

Abstract
The 45-amino acid polypeptide chain of the homodimeric transcriptional repressor, CopG, was chemically synthesized by stepwise solid phase peptide synthesis (SPPS) using a protocol based on Boc-chemistry. The product obtained from the synthesis was readily purified by reversed-phase HPLC to give a good overall yield (21% by weight). Moreover, the synthetic CopG constructs prepared in this work folded into three-dimensional structures similar to the wild-type protein prepared using conventional recombinant methods as judged by far UV-CD spectroscopy. A fluorescent CopG analog, (Y39W)CopG, was also designed and chemically synthesized to facilitate biophysical studies of CopG’s protein folding and assembly reaction. The guanidinium chloride-induced equilibrium unfolding properties of the wild-type CopG and (Y39W)CopG constructs in this work were characterized and used to develop a model for CopG’s equilibrium unfolding reaction. Our results indicate that CopG’s folding and assembly reaction is well modeled by a two-state process involving folded dimer and unfolded monomer. Using this model, ΔGf and m-values of −13.42 ± 0.04 kcal/mole dimer and 1.92 ± 0.01 kcal/(mole M) were calculated for CopG.
Keywords: protein folding/unfolding, total chemical synthesis, protein design
CopG is a homodimeric transcriptional repressor that is involved in the copy number control of the streptococcal plasmid pMV185 (del Solar and Espinosa 1992). X-ray crystallographic data available on CopG have revealed that it is a member of the ribbon-helix-helix family of DNA binding proteins (Acebo et al. 1998; Gomis-Rüth et al. 1998; Costa et al. 2001). Interestingly, there is very little primary sequence homology between CopG and other ribbon-helix-helix superfamily members. For example, CopG and Arc repressor share less than 25% pairwise identity in their amino acid sequence. In this respect, comparative biophysical studies on CopG and other ribbon-helix-helix superfamily members have the potential to provide important insight about how proteins with different primary amino acid sequences fold into the same three-dimensional structures.
Although there have a been a number of studies of the DNA binding properties of ribbon-helix-helix proteins, there have been relatively few studies of the biophysical properties of the protein folding and assembly reactions of ribbon-helix-helix proteins. Only Arc repressor’s folding properties have been well elucidated (Bowie and Sauer 1989; Milla and Sauer 1994). Currently, there are no such biophysical data on CopG’s folding and assembly reaction. Thus, the work described here includes a thermodynamic analysis of the chemical denaturant-induced equilibrium unfolding/folding properties of wild-type CopG and of a fluorescent analog, (Y39W)CopG.
The protein samples used in this study of CopG’s unfolding/folding reaction were prepared by total chemical synthesis. Total chemical synthesis is an attractive means by which to prepare proteins for biophysical studies. The ability to prepare proteins and protein analogs by total chemical synthesis makes it possible to incorporate unnatural amino acids into a protein’s polypeptide chain and to probe a wide range of structure–function relationships. This ability is especially useful in fundamental studies of protein folding and function. For example, the introduction of α-hydroxy amino acids into proteins by total chemical synthesis can be used to generate amide-to-ester bond mutations in the polypeptide backbone of proteins and to acquire information about the role of the polypeptide backbone in protein folding reactions (Lu et al. 1997; Zhou et al. 1998; Beligere and Dawson 2000; Nakhle et al. 2000; Wales and Fitzgerald 2001). Such information is not easily acquired in traditional site-directed mutagenesis experiments on proteins, as the conventional recombinant DNA techniques employed in such experiments do not permit the incorporation of unnatural amino acids into proteins.
The 45-amino acid polypeptide chain of CopG has been previously prepared by total chemical synthesis using a stepwise solid phase peptide synthesis (SPPS) protocol based on 9-fluorenylmethyloxycarbonyl (Fmoc)-chemistry (del Solar et al. 1994). However, the purity and overall yield (5% overall yield from synthesis and purification) of the final 45-amino acid polypeptide product obtained in this earlier Fmoc-based synthesis was limited. This prompted us to explore the utility of Boc-chemistry for synthesizing the 45-amino acid polypeptide chain of CopG. Our results indicate that the use of a SPPS protocol based on Boc-chemistry provides a more efficient synthesis of CopG than does the previously reported Fmoc-chemistry–based SPPS protocol (del Solar et al. 1994). Our results also demonstrate that the CopG and (Y39W)CopG constructs chemically synthesized in this work both folded into three-dimensional structures similar to the wild-type protein prepared using conventional recombinant methods as judged by far UV-CD spectroscopy.
Results
Chemical synthesis and structural analysis
The 45-amino acid polypeptide chains of wild-type CopG and of (Y39W)CopG were prepared by stepwise-SPPS using a highly optimized protocol for Boc-chemistry that has been previously described (Schnölzer et al. 1992). The full-length polypeptide chains of CopG and (Y39W)CopG were readily purified by RP-HPLC to give good yields of high purity polypeptide material with the expected mass for each construct (see Fig. 1 ▶). The overall yield of the pure wild-type CopG and the pure (Y39W)CopG material obtained in this work was 21% and 4% (respectively) by weight.
Figure 1.

Total chemical synthesis of wild-type CopG and (Y39W)CopG. (A) RP-HPLC chromatogram of the purified synthetic CopG product using an acetonitrile gradient of 20%–80% buffer B over 30 min. The major peak at approximately 18 min corresponds to the full-length 45-amino acid monomer unit of CopG. (B) RP-HPLC chromatogram of the purified (Y39W)CopG using the same gradient. The major peak at approximately 18 min corresponds to the full-length 45-amino acid monomer unit of (Y39W)CopG. ESI-MS insets in both A and B are presented for the material collected from the peaks at 18 min in the chromatogram shown for pure wild-type CopG (A) and purified (Y39W)CopG (B). The multiple charge states arising from the distribution of protonated residues are labeled next to each ion signal. The inset represents the hypermass reconstruction of the raw MS data displayed as the calculated mass.
The synthetic polypeptide chains of wild-type CopG and (Y39W)CopG were readily folded by dissolving the pure, lyophilized polypeptide product from each synthesis in a folding buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA. The folded three-dimensional structure of each synthetic protein was characterized by far UV-CD spectroscopy both when folded in folding buffer and when chemically denatured in 7 M GdmCl (see Fig. 2A ▶). The molar ellipticity values obtained for the folded proteins at 222 nm (θ) indicate that the helical content of our folded wild-type and (Y39W)CopG synthetic constructs were 62% and 64%, respectively. This helical content is consistent with that expected from the X-ray crystallographic data (i.e., 66%; Gomis-Rüth et al. 1998).
Figure 2.

Far UV-CD characterization of folded and unfolded synthetic wild-type CopG as well as (Y39W)CopG. (A) Far UV-CD spectra of wild-type CopG (squares) and (Y39W)CopG (circles) with no denaturant (solid shapes) and with 7 M GdmCl (open shapes) were recorded from 195 to 260 nm at 25°C using a 1-mm path length quartz cuvette with a protein concentration of 10 mM in buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA. The data are shown as mean residue molar ellipticity (Θ, deg•cm2•dmole−1) vs. wavelength in 0.5 nm increments. (B) Fluorescence emission spectra of (Y39W)CopG. Fluorescence emission spectra of (Y39W)CopG with no denaturant (solid squares) and with 7 M GdmCl (solid circles) were recorded from 300 to 500 nm, with excitation at 280 nm. Calculated centers of spectral mass for folded and denatured (YW39)CopG were 366 nm and 369 nm, respectively.
The fluorescence properties of wild-type CopG were such that no measurable differences were observed in similar fluorescence emission scans of CopG when it was either folded in folding buffer or unfolded in 7 M GdmCl. This prompted the design of (Y39W)CopG with a buried Trp residue (see Materials and Methods). The fluorescence properties of this analog are shown in Figure 2B ▶. The data show that there is both a decrease in the fluorescence intensity and a 3-nm shift in the center of spectral mass upon chemical denaturation in GdmCl.
Guanidine-induced equilibrium unfolding
The reversibility of CopG’s GdmCl-induced equilibrium unfolding reaction was determined by examining the unfolding and refolding curves generated by monitoring the far UV-CD signal at 222 nm. The unfolding and refolding curves generated for our synthetic CopG are shown in Figure 3 ▶. The unfolding and refolding curves both have a single, cooperative transition with a midpoint of approximately 3.1 M GdmCl. The nearly identical shape and position of the unfolding and refolding curves suggests that the CopG unfolding reaction is reversible.
Figure 3.

Reversibility of the CopG unfolding reaction. Raw ellipticity data (monitored at 222 nm) are presented for CopG refolding (open circles) and unfolding (solid squares) vs. GdmCl concentration at a protein concentration of 5 μM in monomer equivalents. Error bars are calculated from the standard deviations for measurements made at each concentration of GdmCl.
The conformational stability of the wild-type protein was determined using the CD denaturation curves shown in Figure 4 ▶. For this analysis the CD denaturation curves in Figure 4 ▶ were normalized to take into account the GdmCl dependence of the CD signals in the folded and unfolded baselines (see Materials and Methods). The normalized CD denaturation curves obtained at protein concentrations of 10, 50, and 100 μM are shown in Figure 5A ▶. The concentration dependence of the transition midpoints of the denaturation curves in Figure 5A ▶ suggest that the equilibrium unfolding reaction involves oligomeric species.
Figure 4.

GdmCl-induced equilibrium unfolding raw data for CopG. The raw far UV-CD signals that were recorded at protein concentrations of 10 μM (A), 50 μM (B), and 100 μM (C) are shown with the best-fit lines for the data in the folded and unfolded baselines. Far UV-CD signals were monitored at 222 nm (open circles) and 230 nm (open squares) in buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA at 25°C.
Figure 5.

Global analysis of normalized GdmCl equilibrium unfolding data for CopG. Normalized unfolding data (A) are shown for protein concentrations of 10 μM (solid circles), 50 μM (solid squares), and 100 μM (solid triangles). Global analysis of normalized data within the transition region of all three curves, model 1: D↔2 M (B) and model 2: T↔4 M (C), are shown with 10 μM (open squares), 50 μM (open circles), and 100 μM (open triangles).
The equilibrium unfolding data in Figure 5A ▶ were globally fit to two different two-state models involving oligomeric species. In one model, the only species assumed to be populated in the equilibrium unfolding reaction were a folded dimer and an unfolded monomer. In a second model, the only species assumed to be populated in the equilibrium unfolding reaction were a folded tetramer and an unfolded monomer. To evaluate each model, Fapp values in the transition region of each denaturation curve in Figure 5A ▶ were converted to ΔGapp values, and plots of ΔGapp versus [GdmCl] were used to calculate ΔGH2O and m-values, as described in the Materials and Methods section. The ΔGapp versus [GdmCl] plots that were obtained for the two two-state models evaluated in this work are shown in Figure 5, B and C ▶. A visual inspection of the experimental data reveals that the protein concentration dependence of the CopG unfolding/refolding reaction monitored by CD is best described by the two-state model involving folded dimer and unfolded monomer. Using this model, ΔGH2O and m-values of −13.42 ± 0.04 kcal/mole dimer and 1.92 ± 0.01 kcal/ (mole M) can be calculated from the data.
The conformational stability of (Y39W)CopG was measured using the CD and fluorescence denaturation curves in Figure 6 ▶. The normalized equilibrium unfolding data monitored in Figure 6C ▶ show that the fluorescence and CD denaturation curves generated for the (Y39W)CopG analog are nearly coincident, as would be expected for a protein folding reaction that is well modeled by a two-state (folded and unfolded) process. If the normalized data in Figure 6 ▶ are fit to a two-state model involving only folded dimer and unfolded monomer then m-values of 1.9 ± 0.1 kcal/(mole M) and 1.8 ± 0.1 kcal/(mole M) can be extracted from the CD and the fluorescence data, respectively. Using the folded dimer/unfolded monomer model the normalized CD and fluorescence data both yield a ΔGf values of −12.9 ± 0.1 kcal/mole dimer.
Figure 6.

GdmCl-induced equilibrium unfolding of (Y39W)CopG followed by both far UV-CD and fluorescence emission. (A) Raw far UV-CD signals that were recorded at a protein concentration of 10 μM are shown with the best-fit lines for the data in the folded and unfolded baselines. Far UV-CD signals were monitored at 222 nm in buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA at 25°C. (B) Fluorescence emission data with excitation at 280 nm and using an optical filter with a wavelength cutoff of 305 nm recorded at a protein concentration of 10 μM in the above buffer are shown with the best-fit lines for the data in the folded and unfolded baselines. (C) Normalized fluorescence emission data (solid squares) plotted on the same axis as the normalized far UV-CD data (open squares).
Discussion
Chemical synthesis and structural analysis
The chemical synthesis and RP-HPLC purification protocols employed in this work represent a facile approach for the preparation of wild-type CopG and CopG analogs. Previous attempts to chemically synthesize wild-type CopG using other protocols for stepwise-SPPS reportedly yielded little or no protein (del Solar et al. 1994). The most successful chemical synthesis of CopG reported to date involved an Fmoc-chemistry–based SPPS strategy that required 1–2-h coupling times. The Boc-chemistry–based SPPS strategy described here only involved 10-min coupling times. Moreover, the yield previously reported in the Fmoc-chemistry–based SPPS of CopG described above was 5% overall including synthesis and purification. Our yield for wild-type CopG was significantly better, and it should be noted that the final purified product obtained from our Boc-chemistry–based SPPS of CopG appears to be superior to that obtainable by Fmoc-SPPS, as judged by RP-HPLC analysis (see Fig. 1 ▶ in this work and Fig. 2 ▶ in the work of del Solar et al. 1994).
Our results indicate that the overall yield of (Y39W)CopG prepared by the Boc-chemistry–based SPPS protocol in this work was significantly reduced from that of the wild-type protein. We attribute this to the relatively poor quality of the crude synthetic polypeptide product obtained after 45 cycles of peptide synthesis. Presumably, the unprotected tryptophan residue used in our synthesis participates in undesired side reactions during the acidolytic deprotection step in Boc-chemistry (Bodansky and Martinez 1981). However, we note that the final purified product obtained from our syntheses of (Y39W)CopG appears to be comparable in quality to that obtained from our CopG synthesis.
The far UV-CD spectra recorded for the folded CopG and (Y39W)CopG constructs in this work were essentially identical. This result suggests that both of these synthetic constructs folded into very similar three-dimensional structures. The far UV-CD spectra recorded for each construct is also consistent with that expected from the X-ray crystallographic data. The fluorescent properties of (Y39W)CopG were also found to be useful for the chemical denaturant-induced equilibrium unfolding studies in this work (i.e., there was a marked difference between the fluorescence emission of the folded and unfolded states of this analog).
GdmCl-induced equilibrium unfolding
CopG is a multimeric DNA binding protein reportedly composed of two identical subunits that are held together completely by noncovalent interactions. Therefore, the apparent thermodynamic stability of the protein is expected to be protein concentration dependent. As expected, the transition midpoints of the normalized equilibrium unfolding curves recorded for CopG in Figure 5A ▶ were shifted to higher denaturant concentration with increasing protein concentration. The protein concentration dependence of our GdmCl-induced equilibrium unfolding data on CopG was consistent with that expected for a two-state transition involving folded CopG dimer and two unfolded CopG monomers. Using such a two-state model ΔGf and m-values of −13.42 ± 0.04 kcal/mole dimer and 1.92 ± 0.01 kcal/(mole M), respectively, can be extracted. This calculated m-value is in reasonably good agreement with that expected for the cooperative unfolding reaction of a protein of CopG’s size (i.e., 2 × 45 amino acids; Myers et al. 1995).
Typically, the strongest experimental support for two-state folding models comes from the observed coincidence of unfolding transitions monitored by multiple structural probes (e.g., CD and fluorescence spectroscopy). Unfortunately, the native tyrosine residue in the primary structure of CopG was not useful as a structural probe of CopG’s folding reaction because there was no detectable difference in the fluorescence properties of CopG when it was either folded in folding buffer or chemically denatured in 7 M GdmCl. To help substantiate our two-state equilibrium unfolding model, we sought a Trp substitution to provide a suitable fluorescent probe. Design of the (Y39W)CopG analog (see Materials and Methods) predicted that this mutation should have little effect on the three-dimensional structure and thermodynamic stability of CopG. This was confirmed by our far UV-CD and thermodynamic data on (Y39W)CopG. Furthermore, the synthesis and spectroscopic properties of (Y39W)CopG were satisfactory. Significantly, the CD and fluorescence unfolding transitions for (Y39W)CopG were nearly coincident. These data lend additional support for our two-state model for the unfolding reaction of CopG, especially because far UV-CD should respond mainly to helix formation while fluorescence of Trp 39 (in the interface) should respond mainly to dimer formation.
The thermodynamic data collected in this work on CopG suggest that the folding and assembly reactions of this protein are very closely coupled. These thermodynamic results on CopG are very similar to those obtained on Arc repressor, another homodimeric member of the ribbon-helix-helix family. The equilibrium unfolding/refolding reaction of Arc repressor is also well described by a two-state dimer/unfolded monomer model in which folding and assembly are very closely coupled (Bowie and Sauer 1989). Interestingly, the conformational stability of CopG, appears to be approximately 3 kcal/mole dimer more stable than Arc repressor. Site-directed mutagenesis experiments are in progress to investigate the molecular basis for the increased stability of CopG over Arc repressor.
Conclusions
We have successfully synthesized wild-type CopG using established protocols for Boc-SPPS. The crude synthetic product that we obtained from the synthesis was readily purified by RP-HPLC to give a good overall yield. The purified synthetic material also appears to be superior to that which has been previously prepared using an Fmoc-SPPS strategy. A fluorescent analog, (Y39W)CopG, was also designed, synthesized, and found to fold into a three-dimensional structure similar to that of the wild-type protein. The results of our GdmCl-induced equilibrium unfolding studies on CopG and (Y39W)CopG are consistent with a two-state dimer/unfolded monomer model of CopG’s equilibrium unfolding reaction in which the dimer is stabilized by 13.42 ± 0.04 kcal/mole dimer. The CopG system is now well set up to use both natural and unnatural amino acid replacements to dissect primary amino acid sequence effects on the folding and stability of ribbon-helix-helix transcription factors.
Materials and methods
Materials
The Boc-L-amino acids were purchased from Peptide Institute, Inc. and Novabiochem. The Boc-Lys(2-Cl-Z)-OBzl-4-(carboxy-amidomethyl)-resin was obtained from Applied Biosystems. The (N,N)-diisopropylethylamine (DIEA), the neat trifluoroacetic acid (TFA) (biograde), the 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetra-methyluronium hexafluorophosphate (HBTU), and the dimethylformamide (DMF) (spectroscopic grade) were obtained from Sigma-Aldrich, Halocarbon, Quantum Biotechnologies, and J.T. Baker, respectively. Anhydrous HF (ultrahigh purity) was purchased from Matheson Gas. HPLC grade acetonitrile was purchased from Mallinckrodt. The GdmCl was obtained from OmniPur. All other chemicals were of reagent grade or better.
Instrumentation
Reversed-phase high-performance liquid chromatography (RP-HPLC) analyses were performed using a Rainin instrument consisting of a Dynamax SD-200 solvent delivery system and a Dynamax variable wavelength UV/Visible absorbance detector. An analytical reversed-phase C18 column (0.46 × 15.0 cm, 300 Å) was used with 214-nm UV-detection; and a semipreparative reversed-phase C18 column (10 × 250 mm, 300 Å) was used with 230-nm UV-detection. All columns were obtained from Vydac. Chromatographic separations were achieved using linear gradients of buffer B in A (A = 0.1% TFA in water, B = 90% acetonitrile in water containing 0.09% TFA).
Mass spectra were acquired using a PE Sciex 150EX electro-spray mass spectrometer. Samples for ESI-MS were diluted with 20% Buffer B.
Circular dichroism (CD) spectra were acquired on an Applied Photophysics PiStar 180 CDF spectrometer fitted with a temperature-controlled cell holder. Far UV-CD spectra were recorded from 195 nm to 260 nm at 25°C using a 300-μL quartz cuvette with a 1-mm path length and with protein concentrations between 10 and 30 μM in monomer equivalents. The results are presented as a plot of mean molar ellipticity per residue (Θ, deg•cm2•dmole−1) versus wavelength in 0.5-nm increments. The helical content of CopG’s polypeptide chain was determined from Θ222 using equation 1 as described by Chen et al. (1974)
 |
(1) |
where Θmax was estimated to be −37244.1 deg•cm2•dmole−1 for CopG based on the 45-amino acid length of CopG’s polypeptide chain and the data in Chen et al. (1974).
Fluorescence emission spectra were acquired on a Jobin Yvon-SPEX Fluorolog-3 fitted with a temperature-controlled cell holder. Emission spectra were recorded from 300 nm to 500 nm, with excitation at 280 nm at 25°C using a 500 μL quartz cuvette with a 1-cm path length and with a protein concentration of 10 μM in monomer equivalents. The results are presented as a plot of fluorescence intensity (arbitrary units) versus wavelength in 0.5-nm increments. The center of spectral mass for each spectrum was calculated using the equation
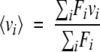 |
(2) |
where Fi is the fluorescence emitted at wave number νi, and the summation is carried out over the full range of the spectrum. Equation 2 yields center-of-mass values in frequency units. Values are reported in wavelengths.
Guanidine-induced equilibrium unfolding data for the wild-type CopG were collected on an Aviv CD spectrometer (model 202) equipped with an automatic titration system and a temperature-controlled cell holder. The solutions used to examine the reversibility of the wild-type CopG unfolding reaction were analyzed on the PiStar 180 CDF Spectrometer from Applied Photophysics. GdmCl-induced equilibrium unfolding data for (Y39W)CopG monitored by CD or total fluorescence were collected on the PiStar 180 CDF Spectrometer from Applied Photophysics equipped with an automatic titrator system and a temperature-controlled cell holder. All UV/Vis absorbance data were collected using a Hewlett Packard 8452A Diode Array UV/vis Spectrometer.
Peptide synthesis, purification, and folding
The 45-amino acid polypeptide chain of CopG (MKKRLTITLSESVLENLEKMAREMGLSKSAMISVALENYKKGQEK) was prepared using manual SPPS methods and optimized protocols for Boc-chemistry that have been described elsewhere (Schnölzer et al. 1992). The synthesis was initiated on Boc-Lys(2-Cl-Z)-4-(oxy-methylphenylacetamidomethyl)-resin where 2-Cl-Z is 2-chloro-benzyloxycarbonyl. Protected Boc-amino acids were preactivated by reaction with HBTU to form hydroxybenzotriazole esters before they were reacted with the TFA salt of the resin bound peptide in the presence of excess DIEA. Side chain protection was as follows: Arg(Tos), Asp(Ochxl), Asn(Xan), Glu(Ochxl), Lys(ClZ), Ser(Bzl), Thr(Bzl), Tyr(BrZ); where Tos is Tosyl, OcHex is cyclohexyl, Xan is xanthyl, ClZ is 2-chlorobenzyloxycarbonyl, Bzl is benzyl, and BrZ is 2-bromobenzyloxycarbonyl. Side-chain deprotection and cleavage of the peptide from the resin was carried out by treatment with anhydrous HF in the presence of 5% v/v p-cresol at 0°C for 1 h. Following HF removal under reduced pressure, the crude peptide product was precipitated and washed with ice-cold anhydrous diethyl ether, dissolved in a minimal amount of 70% acetonitrile containing 0.1% TFA, diluted with water, frozen, and lyophilized. The desired product was purified by semipreparative RP-HPLC using a 35%–55% linear gradient of Buffer B in A. Pure RP-HPLC fractions, as judged by ESI-MS analysis, were pooled, frozen, and lyophilized to a dry, white solid.
The 45-amino acid polypeptide chain of wild-type CopG and (Y39W)CopG was folded by dissolution of 1 mg of the pure, lyophilized polypeptide product in 100-μL folding buffer (50 mM Tris-HCl [pH 7.5], 100 mM KCl, and 0.2 mM EDTA). The resulting protein solution was equilibrated at room temperature for 30 min before any precipitated material was removed by micro-centrifugation. The resulting protein stock concentrations for CopG analogs, in monomer equivalents, were determined using the Waddell (1956) method.
Design of buried Trp substitution
The MAGE/PROBE graphics modeling system (Word et al. 2000), as now enhanced with the penultimate rotamer library (Lovell et al. 2000) built in, was used to provide all-atom contact evaluation of potential side-chain substitutions. All positions in the CopG sequence were examined to see whether a Trp side chain could be accommodated into the existing structure (PDB file 2CPG) with: (1) no backbone motion and no significant shift of neighboring side chains; (2) a near-rotameric Trp side-chain conformation; (3) good all-atom contacts with no bad steric overlaps even for H atoms; and (4) substantial hydrophobic burial of the Trp ring in the folded structure. These are extremely restrictive conditions for the large and rigid Trp side chain, and in general, are impossible to satisfy for most positions that provide enough burial to be useful as fluorescent probes of protein folding. Fortunately, CopG was found to contain one position that satisfies all four criteria: Y39W. As shown in Figure 7 ▶, Trp 39 on the final helix is mostly buried in the dimer interface, and adopts a conformation within 15 degrees of the second most common Trp rotamer; all-atom dot surfaces show excellent contact with six different side chains and with backbone of both monomers. The side chain N atom of the Trp is sufficiently exposed for solvent H-bonding, as was the OH of the original Tyr. The two Trp 39 side chains of a CopG dimer are fairly close together but do not interact directly.
Figure 7.

Stereoview of position and contacts for Trp 39 in the (Y39W)CopG dimer (see Materials and Methods for modeling methods). One chain in the GopG homodimer is in black, and the other chain is in gray, showing the noncompact monomer and intertwined dimer, with Trp 39 at the interface. All-atom contacts (dot patches) and contacting side chains are included for Trp 39 of chain A.
Guanidine-induced equilibrium unfolding of CopG
In experiments to evaluate the reversibility of the CopG unfolding reaction, folded and unfolded stock solutions of CopG were prepared from the same 700-μM solution of the protein dissolved in buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA. In the folded stock solution the final concentration of CopG was 160 μM. The buffer composition was 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA. In the unfolded stock solution the final concentration of CopG was also 160 μM. The buffer composition of this unfolded stock solution was identical to that of the folded stock solution with the exception that it also contained 5.5 M GdmCl. These folded and unfolded stock solutions were each diluted into a series of buffer solutions containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA, and different concentrations of GdmCl. The final concentration of CopG in each series of buffer solutions was 5 μM, and the concentrations of GdmCl ranged from 0.5 M to 5.5 M GdmCl. Ultimately, the far UV-CD signal at 222 nm was measured for each solution; and the data were used to generate the protein’s unfolding and folding curves, respectively.
The conformational stability of the synthetic wild-type CopG was measured using GdmCl to induce unfolding and far UV-CD to monitor the folded state of the protein. In these experiments, a concentrated stock solution of folded CopG was prepared in buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA; it then was diluted into the same buffer to obtain three different folded CopG solutions in which the final protein concentrations were 10, 50, and 100 μM. Similarly, the protein was also diluted in buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA, and 8 M GdmCl to obtain three different unfolded CopG solutions in which the final protein concentrations were also 10, 50, and 100 μM. These folded and unfolded CopG solutions were then used with the automatic titrator system on the Aviv CD spectrometer to record GdmCl-induced equilibrium unfolding curves for CopG at each protein concentration. The CD signal was monitored at 222 nm in the case of the unfolding curve recorded at the 10-μM concentration; and the CD signal was monitored at 230 nm in the case of the unfolding curves at the 50-μM and 100-μM concentrations.
The raw CD signals in the unfolding experiments on the synthetic wild-type CopG were converted to Fapp according to equation 3,
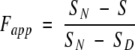 |
(3) |
where S is the observed signal at each GdmCl concentration, and SN and SD are respective signals of the folded and denatured forms of the protein. In our experiments S was linearly dependent on the GdmCl concentration in both the folded and denatured baseline regions. Therefore, linear extrapolations from these baseline were used to estimate SN and SD values in the transition region. The normalized data were then fit to the two-state models (i.e., folded and unfolded) models shown below in equations 4 and 5,
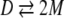 |
(4) |
 |
(5) |
where D represents folded CopG dimer, T represents folded CopG tetramer, and M represents denatured CopG. The equilibrium constants for the unfolding reactions in equations 4 and 5 can be written in the following forms (respectively):
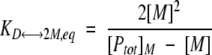 |
(6) |
 |
(7) |
where [Ptot]M is the total protein concentration in terms of monomer and [M] is the concentration of denatured monomer. If the Fapp is defined as [M]/[Ptot]M, the equilibrium constants in equations 6 and 7 can be written in the following forms:
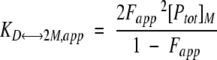 |
(8) |
 |
(9) |
where KD↔2M,app and KT↔4M,app are the apparent equilibrium constants at each [GdmCl] concentration for the two unfolding reactions in equations 4 and 5, respectively. For a two-state unfolding process and at moderate to high denaturant concentrations the apparent free energy of unfolding (ΔGH2O) is linearly dependent on the molar concentration of the denaturant according to equation 10
 |
(10) |
where ΔGapp is the apparent free energy of unfolding at a particular denaturant concentration and at standard state (1M multimer), m is the constant of proportionality (δΔGapp/δ[Denaturant]), and ΔGH2O is the free energy change of unfolding in the absence of denaturant.
In the two-state analyses of the equilibrium unfolding curves for the synthetic wild-type CopG in this work, ΔGH2O and m values were extracted from the data by using equations 8 and 9 and the expression ΔGapp = −RTln(Kapp) to convert Fapp values in the transition region of each denaturation curve (defined as Fapp values from 0.1 to 0.9) to ΔGapp values. These ΔGapp values were then used to generate global ΔGapp versus [GdmCl] plots for each model. Ultimately, a linear least-squares analysis of the data in these global plots was used to determine the model that best described the data and to ultimately determine an m-value and extrapolated ΔGH2O value for CopG.
The conformational stability of (Y39W)CopG was measured using GdmCl to induce unfolding and far UV-CD and total fluorescence to monitor the folded state of (Y39W)CopG. Purified (Y39W)CopG was dissolved in buffer and diluted to a final protein concentration of 10 μM using 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA. Similarly, the protein was also diluted to the above concentration with 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 0.2 mM EDTA containing 8 M GdmCl. The protein was titrated from 0 M to 5.5 M GdmCl, and the CD signal was monitored at 222 nm. For GdmCl-induced equilibrium unfolding experiments monitored by total fluorescence, solutions were prepared as above and titrated from 0.3 M to 5.7 M GdmCl, with excitation at 280 nm and total fluorescence monitored using an optical filter with a wavelength cutoff of 305 nm. The raw signals acquired for both the unfolding curves monitored by CD and fluorescence were normalized as the data for the wild-type protein and then analyzed using a nonlinear least squares fitting routine. Thermodynamic parameters were determined using nonlinear least squares using equation 11
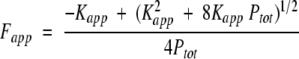 |
(11) |
where Fapp is defined as [M]/[Ptot]M, Kapp is the apparent equilibrium constant at each GdmCl concentration, and
 |
(12) |
Acknowledgments
This work was supported by NIH Grants RO1 GM61680 (to M.C.F.) and RO1 GM15000 (to David C. Richardson in support of J.S.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04671804.
References
- Acebo, P., Garcia de Lacoba, M., Rivas, G., Andreu, J.M., Espinosa, M., and del Solar, G. 1998. Structural features of the plasmid pMV158-encoded transcriptional repressor CopG, a protein sharing similarities with both helix-turn-helix and β-sheet DNA binding proteins. Proteins 32 248–261. [DOI] [PubMed] [Google Scholar]
- Beligere, G.S. and Dawson, P.E. 2000. Design, synthesis, and characterization of 4-ester CI2, a model for backbone hydrogen bonding in protein α-helices. J. Am. Chem. Soc. 122 12079–12082. [Google Scholar]
- Bodansky, M. and Martinez, J. 1981. Side chain reactions in peptide synthesis. Synthesis (Stuttgart) 5 333–356. [Google Scholar]
- Bowie, J.U. and Sauer, R.T. 1989. Equilibrium dissociation and unfolding of the Arc repressor dimer. Biochemistry 28 7139–7143. [DOI] [PubMed] [Google Scholar]
- Chen, Y.H., Yang, J.T., and Chau, K.H. 1974. Determination of the helix and β form of proteins in aqueous solution by circular dichroism. Biochemistry 13 3350–3359. [DOI] [PubMed] [Google Scholar]
- Costa, M., Sola, M., del Solar, G., Eritja, R., Hernandez-Arriaga, A.M., Espinosa, M., Gomis-Rüth, F.X., and Coll, M. 2001. Plasmid transcriptional repressor CopG oligomerises to render helical superstructures unbound and in complexes with oligonucleotides. J. Mol. Biol. 310 403–417. [DOI] [PubMed] [Google Scholar]
- del Solar, G. and Espinosa, M. 1992. The copy number plasmid of pLS1 is regulated by two trans-acting plasmid products: the antisense RNA II and the repressor protein RepA. Mol. Microbiol. 6 83–94. [DOI] [PubMed] [Google Scholar]
- del Solar, G., Albericio, F., Eritja, R., and Espinosa, M. 1994. Chemical synthesis of a fully active transcriptional repressor protein. Proc. Natl. Acad. Sci. 91 5178–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Rüth, F.X., Solà, M., Acebo, P., Párraga, A., Guasch, A., Erijita, R., González, A., Espinosa, M., del Solar, G., and Coll, M. 1998. The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO J. 17 7404–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell, S.C., Word, J.M., Richardson, J.S., and Richardson, D.C. 2000. The penultimate rotamer library. Proteins 40 389–408. [PubMed] [Google Scholar]
- Lu, W.Y., Qasim, M.A., Laskowski Jr., M., and Kent, S.B.H. 1997. Probing intermolecular main chain hydrogen bonding in serine protinase–protein inhibitor complexes: Chemical synthesis of backbone-engineered turkey ovomucoid third domain. Biochemistry 36 673–679. [DOI] [PubMed] [Google Scholar]
- Milla, M.E. and Sauer, R.T. 1994. Arc repressor: Folding kinetics for a single-domain, dimeric protein. Biochemistry 33 1125–1133. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhle, B.M., Silinski, P., and Fitzgerald, M.C. 2000. Identification of an essential backbone amide bond in the folding and stability of a multimeric enzyme. J. Am. Chem. Soc. 122 8105–8111. [Google Scholar]
- Schnölzer, M., Alewood, P., Jones, A., Alewood, D., and Kent, S.B.H. 1992. In situ neutralization in Boc-chemistry solid phase peptide synthesis: Rapid, high yield assembly of difficult sequences. Int. J. Peptide Protein Res. 40 180–193. [DOI] [PubMed] [Google Scholar]
- Waddell, W.J. 1956. A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med. 48 311–314. [PubMed] [Google Scholar]
- Wales, T.E. and Fitzgerald, M.C. 2001. The energetic contribution of backbone–backbone hydrogen bonds to the thermodynamic stability of a hyperstable P22 Arc repressor mutant. J. Am. Chem. Soc. 123 7709–7710. [DOI] [PubMed] [Google Scholar]
- Word, J.M., Bateman Jr., R.C., Presley, B.K., Lovell, S.C., and Richardson, D.C. 2000. Exploring steric constraints on protein mutations using MAGE/ PROBE. Protein Sci. 9 2251–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J., Case, M., Wishart, J.F., and McLendon, G.L. 1998. Thermodynamic and structural effects of a single backbone hydrogen bond deletion in a metal-assembled helical bundle protein. J. Phys. Chem. B 102 9975–9980. [Google Scholar]