary structure assignments were listed in the PDB header file. Each strand was represented as a vector from the second to the eighth Cα, and we inverted the vectors of the odd-numbered strands. By averaging the strand vectors of each protein, we calculated a vector normal to the membrane. If more than one subunit was in a crystal structure, we used all the subunits to determine the membrane normal, but used only one subunit in subsequent calculations. We identified the transmembrane residues by orienting a 30.0 Å-thick slab perpendicular to the membrane normal and positioning it along the membrane normal such that the average hydrophobicity (Fauchere and Pliska 1983) of the residues in the slab were a maximum. A residue was considered to be inside the membrane if its Cα atom was contained in this slab. The 16 structures contain 220 strands and 2042 transmembrane residues. None of the transmembrane strands contains any Cys residues.
We placed each residue into a group based on whether the side chain faced into or out of the center of the β-barrel. A residue was counted as inward facing if its Cβ atom was closer than its Cα atom to the center of mass of the β-barrel. For Gly, we built in and used a Hα2 atom in place of the Cβ atom. We further subdivided the residues according to the location of their Cα atoms in the membrane to the N- or C-terminal end of the strands. We used six 5.0 Å-thick slices or two 15.0 Å-thick slices of the membrane. The frequency of each amino acid in a membrane section is simply the number of counts of the amino acid divided by the total number of amino acids in that section. Error estimates of the frequencies are the square roots of the counts divided by the total number of amino acids. Because the strands of β-barrels are antiparallel, misplacing the membrane toward one end of the β-barrel would add counts of residues to both the N- and C-terminal membrane sections. In this way, our results that show a bias of certain residues for the N- or C-terminal regions of the membrane are not created by errors in the membrane placement.
Statistical test of amino acid bias
To test the statistical significance of the bias of an amino acid to reside in either the N-terminal half or C-terminal half of a trans-membrane β-strand, we constructed a null model in which the amino acid is distributed randomly between the two halves. We then calculated the probability of finding the observed distribution or a more biased distribution, given this random null model. The probability is calculated from the binomial distribution, with the total number of trials equaling the total number of interior-facing or lipid-facing residues. A successful trial occurs when the amino acid is placed in the N terminus. The random frequency that an amino acid will occur in the N terminus is 50.6%, which is the average frequency over all amino acids. A low probability implies that the observed preference of the amino acid for one half is unlikely to occur by random chance.
Measurement of tyrosine side-chain extension
We measured the extension of Tyr side chain along the membrane normal from the Cβatom to the Oη atom in its three most common rotamers. We used the χ1 and χ2 angles from Dunbrack Jr.’s rotamer library (Dunbrack Jr. and Karplus 1993; Dunbrack Jr. and Cohen 1997) and present the average distances using eight positions in two TM barrel structures. These outward-facing positions were residues 223, 274, 292, and 324 in 1E54 and 81, 95, 139, and 164 in 1QJP.
Acknowledgments
We thank Yohan Lee for helping to define the transmembrane strands, and members of the laboratory for their critical reading of the manuscript. This work was supported by NIH grant no. RO1 GM063919. J.U.B. is a Leukemia and Lymphoma Society Scholar.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04777304.
References
- Benz, R. 1994. Permeation of hydrophilic solutes through mitochondrial outer membranes: Review on mitochondrial porins. Biochim. Biophys. Acta 1197 167–196. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K., Lee, Y., Kim, S., and Bowie, J.U. 2004. Snorkeling fosters an amino acid composition bias in transmembrane helices. J. Mol. Biol. 339 471–479. [DOI] [PubMed] [Google Scholar]
- Dunbrack Jr., R.L. and Cohen, F.E. 1997. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack Jr., R.L. and Karplus, M. 1993. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 230 543–574. [DOI] [PubMed] [Google Scholar]
- Engelman, D.M., Steitz, T.A., and Goldman, A. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15 321–353. [DOI] [PubMed] [Google Scholar]
- Fauchere, J.L., and Pliska, V. 1983. Hydrophobic parameters of amino acid side chains from the partitioning of N-acetyl-amino acid amides. Eur. J. Med. Chem.-Chim. Ther. 18 369–375. [Google Scholar]
- Fischer, K., Weber, A., Brink, S., Arbinger, B., Schunemann, D., Borchert, S., Heldt, H.W., Popp, B., Benz, R., Link, T.A., et al. 1994. Porins from plants. Molecular cloning and functional characterization of two new members of the porin family. J. Biol. Chem. 269 25754–25760. [PubMed] [Google Scholar]
- Kleinschmidt, J.H., and Tamm, L.K. 2002. Secondary and tertiary structure formation of the β-barrel membrane protein OmpA is synchronized and depends on membrane thickness. J. Mol. Biol. 324 319–330. [DOI] [PubMed] [Google Scholar]
- Martelli, P.L., Fariselli, P., Krogh, A., and Casadio, R. 2002. A sequence-profile-based HMM for predicting and discriminating β barrel membrane proteins. Bioinformatics 18 (Suppl. 1) S46–S53. [DOI] [PubMed] [Google Scholar]
- Mori, H., and Ito, K. 2001. The Sec protein-translocation pathway. Trends Microbiol. 9 494–500. [DOI] [PubMed] [Google Scholar]
- Schulz, G.E. 2002. The structure of bacterial outer membrane proteins. Biochim. Biophys. Acta 1565 308–317. [DOI] [PubMed] [Google Scholar]
- Sipos, L., and von Heijne, G. 1993. Predicting the topology of eukaryotic membrane proteins. Eur. J. Biochem. 213 1333–1340. [DOI] [PubMed] [Google Scholar]
- Song, L., Hobaugh, M.R., Shustak, C., Cheley, S., Bayley, H., and Gouaux, J.E. 1996. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 274 1859–1866. [DOI] [PubMed] [Google Scholar]
- Ulmschneider, M.B., and Sansom, M.S. 2001. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta 1512 1–14. [DOI] [PubMed] [Google Scholar]
- Wimley, W.C. 2002. Toward genomic identification of β-barrel membrane proteins: Composition and architecture of known structures. Protein Sci. 11 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003. The versatile β-barrel membrane protein. Curr. Opin. Struct. Biol. 13 404–411. [DOI] [PubMed] [Google Scholar]
- Zhai, Y., and Saier Jr., M.H. 2002. The β-barrel finder (BBF) program, allowing identification of outer membrane β-barrel proteins encoded within prokaryotic genomes. Protein Sci. 11 2196–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]