

Abstract
Hunter-killer peptides are chimeric synthetic peptides that selectively target specific cell types for an apoptotic death. These peptides, which are models for potential therapeutics, contain a homing sequence for receptor-mediated interactions and a pro-apoptotic sequence. Homing domains have been designed to target angiogenic tumor cells, prostate cells, arthritic tissue and, most recently, adipose tissue. After a receptor-mediated internalization, the apoptotic sequence, which contains D-enantiomer amino acids, initiates apoptosis through mitochondrial membrane disruption. We have begun structure and functional studies on a peptide (HKP1) that specifically targets angiogenic tumor cells for apoptosis. As a model for mitochondrial membrane disruption, we have examined peptide-induced leakage of a calcein fluorophore from large unilamellar vesicles. These experiments demonstrate more potent leakage activity by HKP1 than the peptide lacking the homing domain. Circular dichroism and 2D homonuclear NMR experiments demonstrate that this tumor-specific HKP adopts a left-handed amphipathic helix in association with dodecylphosphorylcholine micelles in a parallel orientation to the lipid–water interface with the homing domain remaining exposed to solvent. The amphipathic helix of the apoptotic domain orients with nonpolar leucine and alanine residues inserting most deeply into the lipid environment.
Keywords: apoptosis, NMR, circular dichroism, peptide
Peptides that induce programmed cell death, apoptosis, have potential as anticancer (Arap et al. 1998; Ellerby et al. 1999), hypertrophy (Arap et al. 2002), arthritis therapeutics (Gerlag et al. 2001), and adipose tissue (Kolonin et al. 2004). Hunter-killer peptides (HKPs) are short 20- to 40-residue peptides containing a cellular homing domain (Arap et al. 1998) tethered to a pro-apoptotic domain (Fig. 1 ▶). These peptides hunt for tissue-specific cells and then kill through a membrane-disrupting interaction with the mitochondria that induces apoptosis. HKP1 contains a cyclic endothelial cell homing domain. Tumor specificity of HKP1 is attributed to its binding to receptor integrins, which are expressed on the surface of endothelial cells in angiogenic vessels but are absent or barely detectable in normal endothelial cells. Upon integrin binding, the receptors mediate internalization of HKPs. Once internalized, their characteristic positively charged amphipathic death sequence directs the peptide toward the mitochondrial membrane (Ellerby et al. 1999). The mitochondria swell, the membrane barrier is disrupted, and apoptosis is induced (Ellerby et al. 1999). Because cell death activity is mediated via membrane disruption, HKP death sequences can be designed with the D stereochemical configuration, which aids in resistance to protease degradation in the cell.
Figure 1.

Amino acid sequence of HKP1 and KP1. Amino acids in the apoptotic motif are designed with D stereochemical configuration to increase bioavailability. The homing sequence directs the peptide to integrin receptors that are expressed on the surface of angiogenic tumor cells.
The apoptotic sequence of HKPs share a motif with amphipathic, positively charged antibiotic peptides that function by disrupting bacterial cytoplasmic membranes (Blondelle and Houghten 1992). Similarities between bacterial cytoplasmic membranes and eukaryotic mitochondrial membranes, which are both characterized by the presence of negatively charged phospholipids and the maintenance of a large transmembrane potential (Hovius et al. 1993), suggest that the mechanism of mitochondrial membrane disruption by HKPs is similar to membrane barrier disruption by antibiotic peptides. Weak activity toward eukaryotic cytoplasmic membranes, which have lower concentration of negatively charged phospholipids, impart a greater specificity of the HKPs toward their target cells.
It has been suggested that the death sequence of HKP1 will adopt an amphipathic left-handed helix in the presence of membrane mimics (Ellerby et al. 1999); however, at the present time there has been little structural characterization of these peptides. Using a combination of circular dichroism and NMR spectroscopy we demonstrate that HKP1, indeed, adopts a left-handed helix in the presence of detergent micelles. Nitroxide spin labeling suggests a peptide orientation that is parallel to the lipid–water surface in DPC micelles. The peptides studied are functionally active, as verified by leakage studies with unilamellar vesicles, and the biophysical characterization reported here is a first step for the design of more potent HKPs.
Results
As a model system of HKP peptides, we examined two peptides (Fig. 1 ▶) that are based on the hunter-killer peptide with known selectivity to angiogenic tumor cells (Ellerby et al. 1999). The HKP1 peptide consists of a homing domain that is cyclized through a disulfide bond between the N-terminal cysteine and Cys5. This embedded NGR tripeptide is a motif with demonstrated affinity for the integrin αvβ3 as well as the fibronectin receptor,α5β1 (Healy et al. 1995). The NGR tripeptide is derived from the fibronectin human sequence (1401–1403; Healy et al. 1995), where it has been suggested to reside in an exposed flexible loop on the intact protein. Previous studies have linked prevention of tumor cell invasion with peptide motifs binding to α5β1 integrin. The glycinyl glycine linker is designed for flexibility to reduce steric interference of the killer sequence during homing sequence association with cell surface integrins (Ellerby et al. 1999). Residues 8 through 20 are D-amino acids that form the apoptotic domain. This peptide sequence was initially a de novo design for antimicrobial properties with low mammalian cell toxicity (Javadpour et al. 1996). Selectivity of these peptides for bacteriocidal activity over mammalian cytotoxicity is attributed to differential properties of the bacterial membranes. Bacterial membranes and mitochondrial membranes, which share a common ancestry, are characterized by the presence of anionic lipids and a large trans-membrane potential (Hovius et al. 1993). Because the apoptotic activity is suspected to be mediated by an achiral membrane interaction, D-amino acids rather than the natural all L-amino acid sequence can be used to increase bioavailability (Ellerby et al. 1999). For comparison to HKP and to assess binding and structural effects mediated by the homing domain, a truncated peptide KP1 lacking the homing domain was studied as well.
Calcein leakage experiments
HKPs kill cells by disrupting mitochondrial membranes. To confirm this activity of the HKP1 peptide, calcein leakage experiments (Matsuzaki et al. 1993; Medina et al. 2002) were performed using large unilamellar vesicles (LUVs) serving as a membrane mimic. The calcein dye-entrapped in these vesicles exhibit a weaker fluorescence intensity because of self-quenching properties of the calcein fluorophore. Upon escape from the vesicles, fluorescence intensity increases. Leakage occurs quickly and is complete within five minutes of addition of HKP1 peptide (Fig. 2 ▶). Leakage of 100% of the contents is defined by addition of Triton X-100 for the disruption of the vesicles. Interestingly, KP1, a killer peptide lacking the homing domain (Fig. 1 ▶), is less efficient at inducing leakage than HKP1. The homing domain, which directs the peptide to cells expressing receptor integrins, was not expected to contribute to the membrane barrier disruption activity of the peptide.
Figure 2.

Calcein leakage studies with HKP1 and KP1, the peptide lacking its homing domain, demonstrate the peptides induce efflux of the fluorophore from large unilamellar vesicles. HKP1 exhibits higher leakage activity than KP1. The vesicles were composed of 70:30 molar ratio PC:PG phospholipids.
Circular dichroism spectroscopy
Circular dichroism (CD) spectroscopy was used to monitor the incorporation of HKP1 into dodecylphosphorylcholine (DPC) micelles (Fig. 3A ▶). In the absence of detergent micelles (Fig. 3A ▶), the CD spectrum of HKP1 is characteristic of a random coil peptide with weak intensity and λmax near 195 nm. Increasing concentrations of DPC led to conformational changes, as indicated by the development of λmin at 190 nm and a double maximum at 208 nm and 220 nm. These observations are characteristic for left-handed helical peptide conformations (Mortishire-Smith et al. 1991), and are in striking contrast to CD spectra of L-amino acids in helical peptides with a maximum at 190 nm and minima at 208 nm and 220 nm (Mathews et al. 2000). These CD data clearly suggest that the death sequence adopts a left-handed helix. The presence of an isosbestic point at 203 nm is consistent with a two-state transition from random coil to helix. The background CD spectra of DPC micelles at concentrations carried out in the titration have weak intensity and have been subtracted from the peptide spectra. A plot of mean residue ellipticity at 190 nm and 220 nm against concentration of DPC (Fig. 3B ▶) shows a cooperative transition for signals with the midpoint at 1.3 mM. The DPC critical micelle concentration (CMC) is 1 mM (http://www.lipidat.chemistry.ohio-state.edu/cmc_4.html). Because the transition midpoint is close to the CMC, an association constant of the peptide could not be calculated. Formation of helix is probably simultaneous with the cooperative formation of the micelles and subsequent binding.
Figure 3.

Circular dichroism titration experiments with HKP1 show transition of the peptide from random coil to a left-handed helical structure. (A) Random coil features of the spectrum of HKP1, containing D-amino acids, include the low intensity signal at 200 nm and a maxima around 195 nm. In the presence of DPC micelles, the peptide conformation transitions to a structure with CD features consistent with a left-handed helix, a spectral minimum at 190 nm, and maxima at approximately 205 nm and 220 nm. The isosbestic point suggests a transition between two states. (B) Single wavelengths, 190 nm and 220 nm, have been plotted as a function of detergent concentration. The cmc of DPC (1 mM) is close to the concentration of the transition, so a binding constant cannot be determined from the plot.
1H NMR 1D titration of HKP1
Titrations of HKP1 with deuterated DPC (d-DPC) were monitored by 1D 1H NMR. Because of the repetitive sequence of HKP1, there is significant overlap in all regions of the proton spectrum. Initial spectra in the absence of detergent micelles are consistent with the CD spectra, suggesting an unstructured peptide. Virtually all signals in the amide and α-proton regions overlap to form one large signal. Upon addition of d-DPC, the signals in the amide region of the peptide broaden and shift significantly (Fig. 4 ▶). Additions of d-DPC were continued until amide signals of HKP1 ceased shifting. The final spectrum containing 100 mM d-DPC was signal averaged over 16 transients (twice the number of previous spectra); thus, the apparent increase in signal to noise. Amide signal intensities in the final spectrum are roughly 50% the intensity of the first spectrum, even though these signals represent fewer amide protons. Loss of conformational averaging in the bound state and protection from proton exchange with water by hydrogen bonding and insertion into the micelle may contribute to the enhanced signal intensity per amide resonance relative to the free state. Signals in the α-proton region become more disperse, but not to the extent of signals in the amide region. Side-chain proton signals did not appear to shift significantly. This sample was then used for 2D NMR experiments for assignment and characterization of the peptide conformation in the presence of DPC micelles. Side-chain proton signals did not appear to shift significantly. This sample was then used for 2D NMR experiments for assignment and characterization of the peptide conformation in the presence of DPC micelles.
Figure 4.
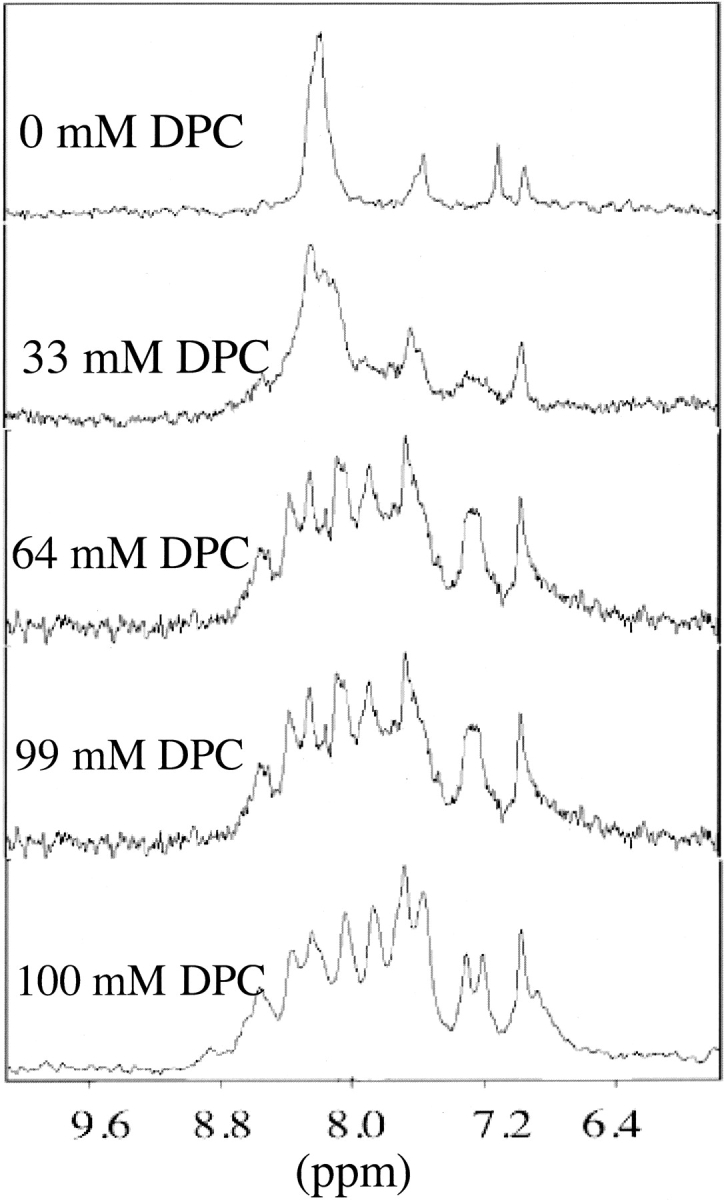
The amide proton region of 1-D NMR experiments of HKP1 in the presence of increasing concentrations of deuterated DPC (dDPC). The initial spectrum in the absence of detergent shows a single amide resonance and side-chain resonances. Minimal chemical dispersion consistent with an unstructured peptide. In the presence of 100 mM dDPC, the region shows increased chemical shift dispersion, consistent with the peptide adopting a defined structure.
1H NMR assignments of HKP1
Homonuclear TOCSY, DQF-COSY, and NOESY NMR experiments were acquired for the purpose of assigning the proton signals of HKP1. Proton resonances of HKP1 were identified by classical methods (Wüthrich 1986). Where possible, spin systems were assigned by combination of DQF-COSY and TOCSY spectra. Spin systems were difficult to identify with the TOCSY spectrum due to the combined effects of broadened signals, poor resolution in the α-proton region, and few cross-peaks beyond three bond coupling (3J). Sequential backbone proton assignments were elucidated via comparison of TOCSY (Fig. 5 ▶) and NOESY spectra in the fingerprint region. The sequential assignments relied heavily upon the presence of amide-to-amide proton (dNN) cross-peaks in the amide region (Fig. 6 ▶) of the NOESY experiment, which span nearly the entire death domain sequence. It was possible to assign the side-chain signals of a few of the alanines, residues in the homing domain, but sequence-specific side-chain signals of the leucines and lysines could not be unambiguously identified. Spectra were acquired at 25°C and 35°C to resolve spectral overlap.
Figure 5.

The fingerprint region of TOCSY experiments of HKP1 in the presence of (A) dDPC, and dDPC with (B) 5-doxyl stearic acid, or (C) 12-doxyl stearic acid. Signals in close proximity to the nitroxide spin labels, which partition into the micelle, will shift or disappear. The remaining signals are distant from the spin labels, and in most cases, in contact with the aqueous environment. To the right of the spectra are idealized helical wheels of the apoptotic sequences. Amino acids with gray circles are not present in the spectrum.
Figure 6.

The amide region of the NOESY spectrum of HKP1 in the presence of dDPC micelles with consecutive amide-amide (dNN) cross-peaks labeled from residues Leu 9 to Lys21. NOEs between adjacent amides are consistent with a helical conformation.
Identification of helical conformation
NOESY spectra provide site-specific information regarding the presence of a left-handed helix suggested by the CD spectrum. A left-handed helix composed of D-amino acids should have the same NOE cross-peak patterns as a right-handed helix composed of L-amino acids (Jourdan et al. 2003). NOE cross-peaks observed between backbone resonances are summarized in Figure 7 ▶. In the amide region of the NOESY spectrum, consecutive dNN cross-peaks were identified spanning the sequence from Leu9 to Lys21 (Fig. 6 ▶). Additionally, seven dNiNi + 2 NOE cross-peaks were identified ranging from Lys11 to Lys21. Crowding in the fingerprint region made identification of long-range NOEs challenging; however, a dαιNi + 2 cross-peak was observed between Leu 9 and Lys11. This cross-peak is the evidence of helix formation closest to the N terminus. Cross-peaks between αι and NHi + 3 were identified at Leu10, Ala13, Ala17, and Lys18. NOEs in the fingerprint and amide regions that could not be observed due to resonance overlap are identified in Figure 7 ▶ with an open box. The identified NOE cross-peaks are consistent with CD spectra prediction of a left-handed α-helix. The chemical shift indexing (CSI) secondary structure prediction algorithm based on α-proton chemical shifts (Wishart et al. 1991, 1992) suggests helix between Ala10 and Leu19. Minimally, these data suggest that HKP1 adopts a left-handed helix between Ala10 and Ala20.
Figure 7.

Summary of NOEs and CSI NMR data of HKP1 in the presence of dDPC. CSI analysis carried out on the α-proton chemical shifts yielded either coil (c) or helix (h) predictions. NOEs are summarized with corresponding bars. Open bars indicate NOEs that cannot be observed due to spectral overlap.
Orientation of the killer sequence
Nitroxide spin-labeled fatty acids were added to NMR samples of HKP1 containing DPC micelles. The nitroxide functional group causes fast relaxation of nearby proton NMR signals. The shifting or disappearance of signals in the fingerprint region are indicative of close proximity to the spin label, and can provide insights to the positioning of the HKP1 backbone in the micelle. 12-Doxyl stearic acid and 5-doxyl stearic acid both partition into the micelle, thus, residues of the peptide that have inserted into the micelle will be most affected by their presence (Brown et al. 1981). The position of the 12-doxyl spin label is the center of DPC micelles, whereas the 5-doxyl stearic acid positions the spin label near the phosphate (Brown et al. 1981) moiety. In the fingerprint region of the TOCSY spectrum of HKP1 in micelles containing 5-doxyl stearic acid (Fig. 5B ▶), the remaining signals included Lys8, Lys11, Ala13, Lys15, Ala17, Lys18, Leu19, and Lys21. The missing cross-peaks are mapped onto a helical wheel with red circles. When 12-doxyl stearic acid was added to the micelles (Fig. 5C ▶), the lysine residues were least affected (Lys8, Lys11, Lys15, Lys18, and Lys21). With the exception of the Leu19, all leucine signals disappeared. The disappearance of signals in response to 12-doxyl stearic acid indicates the portions of the peptide most buried in the micelle. Because the amide proton signals of Ala13 and Ala17 are eliminated by 12-doxyl stearic acid but not 5-doxyl stearic acid, these two amino acids are the most buried in the micelle. The weak signal of the Lys21 in the 5-doxyl stearic acid spectrum and strong signal in the presence of 12-doxyl stearic acid suggests that the backbone of this residue is somewhere near the phosphate group. These studies suggest that Lys21 may be in a position closer to the aqueous environment than illustrated by the helical wheel. However, the spin label experiment results are consistent with the killer sequence adopting an amphipathic helix in association with the micelle surface with the hydrophobic side buried in the lipid and the charged lysines in contact with the aqueous environment.
Homing sequence
There is little information regarding the conformation of the cyclic homing domain from NOESY spectra. The separation of the two α-proton signals of the Gly3 is consistent with the hydrogens being in different environments enforced by the disulfide-linked ring. In the spin label studies, Gly3 and Arg4 α N cross-peaks remained visible, indicating solvent accessibility. Furthermore, a slice through the water resonance in both the TOCSY and NOESY spectra show peaks corresponding to the amide proton chemical shifts for Gly3, Arg4, Cys5, and Gly6 (Fig. 8 ▶). These peaks, which appear at the water resonance in TOCSY and NOESY spectra acquired at different temperatures, indicate rapid chemical exchange between the water protons and these amide protons, consistent with solvent exposure of the homing sequence. Peaks resulting from chemical exchange of amide hydrogens with water for Lys8 and Lys11 of the killer sequence were also observed. Lys11 is in the solvent exposed portion of the helix. Lys8 is likely not part of the helix but is solvent exposed.
Figure 8.

The absence of NOEs between the homing domain and the apoptotic sequence suggest that the homing sequence is either unstructured or in solution. The TOCSY fingerprint region is shown with an overlay of a slice through the water resonance. The slice shows water exchange peaks to residues in the homing domain, suggesting that the homing sequence is exposed to the aqueous environment.
Dynamics at D- and L-amino interface
The first seven amino acids of HKP1, corresponding to the homing and linker sequences are composed of L-amino acids. The apoptotic sequence is composed of D-amino acids. Signals corresponding to the amino acids at the junction between the homing and killer domains of the chimeric peptide (i.e., D- and L-amino acids) were of weak intensity in TOCSY spectra, and there were no NOEs observed to these amino acids. Cys5, Gly6, and Gly7, HNHα resonances were extremely weak. A strong cross-peak in this region was assigned to Ala8 HNHα by the process of elimination, but has no sequential NOE connectivity that confirm this assignment. Ala8 appears to not be part of the helix. An exchange broadened signal in the TOCSY fingerprint region was confirmed to be Gly7 by 1H-15N HSQC (data not shown) of partially labeled peptide. These data suggest that there is conformational dynamics in this region of the peptide, specifically at the junction between D- and L-amino acids, as well as in the homing domain.
Discussion
Hunter-killer peptides have been developed as a potential therapeutic model for treatment of cancers (Arap et al. 1998, 2002) and arthritis (Gerlag et al. 2001). HKP1 was designed to bind specifically to angiogenic tumor cells and induce apoptosis. Studies in vivo with mice models have shown potent activity toward angiogenic tumor cells and relative low toxicity toward healthy tissue (Ellerby et al. 1999) and microscopy experiments have demonstrated that apoptosis is initiated by disruption of the mitochondrial membrane barrier. The positively charged amphipathic sequence of amino acids in the killer sequence was suggestive of a left-handed helix; however, structural characterization of these peptides has been lacking so far. CD studies of the apoptotic sequence made from L-amino acids show a conformational shift toward right-handed helix upon binding DPC micelles (Javadpour et al. 1996). Further structural studies of HKPs in conjunction with biophysical characterization of the binding and membrane interactions will provide insight to the function of these peptides. Design of new potent hunter-killers can be facilitated by insights gained through these experiments.
Although experiments have shown that mitochondrial membrane barrier disruption by HKP1 leads to apoptosis in tumor cells, the mechanism of action at the membrane is unknown. Short amphipathic peptides with membrane disruption activity have been identified that form transiently existing pores (Matsuzaki et al. 1994, 1995b; Hileman 1997; Hara et al. 2001; Medina et al. 2002) or function by detergent disruption of the membrane. Mastoparan, one such peptide from bee venom, forms transiently existing pores and induces apoptosis (Pfieffer et al. 1995; Ellerby et al. 1997; Lin et al. 1997). Magainin, a peptide isolated from frog with antimicrobial activity, also forms transiently existing pores. Both peptides translocate to the interior of vesicles (Matsuzaki et al. 1995a, 1996). Models of pore formation have been proposed for these peptides (Matsuzaki et al. 1995a) as well as for cecropin and dermaseptin (Pouny et al. 1992; Gazit et al. 1995; Shai 1995). Both proposed models are similar, the latter “carpetlike” mechanism proceeds as follows: (1) Peptides associate with the membrane surface in parallel orientation to the interface. (2) Upon reaching a threshold concentration at the bilayer surface the peptides aggregate. These aggregates facilitate formation of channels through the bilayers. In these cases, electrostatic interactions are crucial to peptide association with the membrane (Shai 1995). Common among the peptides that fit this model is the propensity to form helical structures in the presence of bilayers (Faerman and Ripoll 1992; Wakamatsu et al. 1992; Seigneruret and Levy 1995; Gesell et al. 1997; Hori et al. 2001), as do mitochondrial presequences (Roise et al. 1986; Klaus et al. 1996). We have experiments in progress that investigate the appropriateness of the “carpet-like” model for membrane barrier disruption by HKP1, but cannot yet distinguish between this model and a detergent-like disruption of the membrane.
The leakage experiments demonstrate that HKP1 and KP1 can disrupt the membrane barrier to permit leakage of small molecules. Surprisingly, KP1 is less efficient at inducing calcein leakage than HKP1. This result that suggests that the vesicles are not disrupted in a detergentlike mechanism, as the homing domain would not enhance detergent properties of the peptide. It is encouraging for the design of future hunter-killer peptides that the homing domain does not hinder membrane barrier disruption, even though it does not appear to significantly interact with it. This result suggests that a variety of homing domains can be designed to target new tissues without threatening the apoptotic activity of the peptide.
The experiments describe herein demonstrate that HKP1 binds membranes with the killer peptide sequence adopting an amphipathic left-handed helix with the leucine and ala-nine rich sequences inserting into the micelle. There is little structural information regarding the cyclic homing domain; however, it appears to be substantially exposed to the aqueous environment. Coupled with exposure to aqueous environment, broadened and weak signals at the glycine junction (Gly5–Lys8) suggest that there may be conformational dynamics in this region of the peptide. In future experiments, we will examine the role of the homing sequence in determining peptide structure, orientation, and affinity for membranes. Such information will be crucial to design of HKPs that target different tissues.
Materials and methods
Materials
C-terminally amidated HKP1 (H3N+-Cys-Asn-Gly-Arg-Cys-Gly-Gly–(Lys-Leu-Ala-Lys-Leu-Ala-Lys-Lys-Leu-Ala-Lys-Leu-Ala-Lys-CONH2)D) was purchased from Coast Scientific. KP1 was purchased from SynPep. The HKP1 peptide used in the spin label experiments was synthesized in our laboratory. This synthesis was a trial run for incorporation of 15N-D alanine into HKP1 for future experiments. 9-Fluorenylmethyl chloroformate (FMOC-CL) was purchased from Aldrich Chemicals. Fluorenyl-methoxycarbonyl (FMOC) L-amino acids and [O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] (HATU) were purchased from PE Biosystems. 15N-FMOC-glycine and 15N-D-ala-nine were purchased from Cambridge Isotope Labs. All other synthesis reagents were purchased from Fisher Scientific or VWR Scientific, and were used as received. Egg phosphatidylglycerol (PG), egg phosphatidylcholine (PC), and dodecylphosphocholine (DPC) were purchased from Avanti Polar Lipids. Deuterium oxide (D2O), 5-Doxylstearic acid, and 12-doxylstearic acid were purchased from Sigma-Aldrich.
Peptide synthesis
FMOC D-alanine was prepared from FMOC-Cl and D-alanine as described previously (Carpino and Han 1972). HKP1 and its isotopically labeled counterpart was synthesized using FMOC and HATU chemistry on a MilliGen 9050 solid-state peptide synthesizer following a procedure described elsewhere (Meininger et al. 1995).
Vesicle leakage experiments
The ability of HKP1 to induce leakage from LUVs was measured with calceinfilled vesicles composed of phosphatidylcholine (PC) and phosphatidylglycerol (PG) in a 70:30 molar ratio. Vesicles were prepared as described elsewhere (Hope et al. 1985; Medina et al. 2002). Phospholipid concentrations were determined by a phosphate assay that has been modified to use sulfuric acid in place of perchloric acid (Chen 1956). Calcein efflux from vesicles leads to an increase in fluorescence intensity. Fluorescence intensity at 510 nm with an excitation wavelength of 490 nm was monitored on a Jasco FP6600 spectrofluorometer. Leakage was initiated by the addition of peptide. In most cases, calcein efflux was complete in 2 min. Baseline fluorescence (Io) was compared to the increase in fluorescence intensity (Ipeptide) as peptide was added. Complete leakage is the fluorescence intensity upon disruption of the vesicles (Itriton) with addition of 5 μL of 5% Triton X-100. Baselines and 100% leakage was measured for each sample. Leakage (L) is calculated as the percentage change in intensity:
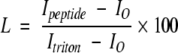 |
CD titration
Circular dichroism spectra were obtained using an Aviv CD Spectrometer Model 202 (Aviv Instruments) purged with nitrogen using a 1-mm rectangular quartz cuvette. Spectra were taken of the HKP1 in the absence and presence of DPC micelles to monitor conformational changes of peptide in the presence of lipid micelles (Medina et al. 2002). Spectra were recorded from 185 nm to 260 nm and appropriate baseline spectra of micelles lacking peptide were collected and subtracted. DPC titrations were performed on 14 μM HKP1 with microliter additions of 68.3 mM DPC. Final concentrations of HKP1-N and DPC were 14 μM and 3.87 mM, respectively. Data points were converted from millidegrees to units of molar ellipticity (deg*cm2/dmole).
1D1H-NMR titration
NMR titrations of HKP1 with dodecylphosphorylcholine (DPC) were performed on a Unity 300 MHz NMR at 30°C to monitor the changes in 1H chemical shift in the amide region upon the addition detergent micelles. A stock solution of 1.18 M DPC was added in 5- to 10-μL increments to a final concentration of 107 mM, yielding a molar ratio of 1:40 (HKP1:DPC). Titrations were carried out on HKP1 with C-terminal amidation and with free C terminus, yielding similar results. 1H 1D NMR experiments were collected with spectral width of 4000 Hz and eight transients with the exception of the spectra data point at 100 mM concentration DPC, which was signal averaged with 16 transients. Spectra were referenced to the water signal at 4.70 ppm (Cavanagh et al. 1996).
2D1H-NMR data collection and processing
The sample described above from the titration experiment was then used for 2D NMR experiments for further structural characterization. For the purpose of initial assignments and identification of NOE constraints, TOCSY (Bax and Davies 1985), DQF-COSY (Rance et al. 1983), and NOESY (Jeener et al. 1979) spectra were acquired on a Bruker DMX 500 MHz NMR. Watergate solvent suppression (Piotto et al. 1992) with gradients was utilized in each experiment. In each experiment States-TPPI phase cycling was used in data collection. Spectral widths were 5482 Hz. NOESY mixing times were 200 msec. TOCSY experiments used a 65-msec mlev17 mixing sequence for Hartman-Hahn transfer (Bax and Davies 1985).
For spin label studies, TOCSY spectra of samples containing 3.5 mM HKP1 and 75 mM DPC were collected on a Bruker Aspect 600 MHz NMR spectrometer at 30°C. For each spin label, the sample was removed and 12-doxylstearic acid or 5-doxylstearic acid was added to a concentration of 1.2 mM, and experiments were repeated with identical data collection parameters. Data were referenced in both dimensions relative to the water signal at 30°C (Hartel et al. 1982; Orbons et al. 1987). Spectral width and reference shift were set to 6127 Hz and 4.70 ppm, respectively. NMR signals that originate from the residues of the HKP1 inserted into the micelle will be diminished in the presence of spin-labeled fatty acids, while the signals in contact with the aqueous environment persist. Peak volumes and peak heights of the HKP1-N sample in DPC micelles were measured in the absence and presence of 5-doxyl and 12-doxyl stearic acid to determine the location of HKP1 residues with respect to the lipid surface.
All data were processed and analyzed using Felix95.0 processing software (Accelrys). Water suppression was enhanced with zero frequency subtraction (Marion et al. 1989). The time domain data were treated with a gaussian multipler function in the direct dimension and a skewed squared sine bell function in the indirect dimension. The first data point in each slice was multiplied one half to reduce T1 noise. For all data, the final processed matrices were 2048 by 2048 points. NOE cross-peaks were identified using a combination of NOESY and TOCSY spectra.
Chemical shift indexing
The position of Hα signals can be indicative of secondary structure (Wishart et al. 1991). The assigned values for Hα were input to the CSI (chemical shift index) program with a smoothing algorithm (Wishart et al. 1992). The output was given in binary code, zero or −1, corresponding to random coil or helical structure, respectively.
Acknowledgments
This work was supported by grants from the UCSD Cancer Center (CCTPJEN-33645G), the NIH (R15 GM068431-01), and a Cotrell College Science Award from Research Corporation (CC5657).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
CSI, chemical shift indexing
dNN, NOE cross-peak between adjacent amide protons
DPC, dodecylphosphocholine
dDPC, dodecylphosphocholine-d38
DQF-COSY, double quantum filtered correlated spectroscopy
HKP, hunter-killer peptide
HKP1, hunter-killer peptide with sequence CNGRCGG(KLAKLAK-KLAKLAK)d
LUV, large unilamellar vesicles
NOESY, nuclear Over-hauser effect spectroscopy
PC, phosphatidylcholine
PG, phosphatidyl-glycerol
SDS, sodium dodecyl sulfate
SFM, serum-free medium
TOCSY, total correlation spectroscopy
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04853204.
References
- Arap, W., Pasqualini, R., and Ruoslahti, E. 1998. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279 377–380. [DOI] [PubMed] [Google Scholar]
- Arap, W., Haedicke, W., Bernasconi, M., Kain, R., Rajotte, D., Krajewski, S., Ellerby, H.M., Bredesen, D., Pasqualini, R., and Ruoslahti, E. 2002. Targeting the prostate for destruction through a vascular address. Proc. Natl. Acad. Sci. 99 1527–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax, A. and Davies, D.G. 1985. Mlev-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65 355–360. [Google Scholar]
- Blondelle, S.E. and Houghten, R.A. 1992. Design of model amphipathic peptides having potent antimicrobial activities. Biochemistry 31 12688–12694. [DOI] [PubMed] [Google Scholar]
- Brown, L., Bosch, C., and Wüthrich, K. 1981. Location and orientation relative to the micelle surface for glucagon in mixed micelles with dodecylphosphocholine: EPR and NMR studies. Biochim. Biophys. Acta 642 296–312. [DOI] [PubMed] [Google Scholar]
- Carpino, L.A. and Han, G.Y. 1972. The 9-fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 37 3404–3409. [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer, A.G., and Skelton, N.J. 1996. Protein NMR spectroscopy: Principles and practice. Academic Press, Inc., San Diego, CA.
- Chen, P.S., Toribara, T.Y., and Warner, H. 1956. Micro determination of phosphorus. Anal. Chem. 28 1756–1758. [Google Scholar]
- Ellerby, H.M., Martin, S.J., Ellerby, L.M., Naiem, S.S., Rabizadeh, S., Salvesen, G.S., Casiano, C.A., Cashman, N.R., Green, D.R., and Bredesen, D.E. 1997. Establishment of a cell-free system of neuronal apoptosis: Comparison of pre-mitochondrial, mitochondrial, and post-mitochondrial phases. J. Neurosci. 17 6165–6178. [PMC free article] [PubMed] [Google Scholar]
- Ellerby, H.M., Arap, W., Ellerby, L.M., Kain, R., Andrusiak, R., Rio, G.D., Krajewski, S., Lombardo, C.R., Rao, R., Ruoslahti, E., et al. 1999. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 5 1032–1038. [DOI] [PubMed] [Google Scholar]
- Faerman, C.H. and Ripoll, D.R. 1992. Conformation analysis of a 12-residue analogue of mastoparan and of mastoparan-X. Proteins 112 111–116. [DOI] [PubMed] [Google Scholar]
- Gazit, E., Boman, A., Boman, H.G., and Shai, Y. 1995. Interaction of the mammalian antibacterial peptide cecropin P1 with phospholipid vesicles. Biochemistry 34 11479–11488. [DOI] [PubMed] [Google Scholar]
- Gerlag, D.M., Borges, E., Tak, P.P., Ellerby, H.M., Bredesen, D.E., Pasqualini, R., Ruoslahti, E., and Firestein, G.S. 2001. Suppression of murine collagen-induced arthritis by targeted apoptosis of synovial neovasculature. Arthritis Res. 3 357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesell, J., Zasloff, M., and Opella, S.J. 1997. Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an α-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. J. Biomol. NMR 9 127–135. [DOI] [PubMed] [Google Scholar]
- Hara, T., Kodama, H., Kondo, M., Wakamatsu, K., Takeda, A., Tachi, T., and Matsuzaki, K. 2001. Effects of peptide dimerization on pore formation: Antiparallel disulfide-dimerized magainin 2 analogue. Biopolymers 58 437–446. [DOI] [PubMed] [Google Scholar]
- Hartel, A.J., Lankhorst, P.P., and Altona, C. 1982. Thermodynamics of stacking and of self-association of dinucleoside monphosphate m2(6)A-U from proton NMR chemical shifts: Differential concentration temperature profile method. Eur. J. Biochem. 129 343–357. [DOI] [PubMed] [Google Scholar]
- Healy, J.M., Murayama, O., Maeda, T., Yoshino, K., Sekiguchi, K., and Kikuchi, M. 1995. Peptide ligands for integrin α v β 3 selected from random phage display libraries. Biochemistry 34 3948–3955. [DOI] [PubMed] [Google Scholar]
- Hileman, M.R., Chapman, B.S., Rabizadeh, S., Krishnan, V.V., Bredesen, D., Assa-Munt, N. and Plesniak, L.A. 1997. A cytoplasmic peptide of the neurotrophin receptor p75NTR: Induction of apoptosis and NMR determined helical conformation. FEBS Lett. 415 145–154. [DOI] [PubMed] [Google Scholar]
- Hope, M.J., Bally, M.B., Webb, G., and Cullis, P.R. 1985. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta 812 55–65. [DOI] [PubMed] [Google Scholar]
- Hori, Y., Demura, M., Iwadate, M., Nidome, T., Aoyagi, H., and Asakur, T. 2001. Interaction of mastoparan with membranes studied by 1H-NMR spectroscopy in detergent micelles and by solid-state 2H-NMR and 15N-NMR spectroscopy in oriented lipid bilayers. Eur. J. Biochem. 268 302–309. [DOI] [PubMed] [Google Scholar]
- Hovius, R., Thijssen, J., van der Linden, P., Nicolay, K., and de Kruijff, B. 1993. Phospholipid asymmetry of the outer membrane of rat liver mitochondria. Evidence for the presence of cardiolipin on the outside of the outer membrane. FEBS Lett. 330 71–76. [DOI] [PubMed] [Google Scholar]
- Javadpour, M.M., Juban, M.M., Lo, W.C., Bishop, S.M., Alberty, J.B., Cowell, S.M., Becker, C.L., and McLaughlin, M.L. 1996. De novo antimicrobial peptides with low mammalian cell toxicity. J. Med. Chem. 39 3107–3113. [DOI] [PubMed] [Google Scholar]
- Jeener, J., Meier, B.H., Bachman, P., and Ernst, R.R. 1979. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 71 4546–4553. [Google Scholar]
- Jourdan, F., Lazzaroni, S., Mendez, B.L., Lo Cantore, P., de Julio, M., Amodeo, P., Iacobellis, N.S., Evidente, A., and Motta, A. 2003. A left-handed α-helix containing both L- and D-amino acids: The solution structure of the antimicrobial lipodepsipeptide tolaasin. Proteins 52 534–543. [DOI] [PubMed] [Google Scholar]
- Klaus, C., Guiard, B., Neupert, W., and Brunner, M. 1996. Determinants in the presequence of cytochrome B2 for import into mitochondria and for proteolytic processing. Eur. J. Biochem. 236 856–861. [DOI] [PubMed] [Google Scholar]
- Kolonin, M., Saha, P.K., Chan, L., Pasqualini, R., and Arap, W. 2004. Reversal of obesity by targeted ablation of adipose tissue. Nat. Med. Epub May 9, 1–8. [DOI] [PubMed]
- Lin, S.Z., Yan, G.M., Koch, K.E., Paul, S.M., and Irwin, R.P. 1997. Mastoparan-induced apoptosis of cultured cerebellar granule neurons is initiated by calcium release from intracellular stores. Brain Res. 771 184–195. [DOI] [PubMed] [Google Scholar]
- Marion, D., Ikura, M., and Bax, A. 1989. Improved solvent suppression in 1-dimensional and 2-dimensional NMR spectra by convolution of time-domain data. J. Magn. Reson 84 425–430. [Google Scholar]
- Mathews, C.K., van Holde, K.E., and Ahern, K.G. 2000. Biochemistry, 3rd ed, p. 205. Benjamin/Cummings, New York.
- Matsuzaki, K., Fujii, N., Fujii, N., and Miyajima, K. 1993. Permeabilization and morphological changes in phosphatidylglycerol bilayers induced by an antimicrobial peptide, tachyplesin I. Colloid Polym. Sci. 271 901–908. [Google Scholar]
- Matsuzaki, K., Murase, O., Tokuda, H., Funakoshi, S., Fujii, N., and Miyahima, K. 1994. Orientational and aggregational states of magainin 2 in phospholipid bilayers. Biochemistry 33 3342–3349. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., Murase, O., Fujii, N., and Miyajima, K. 1995a. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 34 6521–6526. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., Murase, O., and Miyahima, K. 1995b. Kinetics of pore formation by an antimicrobial peptide, magainin 2, in phospholipid bilayers. Biochemistry 34 12553–12559. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., Yoneyama, S., Murase, O., and Miyajima, K. 1996. Transbilayer transport of ions and lipids coupled with mastoparan X translocation. Biochemistry 35 8450–8456. [DOI] [PubMed] [Google Scholar]
- Medina, M.L., Chapman, B.S., Bolender, J.P., and Plesniak, L.A. 2002. Transient vesicle leakage initiated by a synthetic apoptotic peptide derived from the death domain of neurotrophin receptor, p75NTR. J. Peptide Res. 59 149–158. [DOI] [PubMed] [Google Scholar]
- Meininger, D.P., Hunter, M.J., and Komives, E.A. 1995. Synthesis, activity, and preliminary structure of the fourth EGF-like domain of thrombomodulin. Protein Sci. 4 1683–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortishire-Smith, R.J., Drake, A.F., Nutkins, J.C., and Williams, D.H. 1991. Left handed α-helix formation by a bacterial peptide. FEBS Lett. 278 244–246. [DOI] [PubMed] [Google Scholar]
- Orbons, L.P.M., van der Marel, G.A., van Boom, J.H., and Altona, C. 1987. An NMR study of polymorphous behaviour of the mismatched DNA octamer d(m5C-6-m5C-G-A-G-m5C-6) in solution. The B-duplex and hairpin forms. Eur. J. Biochem. 170 225–239. [DOI] [PubMed] [Google Scholar]
- Pfieffer, D.R., Gudz, T.I., Novgorodov, S.A., and Erdahl, W.L. 1995. The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition. J. Biol. Chem. 270 4923–4932. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2661–665. [DOI] [PubMed] [Google Scholar]
- Pouny, Y., Rapaport, D., Mor, A., Nicolas, P., and Shai, Y. 1992. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 31 12416–12423. [DOI] [PubMed] [Google Scholar]
- Rance, M., Sørensen, O.W., Bodenhausen, G., Wagner, G., Ernst, R.R., and Wüthrich, K. 1983. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 117 479–485. [DOI] [PubMed] [Google Scholar]
- Roise, D., Horvath, S.J., Tomich, J.M., Richards, J.H., and Schatz, G. 1986. A chemically synthesized pre-sequence of an imported mitochondrial protein can form an amphiphilic helix and perturb natural and artificial phospholipid bilayers. EMBO J. 5 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneruret, M. and Levy, D. 1995. A high-resolution 1H NMR approach for structure determination of membrane peptides and proteins in non-deuterated detergent: Application to mastoparan X solubilized in n-ocylglucoside. J. Biomol. NMR 5 345–352. [DOI] [PubMed] [Google Scholar]
- Shai, Y. 1995. Molecular recognition between membrane-spanning polypeptides. Trends Biochem. Sci. 20 460–464. [DOI] [PubMed] [Google Scholar]
- Wakamatsu, K., Okada, A., Miyazawa, T., Ohya, M., and Higashijima, T. 1992. Membrane-bound conformation of mastoparan-X, a G-protein-activating peptide. Biochemistry 31 5654–5660. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222 311–333. [DOI] [PubMed] [Google Scholar]
- ———. 1992. The chemical shift index—A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31 1647–1651. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. John Wiley & Sons, New York.