

Abstract
Engineering disulfide bridges is a common technique to lock a protein movement in a defined conformational state. We have designed two double mutants of Escherichia coli 5′-nucleotidase to trap the enzyme in both an open (S228C, P513C) and a closed (P90C, L424C) conformation by the formation of disulfide bridges. The mutant proteins have been expressed, purified, and crystallized, to structurally characterize the designed variants. The S228C, P513C is a double mutant crystallized in two different crystal forms with three independent conformers, which differ from each other by a rotation of up to 12° of the C-terminal domain with respect to the N-terminal domain. This finding, as well as an analysis of the domain motion in the crystal, indicates that the enzyme still exhibits considerable residual domain flexibility. In the double mutant that was designed to trap the enzyme in the closed conformation, the structure analysis reveals an unexpected intermediate conformation along the 96° rotation trajectory between the open and closed enzyme forms. A comparison of the five independent conformers analyzed in this study shows that the domain movement of the variant enzymes is characterized by a sliding movement of the residues of the domain interface along the interface, which is in contrast to a classical closure motion where the residues of the domain interface move perpendicular to the interface.
Keywords: X-ray crystallography, 5′-nucleotidase, protein engineering, disulfide trapping, disulfide geometry, domain rotation, protein movement, protein flexibility, TLS refinement, protein conformation
Many extracellular proteins contain disulfide bridges that provide the protein with extra rigidity (Thornton 1981). Consequently, the stability of proteins can be greatly enhanced with the help of genetically introduced disulfide bridges, including an improved thermal stability (Velanker et al. 1999; Almog et al. 2002). Engineering disulfide bonds into proteins became an important tool for an analysis of the role of protein motions for enzyme catalysis (Matsumura and Matthews 1989), and in general, for detecting functional protein conformational changes (Lee et al. 1995; Kawate and Gouaux 2003). Trapping of a protein motion does not only demonstrate the importance of the movement but also enables an analysis of individual protein conformers in solution where otherwise different conformational states are likely to be present. The presence of the open and closed forms of the enzyme in equilibrium has been demonstrated for citrate synthase, for which both an open and a closed conformer were crystallographically observed, even in crystals grown in the same crystallization drop (Liao et al. 1991). Locking a protein in a specific conformation may allow for an unambiguous assignment of experimental signals (e.g., from spectroscopic studies) to the respective conformation.
In Escherichia coli 5′-nucleotidase (5′-NT), a versatile enzyme that hydrolyses mono-, di-, and trinucleotides as well as nucleotide sugars (Neu 1967) and bis(5′-nucleosidyl) polyphosphates (Ruiz et al. 1989), a domain rotation has been hypothesized as a crucial step in the catalytic mechanism of the enzyme (Knöfel and Sträter 2001). Previous structure analysis showed that the protein consists of two domains linked by an α-helix. The active site of 5′-NT is located at the interface between the two domains. The N-terminal domain bears the core catalytic residues including the two metal ions and an Asp-His dyad, whereas the C-terminal domain contains the substrate specificity pocket where the nucleosyl group is bound. In the open conformation, ATP binds more than 20 Å away from the catalytic center (Knöfel and Sträter 1999,Knöfel and Sträter 2001b). This enzyme conformer is obviously inactive. After a domain rotation of about 96° to the closed conformation, the substrate analog inhibitor AMPCP is bound to the same residues of the C-terminal domain but positioned within the active site in a catalytically competent position. It has been hypothesized that the domain rotation is a crucial step in the catalytic mechanism of the enzyme, most likely to enable substrate binding and product leaving (Knöfel and Sträter 2001a).
An analysis of nine independent conformers in four crystal forms showed that the protein crystallizes either in an open or in a closed conformation with an interdomain rotation angle (rotational difference of the relative orientation of the two domains) of at least 80° between the two states and a maximum rotation of more than 96° (Knöfel and Sträter 1999,Knöfel and Sträter 2001a,b). The 96° movement can be described as a rotation of the domains around an axis that passes through the domain center and residues of the helix that connects the two domains (the hinge region). A comparison of nine independent conformers of wild-type 5′-NT in four crystal forms showed that all interdomain rotation axes are located approximately in one plane, which includes the domain centers and the hinge region in the interdomain helix. All movements are such that residues of the domain interface move along the interface. This type of movement is in contrast to the common classical closure motion where the rotation axis is perpendicular to the axis connecting the mass centers of both domains and the residues of the domain interface move perpendicular to the interface.
The fact that 5′-NT crystallizes in the closed state both in the presence and in the absence of an inhibitor indicates that the conformers have similar energy, and that there is no large barrier to rotation even in the absence of a bound substrate or inhibitor. It is therefore likely that the protein assumes different conformations in solution. We therefore decided to trap the protein in a closed and in an open conformation to pursue analysis in solution. The trapped proteins can serve as conformational reference states for the interpretation of spectroscopic data of proteins containing EPR spin labels or fluorescent labels. With the help of these reference states the assignment of the conformation(s) and mobility of the wild-type protein in solution will be greatly facilitated. Furthermore, a kinetic analysis of the trapped conformers in comparison to the wild-type enzyme will demonstrate to what extent the domain rotation is necessary for the catalytic function of the enzyme. The present crystallographic study defines the structure and mobility of the trapped variants and turns out to be of crucial importance to the interpretation of the ongoing kinetic and spectroscopic analysis. To assess the success of this approach, especially with respect to the orientation and remaining flexibility of the domains, we determined the structures of the trapped 5′-NT enzymes.
Here, we report on three crystal structures of two trapped variants of E. coli 5′-nucleotidase: one crystal structure of the double mutant P90C, L424C that was designed to lock the enzyme in a closed state (designated 5′NT–C), and two crystal structures, further on abbreviated as 5′NT–O1 and 5′NT–O2, of the double mutant S228C, P513C that was designed to lock the enzyme in an open state. Our study strengthens earlier conclusions that appropriately engineered disulfide bridges do not interfere with the structural integrity of the protein but also demonstrates that the disulfide constrained proteins can retain considerable residual flexibility.
Results and Discussion
Domain cross-link
Two disulfide variants of 5′-NT were produced and purified. In both proteins the disulfide bridge forms spontaneously during expression in E. coli without the need to add oxidizing agents. This was demonstrated by adding the substrate bis-p-nitrophenyl phosphate (BPNP) to the culture medium. Because 5′-NT is a periplasmic enzyme, the enzyme activity can be monitored without lysis of the cells. BPNP is hydrolyzed to p-nitrophenyl phosphate and p-nitrophenol, resulting in a yellow color. Upon addition of DTT, BPNP is hydrolyzed more rapidly (data not shown). Because DTT reduces the disulfide bridge to form free cysteine thiols, this finding indicates a successful formation of the cystine bridges. Moreover, the two purified disulfide proteins were shown, by Ellman’s assay (1959), to have no free thiols and by gel chromatography to elute as monomers (no intermolecular cross-linking). The kinetic and spectroscopic analysis of the variants will be published elsewhere.
The P90C, L424C double mutant (5′NT–C) crystallized with two molecules in the asymmetric unit (5′NT–CA and 5′NT–CB). The S228C, P513C double mutant crystallized in two different crystal forms. The first crystal form has two molecules in the asymmetric unit, which are referred to as 5′NT–O1A and 5′NT–O1B, while the second has only one molecule in the asymmetric unit and is called 5′NT–O2.
To unbiasedly establish the presence of the intended disulfide bonds in the crystallized proteins, electron density maps have been calculated omitting the coordinates of the cysteines and the two adjacent amino acids (Fig. 1 ▶). These omit maps clearly show electron density for interdomain disulfide bridges at the positions of the introduced cysteines. The 5′NT–C crystal, for which data were collected at a synchrotron beamline, shows a break in the density of the disulfide bond and an alternate open conformation for the Cys90 residue (Fig. 1A ▶). The density features early signs of radiation damage at the cystine bridge (Ravelli and McSweeney 2000). Data sets of the two structures of the open form of 5′-NT were taken at a rotating anode home source and the disulfide bridges appear unaffected by radiation damage (Fig. 1B,C ▶).
Figure 1.
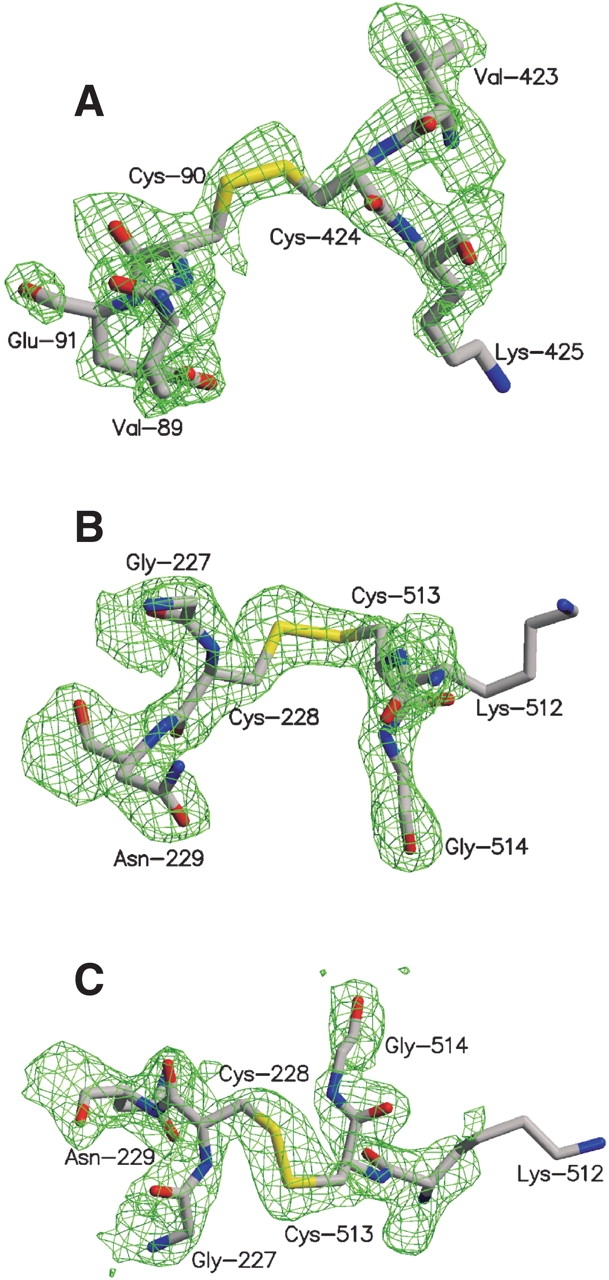
2Fo–Fc map around the disulfide bridges. The residues shown have been omitted from map calculation. (A) Chain A of the 5′NT–C structure. (B) Chain A of the 5′NT–O1 structure. (C) 5′NT–O2 structure.
Geometry of the disulfide bridges
The positions of the engineered disulfide bonds used for trapping the enzyme conformers were predicted with the program SSBOND (Hazes and Dijkstra 1988) using the most open (PDB ID 1hp1) and the most closed (PDB ID 1hpu chain C) 5′-NT structures found so far (Knöfel and Sträter 1999,Knöfel and Sträter 2001a). SSBOND predicts possible cystine bridges to engineer into a protein on the basis of the distance between Cβ positions and the energies of possible disulfide bond conformations.
The disulfide geometry has been analyzed to assess the flexibility of the bonds and to compare the actual disulfide bridges with the predicted ones. The parameters describing the geometry of the engineered disulfide bridges in the refined structures, together with the parameters for the predicted “optimal” disulfide bridges, are summarized in Table 1.
Table 1.
Geometry of engineered disulfide bonds
| χ1 | χ2 | χ3 | χ′2 | χ′1 | Cα-Cα | Cβ-Cβ | Energy (kcal/mole)a | |
| 1hpu Cb1 | −44.3 | −136.5 | 90.9 | 95.8 | 42.7 | 5.83 | 3.95 | 3.10 |
| 2 | −147.2 | 120.4 | −109.6 | 157.3 | 41.9 | 5.83 | 3.95 | 5.49 |
| 3 | −145.0 | 65.9 | 112.3 | −155.7 | 156.7 | 5.83 | 3.95 | 5.55 |
| 5′NT-C chain A | −103.6 | 169.2 | −78.1 | −126.0 | −60.3 | 6.11 | 3.60 | 11.69 |
| 5′NT-C chain B | −91.9 | 177.5 | −94.0 | −120.6 | −62.1 | 6.16 | 3.59 | 8.91 |
| 1hp1c | −73.35 | 66.3 | 127.6 | −83.5 | −146.1 | 5.60 | 4.39 | 5.47 |
| 5′NT-O1 chain A | −67.4 | 178.6 | −96.2 | 74.5 | −158.1 | 5.29 | 4.22 | 9.77 |
| 5′NT-O1 chain B | −70.4 | 174.3 | −94.7 | 78.2 | −166.4 | 5.35 | 4.13 | 4.04 |
| 5′NT-O2 | −65.3 | 65.4 | 108.2 | −80.4 | −149.8 | 5.27 | 4.24 | 4.69 |
a Total energy of the energy bond according to the algorithm of Hazes and Dijkstra (1988).
b 1hpu C: chain C of PDB ID 1hpu used as model for the most closed form of bacterial 5′NT, three possible conformations were predicted.
c 1hp1 PDB ID for the most open form of bacterial 5′NT
For the disulfide bridge between Cys-90 and Cys-424 in the closed enzyme form, three energetically similar disulfide conformations were predicted by SSBOND (Table 1). Interestingly, the disulfide conformers found in both molecules in the asymmetric unit (5′NT–CA and 5′NT–CB) are similar to each other but adopt nonstandard conformations and do not match any of these predicted conformers. The unfavorable bond geometry is also reflected in the higher energy content of the disulfide bridges in the crystal structures compared to the predicted ones (Table 1). The 5′NT–O2 structure, in contrast, matches closely the target model. In the 5′NT–O1 crystal, the disulfide conformers of the two molecules in the asymmetric unit are again similar to each other but do not match the predicted conformers. The difference of the disulfide conformations in the 5′NT–O structures indicates that there might be an equilibrium between at least these two conformational states in solution. A remarkable difference between the two forms of the 5′NT–O structures is the switch from a left-handed disulfide bond in structure 1 to the right-handed disulfide bond in structure 2 (Fig. 2 ▶).
Figure 2.

Geometries of the disulfide bridges. Shown is a superposition based on the cysteine backbone and the Cβ atoms of the N-terminal domain. Green, chain A of 5′NT–C; blue, chain A of 5′NT–O1; red, 5′NT–O2. Carbon atoms are depicted in gray; nitrogen atoms, in blue; and sulfur atoms, in yellow. For 5′NT–O2 the denotations of atoms and dihedral angles are shown (cf. Table 1). The descriptors for the C-terminal cysteine are labeled by a prime.
In total, three different geometries were observed for the disulfide bridges (Fig. 2 ▶). Based on the Cα–Cα and Cβ–Cβ interatomic distances, and the torsion angles χ3, χ1, and χ′1 (see Fig. 2 ▶ for definition of the angles), the 5′NT–C disulfide linkage may be classified as a left-handed spiral, as first identified by Richardson (1981; Srinivasan et al. 1990). The χ2 in the trans position is, however, highly unusual (Petersen et al. 1999). Likewise, the χ2 torsion angle in the left-handed 5′NT–O1 disulfide bond and the χ ′1 torsion angle in the right-handed 5′NT–O2 disulfide bond assume unusual conformations that strain the disulfide bridges (Petersen et al. 1999).
These atypical conformations of the disulfide bonds most likely reflect the fact that the energetic benefits of favorable interdomain contacts and crystal packing outweigh the relatively minor energy penalties inflicted by an atypical disulfide bond geometry (Gorbitz 1994). It has been shown previously that atypical disulfide bridges can be built into a protein and even stabilize it (Katz and Kossiakoff 1986), and indeed, in many proteins with engineered disulfide bridges the disulfide bond geometry is atypical without distorting or destabilizing the structure of the protein (Villafranca et al. 1987; Clarke et al. 1995). Nevertheless, natural disulfide bridges in proteins are usually present in the low energy conformations. Possibly, in these evolutionarily optimized structures, the cystine bridge has optimal stability with respect to its reduction to two free cysteines.
Conformational analysis of the trapped mutants
The computer program DYNDOM (Hayward and Berendsen 1998) was used to compare the targeted structures and the experimental crystal structures. For a given pair of conformers, the program determines the domains that move as rigid bodies, and calculates the screw axis that describes the domain motion.
The most surprising observation in this analysis was the 43.2° deviation of the 5′NT–C double mutant from its targeted model (1hpu chain C; Fig. 3A,B ▶). As the nine previously described conformers of 5′-NT have been found either in an open or in a closed conformation (Knöfel and Sträter 2001a) the 5′NT–C structure represents the first intermediate conformation along the rotation trajectory. The axis corresponding to the large 96° rotation passes through the center of the C-terminal domain and residues of the C-terminal end of the domain-connecting helix, where the hinge region is located. This axis has shifted slightly so that it passes through residues at the N-terminal side of the helix (Fig. 3A,B ▶). There are two pieces of evidence that support the idea that this structure is a true intermediate conformation and not an artifact due to the incorporation of the cysteines. First, the rotation axis relating the 5′NT–C structure (Fig. 3B ▶) and the most closed wild-type structure (1hpu chain C; Fig. 3A ▶) coincides with the overall axis of rotation calculated from the most closed and most open (1hp1, Fig. 3F ▶) structures. Second, in comparison with the closed wild-type structure three new interdomain contacts have been formed, which are likely to stabilize the conformation: two polar contacts between Ile-178 and Gly-179 on the N-terminal domain and Asn-497 and Asn-517, respectively, on the C-terminal domain, and one hydrophobic contact between Asn-180 and Gly-519. On the other hand, only one polar contact, between Gly-88 and Lys-425, has been lost.
Figure 3.
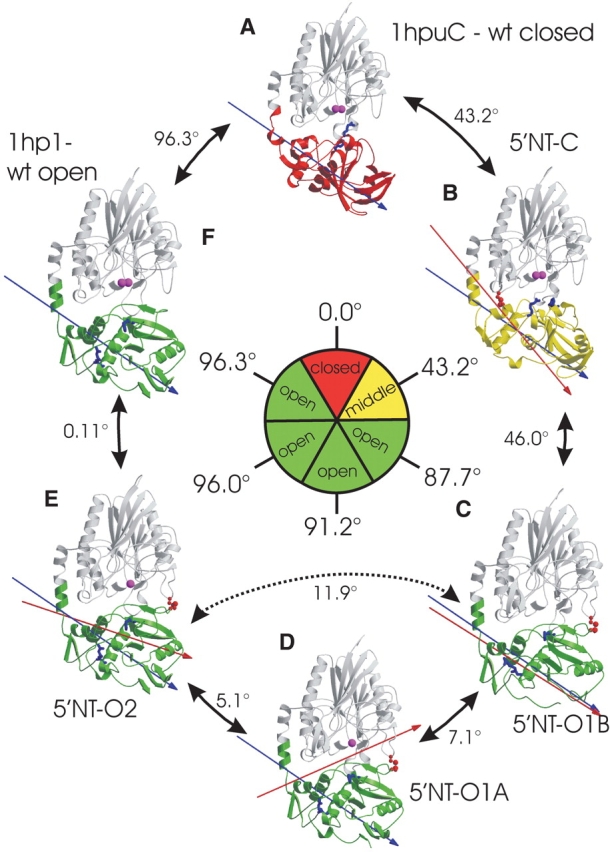
Comparison between wild-type and mutant structures, based on a superposition of the N-terminal domains, shown in gray. The moving C-terminal domains are color-coded according to their position with respect to the N-terminal domain. The closed conformation is depicted in red, and chosen as reference point (0.0°). The intermediate conformation is shown in yellow, and the open conformations, in green. Arrows between two structures indicate the degree of rotation of the C-terminal domain between these structures. The rotation of the C-terminal domain can be followed by the residues Arg 375 and Phe 498 (light-blue stick models) that belong to the binding pocket and point in the closed conformation towards the active site (indicated by the metal ions shown in magenta). The inner circle gives the degree of rotation relative to the reference structure. Because the interdomain rotation axes are not colinear, the numbers in the outer circle do not add up to the overall rotation shown in the inner circle. The engineered disulfide bonds are shown in red. The blue arrow represents the 96° axis of rotation between the most closed and the most open conformation, and the red arrow marks the interdomain rotation axis between a structure and its counterclockwise neighbor.
The large deviation from the predicted conformation is made possible by the location of the interdomain link and the nature of the domain rotation. The residues at the domain interface slide along the interface instead of moving apart from each other. In the case of the 5′-NT–C structure, the introduced disulfide bridge lies parallel to the axis of rotation (Fig. 3B ▶) and is located in close proximity to the axis so that a large rotational movement is possible with little distortion of the disulfide bridge. It is therefore likely that the disulfide bridge allows such flexibility that the protein can occur in a closed conformation (for which the disulfide bond was designed) as well as in the intermediate conformation in solution.
In contrast to the two virtually identical molecules in the 5′NT–C structure, the two proteins in the 5′NT–O1 structure differ by an interdomain rotation angle of 7.1° (Fig. 3C,D ▶). Molecule B (Fig. 3C ▶) differs from the most open conformation (Fig. 3F ▶) by 11.9°, whereas molecule A (Fig. 3D ▶) has a rotational difference of 5.6° to the open conformation. The axis of rotation in Figure 3C ▶, for the transition between 5′NT–O1B and 5′NT–CA, coincides closely with the overall 96° rotation axis between the most closed and most open 5′-NT conformers. The 7.1° domain rotation between 5′NT–O1B and 5′NT–O1A is described by an axis that passes through residues close to the active site at the domain interface and through the hinge region at the C-terminal end of the domain-connecting helix. This movement is in agreement with previous data on the wild-type enzyme showing that all rotation axes derived from a comparison of nine independent conformers lie approximately in one plane, which goes through the centers of the two domains and the interdomain helix (Knöfel and Sträter 2001a). All these rotation axes describe movements, which either maintain the domain interface or in which residues of the domain interface slide along the interface. No movements have been observed so far, which correspond to the classical domain closure motion, in which residues of the domain interface move perpendicular to the interface.
The second structure (5′NT–O2) that was determined for the 5′NT–O enzyme is isomorphous to a crystal form of the wild-type enzyme in the open state (1hp1). This structure matches with an interdomain rotation angle of 0.1° almost exactly the model (1hp1) it was designed to reproduce (Fig. 3E,F ▶), which indicates that both structures are virtually identical.
The maximum interdomain rotation angle between the three conformers of the 5′NT–O enzyme is a rotation of 11.9° between 5′NT–O1B and 5′NT–O2, suggesting that incorporation of the disulfide bond still leaves the protein with a considerable flexibility. The rotation angle of 11.9° is even slightly more than the maximum interdomain rotation angle of 10.4° for a comparison of the three open conformers of the wild-type enzyme (Knöfel and Sträter 2001a).
Analysis of domain motion in the crystal
Due to crystal packing constraints, only a limited mobility of the domains is generally present in crystals. Usually the whole protein as well as the domains is more or less fixed by the intermolecular interactions. Sometimes minor rotational movement is possible and results in a weak and ill-defined electron density for the whole domain and in large crystallographic temperature factors (B-values).
It has been shown that a protein in different packing environments can retain similar thermal factors, suggesting that mobility as estimated by the B-factors is related to mobility in solution (Artymiuk et al. 1979). In the case of the nucleotidase, it could be shown that a limited mobility of the C-terminal domain is present in one crystal form as indicated by high B-values of the whole domain (Knöfel and Sträter 2001a). An analysis of motion in the crystal as inferred from static disorder (all data collections were at 100 K), thus seemed appropriate to investigate the flexibility of the disulfide constrained proteins. We analyzed all three crystal forms by a TLS refinement, which yields translation, libration, and screw tensors that describe the mobility of the domains (residues 26–340 and 360–550) in the crystal.
The eigenvalues of the libration tensor describe the rotational mobility around three perpendicular axes. The libration tensors of the N- and C-terminal domains of the molecules in the asymmetric units of the 5′NT–C as well as 5′NT–O2 crystals are similar with respect to the relative magnitude of the eigenvalues (Fig. 4 ▶). However, only for 5′;NT–CA are also the eigenvectors of the librational tensors of the two domains similarly oriented (with a maximum angle of 23° between the eigenvectors), implying that the tensors describe a rigid body movement of the whole protein with little interdomain movement. For molecules 5′NT–CB and 5′NT–O2, the librational eigenvector directions of the two domains differ by more than 50°, indicating that an individual movement of the domains takes place in these two molecules.
Figure 4.

Eigenvalues of the libration tensors of the individual TLS groups. To visualize the libration magnitude and anisotropy in each TLS group the eigenvalues are shown as a cumulative stack bar. Anisotropic movement is present if the eigenvalues are of significantly different magnitude. The axis with the lowest eigenvalue (black) is at the bottom; the one with the highest (gray), at the top.
Generally, the librational motion is of low magnitude in these TLS groups. This situation is different for the two molecules in the 5′NT–O1 crystal. Here the librational motion is in the C-terminal domain of higher magnitude than in the N-terminal domain, and it is highly anisotropic. This is due to a low number of contacts between the C-terminal domain and the N-terminal domain or other molecules in the crystal. There are 12 polar contacts, of which six are contacts to the C-terminal domain of the second molecule in the asymmetric unit, four are contacts to the equally mobile start sequence of the protein, and only two are contacts to the N-terminal domain, which shows less movement. When the Cα atoms of this molecule are drawn as thermal ellipsoids (Fig. 5 ▶), it can be seen that the C-terminal domain displays a stronger and more anisotropic motion than the N-terminal domain. The B-values of the main chain atoms of the C-terminal domain have a mean value of 63.6 Å2 compared to a mean value in the N-terminal region of 31.2 Å2. Because the usually rigid secondary structural elements show high B-values and anisotropic motion as well, it is reasonable to assume that the thermal motion seen in the crystal reflects a concerted motion of the C-terminal domain. The axis corresponding to the main libration component (5.13 deg2) passes through the interdomain helix, runs along the interface between the two domains, and passes through a point close to the disulfide bond between the two domains. Interestingly, the orientation of this axis is similar to the rotation axis between the conformers 5′NT–O1A and 5′NT–O1B (Figs. 3 ▶,5 ▶). Furthermore, this axis as well as the axis corresponding to the second largest libration component of the tensor lies almost perfectly in the preferred plane for interdomain rotation axes in 5′NT (see introduction).
Figure 5.

Flexibility in the crystal. Shown is molecule B of the 5′NT–O1 structure, color-coded according to the B-values from 30 Å to 100 Å (cf. color-code bar in inset). (A) The Cα atoms are depicted as thermal ellipsoids. The ellipsoids are derived from rigid-body movement of the two domains described by the libration tensors. The domain-bridging disulfide bond is shown in yellow as a stick model. (B) The molecule is shown as a Cα-trace. The rotation axis between chain A and chain B of the 5′NT–O1 structure is shown in black (cf. Fig. 3 ▶). The libration tensor of the C-terminal domain is shown in purple. The engineered disulfide bridge is shown in yellow. (C) Same as in B, but the figure is rotated by 90°.
Of special interest is the question of whether the introduction of disulfide bridge reduces disorder in the crystal relative to the wild-type conformations. The isomorphous crystals of the most open conformation of the wild-type enzyme (1hp1) and the 5′NT–O2 crystal are ideally suited for such a comparison (Fig. 6A ▶). Likewise, the 5′-NT crystal form 1ho5 chain B (closed wild-type conformation) (Knöfel and Sträter 2001a) of which the C-terminal domain has only eight polar crystal contacts with C-terminal domains of neighboring molecules, can be compared to the 5′NT–O1B structure that is also only weakly restrained in the crystal (see above).
Figure 6.

Comparison of B-values. (A) Open form of wt 5′-NT (PDB ID 1hp1) and 5′NT–O2. The interruption of the X-axis is due to missing residues in the structures. The B-value distribution of 5′NT–O2 has been scaled by a factor of 1.9 to that of 1hp1. (B) Closed form of wt 5′-NT (PDB ID 1ho5, chain B) and 5′NT–O2B.
The B-value distribution of these two domains overlay almost exactly (Fig. 6B ▶), which again, corroborates the conclusion that the thermal factors are likely to reflect motions in solution rather than crystal packing artifacts, because both crystals are packed differently. Both comparisons shown in Figure 6 ▶ indicate that the incorporation of the disulfide bridge does not restrict the domain flexibility in the crystal to any extent. This is in agreement with the findings of many other studies (Pjura et al. 1990a,b; Jacobson et al. 1992; van den Akker et al. 1997). However, in contrast to these studies, which have all found a marked increase of the thermal factors at and/or around the sites of the cysteine mutations, the 5′-NT variants show no comparably elevated B-values at the sites of the mutations.
Structural integrity of the mutant proteins
Although disulfide groups can assume a number of different conformations, insertion of a disulfide between two segments of a polypeptide chain always bears the risk of inducing structural distortions both in the flanking sections of the disulfide bond and, through long-range effects, in the protein as a whole.
To determine the extent of the structural distortions of the main chain, the N- and C-terminal domains from the disulfide-linked structures were superimposed on their respective target structures (Fig. 7 ▶). Considering the high overall B-values of the structures the overall displacement is within the limits of variation due to disorder of loop regions, crystallization effects, and data and refinement error limits. This view is supported by the fact that the average atomic displacement for the individual domains of the disulfide-linked proteins is not greater than that for other 5′-NT wild-type structures (data not shown). Furthermore, in all obtained crystal forms a metal ion was found in the high affinity binding site (as defined by McMillen et al. 2003) of the active center. Uptake of this metal ion from the medium or during purification argues for the functional integrity of the proteins. Although small systematic long-range displacements have been detected in subtilisin disulfide variants (Katz and Kossiakoff 1990), our results confirm the general observation from structural studies that an engineered disulfide cross-link does not perturb the protein structure as a whole (Clarke et al. 1995; Ivens et al. 2002).
Figure 7.

Root-mean-square deviations (RMSD) between backbone atoms of the mutant crystal structures and their respective target structures (1hpu chain C for the closed conformation, and 1hp1 for the open conformation). For calculation of the RMSD, the N- and C-terminal domains were superimposed separately. Only the A-chains of the 5′NT–C and 5′NT–O1 crystal structures are shown.
The distortions around the disulfide bridges themselves in the mutant structures are slightly above the noise level (Fig. 7 ▶). These differences are partly due to the fact that the Cα-atoms of the mutated residues are pulled towards each other when the disulfide bridges form (Table 1). Another effect might be that the hydrogen bonding pattern is disturbed as a cause of the mutations. In the wild-type structures there are polar contacts between Ser-228 and Gly-514 in the open conformation and between Gly-88, Gly-91 and Leu-424, Lys-425 in the closed conformation. These contacts are lost in the mutants either through the mutations themselves or through a different orientation of the domains to each other due to the domain rotation. The large RMSDs around residue 180 for 5′NT–O1A seen in Figure 7 ▶ concern a flexible loop that is rearranged in this structure due to crystal contacts of the residues Glu-182 and Tyr-183. These contacts have not been observed in other 5′-NT structures.
Our results are in contrast to most structural studies with engineered disulfide bonds where normally significant deviations around the disulfide linkage are found (Jacobson et al. 1992; Wakarchuk et al. 1994; Almog et al. 1998). The domain rotation of 5′-NT might assist to accommodate the local packing restraints imposed by the inserted cross-link by augmenting the flexibility of the disulfide bond.
Conclusions
Disulfide bridges are often incorporated into proteins to analyze or prove the relationship between protein motion and function. These studies are normally not assisted by structural information. Our investigation of three structures of disulfide-trapped 5′-NT variants implies significant generalizations for the analysis of proteins with engineered disulfide bridges in solution:
The actual structure might differ considerable from the disulfide-linked target structures, both in the conformation of the disulfide linkage but also in the global protein conformation. In the case of the 5′NT–C disulfide-trapped enzyme, the position of the C-terminal domain differs by more than 40° from the predicted position.
The residual flexibility of the disulfide linked polypeptide chains might be larger than expected. A disulfide bridge is highly flexible, and can assume different conformations in the same protein. This, in turn, helps to accommodate different positions of the polypeptide chains to be cross-linked.
The same flexibility of the disulfide bridge allows for cross-linking polypeptide chains without distorting the overall structure of the protein.
Furthermore, our analysis demonstrates that an incorporated disulfide bridge not necessarily restricts thermal motion in the crystal. However, the residual flexibility of the disulfide linked domains is likely to be dependent on the particular location and orientation of the linkage.
The new conformers of 5′-NT characterized in this study corroborate previous results on the nature of the domain rotation. All interdomain rotation axes are located approximately in one plane, including the domain centers and the hinge region. As a result, residues of the domain interface slide along the interface in contrast to a classical closure motion where they move perpendicular to the interface to open the binding cleft. This includes not only motions derived from a comparison of conformers but also the mobility of the C-terminal domain within the crystal lattice of one crystal form. The novel intermediate conformation between the open and closed enzyme forms not only agrees with this description of the domain mobility but is almost perfectly on the linear rotational path between the open and closed conformers described by a rotation axis through the hinge region and the center of the C-terminal domain. These findings indicate that the conformations assumed by the engineered proteins represent plausible approximations to the transitions happening in the wild-type enzyme, although this notion is difficult to prove experimentally. Ongoing kinetic and spectroscopic studies on these variants show that the information on the conformational flexibility of the disulfide-trapped variants obtained from these crystallographic structures is crucial for an understanding and interpretation of the data.
Materials and methods
Cloning and mutagenesis
To avoid contamination of chromosomally expressed wild-type protein, the ushA gene encoding 5′-nucleotidase was subcloned from the pTRC99A plasmid (Knöfel and Sträter 1999) into a pET 28a vector (Novagen) to attach a 6× His tag to the protein. Using the oligonucleotides 5′-CCGGCTCGTATAATGTGTGG-3′and 5′-ACAATAGGCCGCTCGAGCTGCCAGCTCACCTCACC-3′the gene was PCR amplified and digested with NcoI and XhoI (New England Biolabs), and cloned into the new vector according to standard techniques.
The mutations leading to disulfide bridge formation were designed using the computer program SSBOND (Hazes and Dijkstra 1988). The PDB entries 1hp1 and 1hpu chain C previously described to correspond to the most open and most closed conformation of the protein (Knöfel and Sträter 2001a) were used as input coordinates for the program. SSBOND suggested the mutations P90C, L424C and P181C, T501C to trap the enzyme in the closed conformation and N180C, G398C and S228C, P513C for the open conformation. The N180C, G398C double mutant could not be expressed, the P181C, T501C double mutant did not fully close the disulfide bridge, so that work with both these mutants was discontinued. Mutagenesis was performed following the instructions of the Quick-Change-Mutagenesis Kit (Stratagene) with the primers as listed in Table 2.
Table 2.
Oligonucleotides used for mutagenesis
| Mutation | Oligonucleotide 5′−>3′ |
| P90C forward | GCG ACA TTA ACA CTG GCG TGT GCG AGT CTG ACT TAC AGG ATG CC |
| P90C reverse | GGC ATC CTG TAA GTC AGA CTC GCA CAC GCC AGT GTT AAT GTC GC |
| L424C forward | GGC GAT ATC AGC TCT AAA AAC GTG TGT AAA GTG CAG CCA TTC GGC |
| L424C reverse | GCC GAA TGG CTG CAC TTT ACA CAC GTT TTT ATA GCT GAT ATC GCC |
| S228C forward | CGA TAA TGG TGA GCA CGG CTG TAA CGC ACC GGG CGA TGT GG |
| S228C reverse | CCA CAT CGC CCG GTG CGT TAC AGC CGT GCT CAC CAT TAT CG |
| P513C forward | GGA TAT CCG CGC CTT GAT AAC AAA TGT GGC TAT GTG AAT ACC GGC |
| P513C reverse | GCC GGT ATT CACATA GCC ACA TTT GTT ATC AAG GCG CGG ATA TCC |
The constructs were sequenced (GATC Biotech GmbH) to confirm the mutations and to check for second site mutations.
Expression and purification
The proteins were transformed in E. coli ER2566 cells (New England Biolabs) and grown in LB medium. Expression was induced at A600 = 0.8 with 3 μM IPTG. Cells were grown for 5.5 h after induction. Cells were harvested and resuspended in 20 mM K2HPO4 (pH 7.5) and 250 mM NaCl. Cells were lysed with a French Press and centrifuged. The protein in the supernatant was subsequently purified to homogeneity according to the following procedure: The protein solution was loaded onto a metal-chelate column (Poros MC 20, Applied Biosystems), washed with 25 mM imidazole in 20 mM Tris (pH 8.0), and eluted with a gradient to 250 mM imidazole in the same buffer over 10 column volumes. The protein was buffer exchanged in 20 mM Tris-HCl (pH 8.5), 10 mM betaine, 0.5 mM EDTA with a size-exclusion column (Sephadex G25 fine, Amersham Bioscience). The eluted fractions were loaded onto an anion exchange column (PLSAX 1000 Å/10 μm, Polymer Laboratories) and eluted with a gradient from 20 mM Tris-HCl (pH 8.5), 10 mM betaine, 0.5 mM EDTA to 500 mM KCl in the same buffer over 20 column volumes. The final purification step was a size exclusion column (Superdex 75, Amersham Bioscience) equilibrated with 20 mM Tris-HCl (pH 7.5), 50 mM KCl, 0.5 mM EDTA. The protein was concentrated to 15 mg/mL as quantified by a Bradford assay (Bradford 1976).
Crystallization
Crystallization was carried out in hanging drops by vapor diffusion at 18°C. The double mutant S228C, P513C was obtained both in a monoclinic (here referred to as 5′NT–O1) and a tetragonal (referred to as 5′NT–O2) crystal form containing one or two molecules per asymmetric unit, respectively. The former crystals were grown in 26% PEG 400 and 100 mM cacodylate (pH 6.5), whereas 2.1 M malonate (pH 6.5) was used as a buffer for the latter crystals. The double mutant P90C, L424C crystallized in a tetragonal crystal form (referred to as 5′NT–C) with two molecules per asymmetric unit. The crystallization buffer contained 1.8 M Li2SO4, 100 mM Tris, 1 mM MnCl, 10 mM betaine, and 10% 2-methyl-2,4-pentandiole. A droplet of 2 μL protein solution (15 mg/mL) and 2 μL of reservoir solution was mixed and equilibrated against 0.5 mL of reservoir solution. It is noteworthy that a change in the crystallization reagent for the 5′NT–C enzyme variant from (NH4)2SO4 to Li2SO4 was necessary to overcome perfect twinning of the crystals observed with ammonium sulfate.
Data collection
All data were collected at 100 K. Due to rapid radiation damage, it was not possible to collect data sets for the 5′NT–O crystals at the synchrotron beamline ID29 in Grenoble. Both data sets for the 5′NT–O variants were therefore collected at home with an Enraf-Nonius rotating anode generator with copper Kα radiation. The data set for the 5′NT–C variant was collected in Hamburg at the EMBL/DESY beamline BW7B running at a wavelength of 0.8453 Å. The crystal was soaked in 3 M malonate prior to cryocooling. All data sets were recorded with a MAR 345 imaging plate detector system, and were processed using the programs DENZO and SCALEPACK (Otwinowski and Minor 1997). Details of the data collection statistics are summarized in Table 3.
Table 3.
Data collection statistics
| 5′NT-01 | 5′NT-02 | 5′NT-C | |
| Space group | P21 | P41212 | P41212 |
| Cell dimensions (Å) | 80.49, 93.70, 82.90 | 83.13, 83.13, 180.66 | 97.68, 97.68, 312.05 |
| Cell angles (°) | 90, 97.71, 90 | 90, 90, 90 | 90, 90, 90 |
| Mole/asym. unit | 2 | 1 | 2 |
| Resolution range (Å) | 20–2.1 (2.17–2.10)a | 20–2.33 (2.41–2.33) | 30–2.1 (2.18–2.1) |
| Unique reflections | 194,970 | 193,519 | 89,316 |
| Completeness overall (%) | 98.4 (90.8) | 99.7 (100) | 95.1 (95.6) |
| Overall I/(I) | 11.3 (2.1) | 28.4 (11.8) | 17.2 (4.7) |
| Overall Rsym (%) | 5.5 (32.0) | 6.6 (16.5) | 6.9 (26.7) |
| Data redundancy | 2.8 (2.4) | 7.0 (6.8) | 3.2 (3.3) |
a Numbers in parentheses refer to the outer resolution shell.
Structure determination and refinement
All three structures could be determined by molecular replacement. The PDB entry 1hp1 was used as a search model for the 5′NT–O structures, whereas the PDB entry 1hpu chain C (the most closed of the four conformers in this crystal form) was used for the 5′NT–C structure. With the exception of 5′NT–O2 the search needed to be carried out with the N-terminal residues 26–355 only to obtain prominent solutions for the rotation function with the AMoRe program (Navaza 1994). The C-terminal domain (residues 356–550) of the A chain of the 5′NT–O1 structure was subsequently found with the AMoRe program, the second C-terminal domain could then be built by exploiting symmetry to chain A. In the case of the 5′NT–C mutant the first C-terminal domain could only be found with the program BEAST (Read 2001). The second C-terminal domain was again built by symmetry to the first chain. In the case of 5′NT–O2, unambiguous rotational and translational solutions were found with the program MolRep (Vagin and Teplyakov 1997) using the entire search model. After a first complete model has been established, each model was automatically improved with the program ArpWarp (Perrakis et al. 1999).
Mutations were introduced manually with the program O (Jones et al. 1991), which was also used for manual rebuilding against the electron density maps.
The refinement and building of a loop (residues 322–331) missing in the 1hp1 search model was carried out with the CNS package using a simulated annealing protocol (Brunger et al. 1998). The loop could be built in the 5′NT–O1 structure but not completely in the 5′NT–O2 structure where residues 324–329 have been omitted due to weakly defined electron density. For refinement the CNS package was used (Brunger et al. 1998). Water molecules were added automatically in CNS according to the criterion that they had a peak height above 2.8 σ in the Fobs–Fcalc map and at least one hydrogen bond (between 2.0 and 3.5 Å) connecting water and any other atom. All water molecules were checked manually. After convergence of the restrained refinement the coordinates were further refined by 20 cycles of TLS refinement followed by 15 cycles of restrained refinement with REFMAC (Murshudov et al. 1997). In between iterative refinement cycles the following heteroatoms could be defined: two sulfate, two carbonate, and two manganese ions in the 5′NT–C structure, and one in each 5′NT–O structure. For unknown reasons, the Ni2+ ion was only seen in chain A of the 5′NT–O1 structure but not in chain B. The refinement summaries are presented in Table 4. Residues His-252 (metal coordinating residue) and His-289 have in all structures consistently been found to assume disallowed positions in a Ramachandran plot. Thr-87 lies outside the allowed region in 5′NT–C as well as residue Gln-161 in the structures 5′NT–O2 and 5′NT–O1A. All coordinates were deposited in the PDB with the following codes: 1OI8 (5′NT–C), 1OD1 (5′NT–O1), and 1OE1 (5′NT–O2).
Table 4.
Refinement summary
| 5′ NT-01 | 5′ NT-02 | 5′ NT-C | |
| Resolution range (Å) | 20.0–2.1 | 20.0–2.3 | 20.0–2.1 |
| R-factor | 16.6 | 16.8 | 15.6 |
| Rfreea | 21.5 | 22.0 | 20.5 |
| Protein atomsb | 8174 | 4048 | 8194 |
| Water molecules | 746 | 389 | 921 |
| RMSD | |||
| Bond length (Å) | 0.029 | 0.024 | 0.022 |
| Bond angles (°) | 2.17 | 1.74 | 1.74 |
| Average B-factor (Å2) | 42.0 (B = 44.4) (A = 36.8) | 32.0 | 26.1 |
| Main chains | 39.2 | 30.4 | 23.5 |
| Side chains and waters | 44.5 | 33.7 | 28.4 |
| Ramachandran plotc (% residues) | |||
| In most favorable regions | 87.9 (b) 88.4 (a) | 89.6 | 89.6 (b) 90.4 (a) |
| In allowed regions | 10.5 (b) 10.2 (a) | 9.3 | 9.6 (b) 8.4 (a) |
| In regions generously allowed | 1.1 (b) 0.4 (a) | 0.5 | 0.2 (b) |
| In disallowed regions | 0.4 (b) 0.9 (a) | 0.7 | 0.7 (b) 0.7 (a) |
aRfree calculated using 5% of the reflections.
b Nonhydrogen protein atoms inserted in the model/asymmetric unit.
c Calculated with Procheck (Laskowski et al. 1993).
Structure analysis and generation of figures
Omit maps were calculated using the CNS software package (Brunger et al. 1998). For coordinate superpositions the program LSQKAB was used (Kabsch et al. 1976). TLS analysis was performed with the program TLSANL (Howlin 1993). The rigid domains were chosen as indicated in the text. The interdomain screw axes were calculated using the program Dyndom (version 1.5) by a pairwise comparison of the structures (de Groot et al. 1998; Hayward and Berendsen 1998; Hayward and Lee 2002). The CCP4 program suite was also used to calcultate crystal contacts with the program CONTACT, and B-values were extracted from the PDB files with the help of the program BAVERAGE (Bailey 1994).
Figures 1 ▶–3 ▶ were generated using the programs MOLSCRIPT (Kraulis 1991) and Raster3d (Merritt and Murphy 1994) with the help of the graphical user interface MOLDRAW (N. Sträter, unpubl.). Figure 5 ▶ was prepared with the program MOLSCRIPT, POVScript+ (Fenn et al. 2003), and POV-ray. Figures 4 ▶, 6 ▶, and 7 ▶ were plotted with the program Origin (Microcal Software).
Acknowledgments
We thank W. Saenger for generous support and J. Lodge for collecting the data set for the 5′NT-C structure. P. Tucker and M. Weiss are acknowledged for support with the data collection at the EMBL at DESY, Hamburg. We also thank Sascha Marek for assistance in crystallization. This work was supported by the Deutsche Forschungsgemeinschaft.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
5′-NT, 5′-nucleotidase from E. coli
5′NT–C, 5′-nucleo-tidase designed to be trapped in a closed conformation
5′NT–O, designed to be trapped in an open conformation
BPNP, bis-para-nitrophenyl phosphate
DTT, dithiothreitol
EDTA, ethylene-di-amine-tetra-acetic acid
IPTG, isopropylthiogalactosid
LB, Luria-Bertani broth
PEG, polyethylene glycol
PNPP, para-nitrophenyl phosphate
RMSD, root-mean-square deviation
TLS, translation libration screw.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04629604.
References
- Almog, O., Benhar, I., Vasmatzis, G., Tordova, M., Lee, B., Pastan, I., and Gilliland, G.L. 1998. Crystal structure of the disulfide-stabilized Fv fragment of anticancer antibody B1: Conformational influence of an engineered disulfide bond. Proteins 31 128–138. [PubMed] [Google Scholar]
- Almog, O., Gallagher, D.T., Ladner, J.E., Strausberg, S., Alexander, P., Bryan, P., and Gilliland, G.L. 2002. Structural basis of thermostability—Analysis of stabilizing mutations in subtilisin BPN′. J. Biol. Chem. 277 27553–27558. [DOI] [PubMed] [Google Scholar]
- Artymiuk, P.J., Blake, C.C., Grace, D.E., Oatley, S.J., Phillips, D.C., and Sternberg, M.J. 1979. Crystallographic studies of the dynamic properties of lysozyme. Nature 280 563–568. [DOI] [PubMed] [Google Scholar]
- Bailey, S. 1994. The Ccp4 suite—Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macro-molecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Clarke, J., Henrick, K., and Fersht, A.R. 1995. Disulfide mutants of barnase. 1. Changes in stability and structure assessed by biophysical methods and X-ray crystallography. J. Mol. Biol. 253 493–504. [DOI] [PubMed] [Google Scholar]
- de Groot, B.L., Hayward, S., van Aalten, D.M.F., Amadei, A., and Berendsen, H.J.C. 1998. Domain motions in bacteriophage T4 lysozyme: A comparison between molecular dynamics and crystallographic data. Proteins 31 116–127. [DOI] [PubMed] [Google Scholar]
- Ellman, G.L. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82 70–77. [DOI] [PubMed] [Google Scholar]
- Fenn, T.D., Ringe, D., and Petsko, G.A. 2003. POVScript+: A program for model and data visualization using persistence of vision ray-tracing. J. Appl. Cryst. 36 944–947. [Google Scholar]
- Gorbitz, C.H. 1994. Conformational properties of disulfide bridges. 3. An estimation of structural flexibility from a theoretical-study of diethyl disulfide. J. Chem. Soc. [Perkin. 2]: 259–263.
- Hayward, S. and Berendsen, H.J. 1998. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 30 144–154. [PubMed] [Google Scholar]
- Hayward, S. and Lee, R.A. 2002. Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J. Mol. Graph. Model. 21 181–183. [DOI] [PubMed] [Google Scholar]
- Hazes, B. and Dijkstra, B.W. 1988. Model-building of disulfide bonds in proteins with known three-dimensional structure. Protein Eng. 2 119–125. [DOI] [PubMed] [Google Scholar]
- Howlin, B., Butler, S.A., Moss, D.S., Harris, G.W., and Driessen, H.P.C. 1993. Tlsanl—Tls parameter-analysis program for segmented anisotropic refinement of macromolecular structures. J. Appl. Crystallogr. 36 944–947. [Google Scholar]
- Ivens, A., Mayans, O., Szadkowski, H., Jurgens, C., Wilmanns, M., and Kirschner, K. 2002. Stabilization of a (βα)8-barrel protein by an engineered disulfide bridge. Eur. J. Biochem. 269 1145–1153. [DOI] [PubMed] [Google Scholar]
- Jacobson, R.H., Matsumura, M., Faber, H.R., and Matthews, B.W. 1992. Structure of a stabilizing disulfide bridge mutant that closes the active-site cleft of T4-lysozyme. Protein Sci. 1 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W., Kabsch, H., and Eisenberg, D. 1976. Packing in a new crystalline form of glutamine synthetase from Escherichia coli. J. Mol. Biol. 100 283–291. [DOI] [PubMed] [Google Scholar]
- Katz, B.A. and Kossiakoff, A. 1986. The crystallographically determined structures of atypical strained disulfides engineered into subtilisin. J. Biol. Chem. 261 15480–15485. [PubMed] [Google Scholar]
- ———. 1990. Crystal structures of subtilisin BPN′ variants containing disulfide bonds and cavities: Concerted structural rearrangements induced by muta-genesis. Proteins 7 343–357. [DOI] [PubMed] [Google Scholar]
- Kawate, T. and Gouaux, E. 2003. Arresting and releasing staphylococcal α-hemolysin at intermediate stages of pore formation by engineered disulfide bonds. Protein Sci. 12 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöfel, T. and Sträter, N. 1999. X-ray structure of the Escherichia coli periplasmic 5′-nucleotidase containing a dimetal catalytic site. Nat. Struct. Biol. 6 448–453. [DOI] [PubMed] [Google Scholar]
- ———. 2001a. E. coli 5′-nucleotidase undergoes a hinge-bending domain rotation resembling a ball-and-socket motion. J. Mol. Biol. 309 255–266. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Mechanism of hydrolysis of phosphate esters by the dimetal center of 5′-nucleotidase based on crystal structures. J. Mol. Biol. 309 239–254. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. Molscript—A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lee, G.F., Lebert, M.R., Lilly, A.A., and Hazelbauer, G.L. 1995. Transmembrane signaling characterized in bacterial chemoreceptors by using sulfhydryl cross-linking in-vivo. Proc. Natl. Acad. Sci. 92 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, D.I., Karpusas, M., and Remington, S.J. 1991. Crystal-structure of an open conformation of citrate synthase from chicken heart at 2.8-Å resolution. Biochemistry 30 6031–6036. [DOI] [PubMed] [Google Scholar]
- Matsumura, M. and Matthews, B.W. 1989. Control of enzyme-activity by an engineered disulfide bond. Science 243 792–794. [DOI] [PubMed] [Google Scholar]
- McMillen, L., Beacham, I.R., and Burns, D.M. 2003. Cobalt activation of Escherichia coli 5′-nucleotidase is due to zinc ion displacement at only one of two metal-ion-binding sites. Biochem. J. 372 625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster3d version 2.0—A program for photorealistic molecular graphics. Acta Crystallogr. D 50 869–873. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr. D 53 240–255. [DOI] [PubMed] [Google Scholar]
- Neu, H.C. 1967. The 5′-nucleotidase of Escherichia coli. I. Purification and properties. J. Biol. Chem. 242 3896–3904. [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis, A., Morris, R., and Lamzin, V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6 458–463. [DOI] [PubMed] [Google Scholar]
- Petersen, M.T.N., Jonson, P.H., and Petersen, S.B. 1999. Amino acid neighbours and detailed conformational analysis of cysteines in proteins. Protein Eng. 12 535–548. [DOI] [PubMed] [Google Scholar]
- Pjura, P.E., Matsumura, M., Wozniak, J.A., and Matthews, B.W. 1990a. Structure of a thermostable disulfide-bridge mutant of phage T4 lysozyme shows that an engineered cross-link in a flexible region does not increase the rigidity of the folded protein. Biochemistry 29 2592–2598. [DOI] [PubMed] [Google Scholar]
- ———. 1990b. Structure of a thermostable disulfide-bridge mutant of phage-t4 lysozyme shows that an engineered cross-link in a flexible region does not increase the rigidity of the folded protein. Biochemistry 29 2592–2598. [DOI] [PubMed] [Google Scholar]
- Ravelli, R.B. and McSweeney, S.M. 2000. The “fingerprint” that X-rays can leave on structures. Structure 8 315–328. [DOI] [PubMed] [Google Scholar]
- Read, R.J. 2001. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D 57 1373–1382. [DOI] [PubMed] [Google Scholar]
- Richardson, J.S. 1981. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 34 167–339. [DOI] [PubMed] [Google Scholar]
- Ruiz, A., Hurtado, C., Meireles Ribeiro, J., Sillero, A., and Gunther Sillero, M.A. 1989. Hydrolysis of bis(5′-nucleosidyl) polyphosphates by Escherichia coli 5′-nucleotidase. J. Bacteriol. 171 6703–6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, N., Sowdhamini, R., Ramakrishnan, C., and Balaram, P. 1990. Conformations of disulfide bridges in proteins. Int. J. Pept. Protein Res. 36 147–155. [DOI] [PubMed] [Google Scholar]
- Thornton, J.M. 1981. Disulphide bridges in globular proteins. J. Mol. Biol. 151 261–287. [DOI] [PubMed] [Google Scholar]
- Vagin, A. and Teplyakov, A. 1997. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 30 1022–1025. [Google Scholar]
- van den Akker, F., Feil, I.K., Roach, C., Platas, A.A., Merritt, E.A., and Hol, W.G. 1997. Crystal structure of heat-labile enterotoxin from Escherichia coli with increased thermostability introduced by an engineered disulfide bond in the A subunit. Protein Sci. 6 2644–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velanker, S.S., Gokhale, R.S., Ray, S.S., Gopal, B., Parthasarathy, S., Santi, D.V., Balaram, P., and Murthy, M.R. 1999. Disulfide engineering at the dimer interface of Lactobacillus casei thymidylate synthase: Crystal structure of the T155C/E188C/C244T mutant. Protein Sci. 8 930–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villafranca, J.E., Howell, E.E., Oatley, S.J., Xuong, N.H., and Kraut, J. 1987. An engineered disulfide bond in dihydrofolate reductase. Biochemistry 26 2182–2189. [DOI] [PubMed] [Google Scholar]
- Wakarchuk, W.W., Sung, W.L., Campbell, R.L., Cunningham, A., Watson, D.C., and Yaguchi, M. 1994. Thermostabilization of the Bacillus circulans xylanase by the introduction of disulfide bonds. Protein Eng. 7 1379–1386. [DOI] [PubMed] [Google Scholar]