

Abstract
Methods that predict the topology of helical membrane proteins are standard tools when analyzing any proteome. Therefore, it is important to improve the performance of such methods. Here we introduce a novel method, PRODIV-TMHMM, which is a profile-based hidden Markov model (HMM) that also incorporates the best features of earlier HMM methods. In our tests, PRODIV-TMHMM outperforms earlier methods both when evaluated on “low-resolution” topology data and on high-resolution 3D structures. The results presented here indicate that the topology could be correctly predicted for approximately two-thirds of all membrane proteins using PRODIV-TMHMM. The importance of evolutionary information for topology prediction is emphasized by the fact that compared with using single sequences, the performance of PRODIV-TMHMM (as well as two other methods) is increased by approximately 10 percentage units by the use of homologous sequences. On a more general level, we also show that HMM-based (or similar) methods perform superiorly to methods that focus mainly on identification of the membrane regions.
Keywords: transmembrane protein, α-helix, topology prediction, hidden Markov model, sequence profile
Integral α-helical membrane proteins constitute an important subset of the proteins encoded by a genome, making up ~20%–25% of the proteome (Krogh et al. 2001). These proteins are crucial for many cellular processes including signaling and transport processes. They are also the target for the majority of all drugs, making them important for the pharmacological industry (Chen et al. 2002). For several experimental reasons, it is more difficult to obtain the structures of TM proteins than of globular proteins. A consequence of this is that <1% of the 3D structures in the Protein Data Bank (Berman et al. 2000) are of TM proteins. However, “low-resolution” information about the structures of TM proteins can be obtained by determining the topology, that is, the location in the sequence of the TM regions and the orientation of the protein relative to the membrane. This can be done protein by protein using experimental methods such as gene fusion, proteolytic digestion in situ, antibody binding, and chemical modification; on a larger scale using automated prediction methods (Krogh et al. 2001); or via a combination of the two (Kim et al. 2003). Today, such experimental low-resolution information is available for ~500 proteins. A correctly determined topology can provide important knowledge in further structural and functional studies, including the detection of homologous membrane proteins (Hedman et al. 2002).
The first predictors for α-helical TM proteins only used the fact that TM helices are on average more hydrophobic than the loop regions of TM proteins and all regions of globular proteins. These methods classified each segment that was sufficiently hydrophobic as a TM helix. Although these simple methods worked surprisingly well, many regions were wrongly classified. A significant improvement was obtained with the observation that positively charged amino acids are more common in the cytoplasmic than in the external loop regions (the positive inside rule; von Heijne 1986 von Heijne 1994). This was included in the Toppred prediction method (von Heijne 1992; Claros and von Heijne 1994). In Toppred, regions that are of intermediate hydrophobicity can be classified either as membrane regions or as loop regions to optimize the number of positive residues in the cytoplasmic loops. Later, the same information was included in statistical optimization methods such as MEMSAT (Jones et al. 1994), which is built on propensity scales and uses a dynamic programming algorithm to find the optimal topology. Information from multiple sequence alignments has been included in machine learning methods (for example, PHD_htm; Rost et al. 1996) and statistical methods (TMAP; Persson and Argos 1994). Recently, HMMs (HMMTOP [Tusnády and Simon 1998 HMMTOP [Tusnády and Simon 2001]; TMHMM [Sonnhammer et al. 1998; Krogh et al. 2001]) have been used to extract the significant features of different regions in TM proteins.
The predictors can roughly be divided into two classes, those that primarily focus on a residue-based evaluation of the propensity of each amino acid to be in a particular region (Toppred, TMAP, THUMBUP [Zhou and Zhou 2003], PHD_htm) and those that focus on aligning the sequence with a membrane protein model (the HMM methods and MEMSAT). As the topology of a membrane protein is determined both by the composition of the membrane regions and the loops, it could be assumed that the best performance should be achieved by methods that fully use this information.
Several groups have published independent measurements of the performance of different predictors. But as of yet, no prediction method has distinguished itself as being indisputably better than all others in all tests. Chen et al. (2002) concluded that “no method(s) performed consistently better than all others by more than one standard error.” Differences between the evaluations are due to what is being measured (per residue accuracy, per protein accuracy, etc.) and perhaps more important, the composition of the data set used in the comparison, which may be more or less similar to the data set for which a particular method has been optimized. However, Ikeda et al. (2002) observed that “model-based methods perform better than non-model based ones,” and Chen et al. concluded that “methods based on alignments were superior to those based on single sequences.” As far as we can tell, neither of these studies tested the multiple sequence version of HMMTOP, nor was this done by Möller et al. (2001) or Jayasinghe et al. (2001).
In this study, we will focus on the evaluation of topology prediction, that is, the ability to correctly identify all membrane regions as well as to correctly predict the orientation of the protein in the membrane. There are two reasons for this: First, we think that this is the type of information that will provide the most useful information about an unknown membrane protein. Second, it seems as if this is the most challenging part of membrane protein predictions and that for this reason, differences in performance might only be seen by studying topology prediction accuracies.
A common property of most TM protein prediction methods is that they depend on statistical information from sequences with known topology for parameter optimization. A well-established method to improve the statistical stability of protein sequence data is to use evolutionary information in the form of multiple sequence alignments instead of single sequences. For instance, this is known to improve prediction performance of secondary structure prediction for globular proteins by several percentage units (Rost and Sander 1993). Of these methods, TMAP and PHD_htm are the only methods that use multiple sequence alignment data. Further, HMMTOP can use the information from a set of homologous sequences when predicting the topology of a particular query sequence. However, these sequences are used as single sequences and not in the format of a profile.
Recently, sequence profiles created from multiple sequence alignments have been used as inputs to HMMs (Martelli et al. 2002; Edgar and Sjölander 2003). However, for α-helical TM proteins, it is not obvious a priori that multiple sequence alignment information should improve the predictions, because it is not certain that membrane proteins from the same family all share the same topology. For instance, homologous sequences of the same protein family that have inverted topologies have been shown to exist (Sääf et al. 1999). It is also well known that homology detection of membrane proteins is more difficult than for globular proteins (Hedman et al. 2002). Finally, the optimal way to use sequence profiles with HMMs is still not well understood. The two implementations by Martelli et al. (2002) and Edgar and Sjölander (2003) are quite different from each other and it is our belief that the optimal way to use sequence profiles with HMMs depends on the properties of the problem to which the HMM is applied.
In this study, we show that a novel HMM-based method, PRODIV-TMHMM, which is an alignment-based HMM using sequence profiles that includes the best features of TMHMM and HMMTOP, achieves ~66% accuracy in topology predictions, whereas methods that do not use both HMMs and evolutionary information achieve at best 52% accuracy.
PRO-TMHMM and PRODIV-TMHMM
An HMM is a first-order Markov chain, where in each state a symbol is emitted. Transition probabilities are the probabilities of moving from a model state s to some other model state s′, and the emission probabilities are the probabilities of emitting an alphabet symbol a when in state s of the model. Given an HMM with model parameters θ, the probability of a sequence x being produced by the HMM using a particular state path p, can be written P(x,p|θ = ∏l−1i=0tpi,pi+1 × ∏li = 1epi (xi) where tj,k is the transition probability from state j to state k, ej(a) is the emission probability for letter a in state j, and l is the sequence length. When using HMMs for topology prediction, the basic idea is to label each state as either “Membrane,” “Inside,” or “Outside.” Then, using sequences with experimentally known topology, the objective is to adjust the transition and emission probabilities in such way that the most likely path for a TM protein through the HMM is one where its TM residues are emitted in states labeled “Membrane,” and so forth.
The principle for the architecture of both TMHMM (Fig. 1 ▶; Sonnhammer et al. 1998; Krogh et al. 2001) and HMMTOP (Tusnády and Simon 1998,Tusnády and Simon 2001) is that there are separate compartments (sets of states and state transitions) for modeling the TM regions, the loop regions on the cytoplasmic side (inside), and the loop regions on the periplasmic side (outside). The composition and arrangement of the compartments differs slightly between the two methods, but the principle remains the same. The transitions within the TM compartments limit the lengths of these regions to somewhere around 15–35 residues. Inside and outside regions are built so that arbitrarily long loops are allowed but somewhat less likely than shorter ones. Intercompartmental transitions are restricted so that transitions directly between inside and outside regions are not allowed. An outside region followed by a TM region must be followed by an inside region and vice versa. Using the architecture of TMHMM2.0, we have created two new variants of HMM-based prediction methods, PRO-TMHMM and PRODIV-TMHMM, as well as a retrained version of TMHMM, referred to as S-TMHMM. The reason for using a retrained version of TMHMM and not TMHMM2.0 is to get a cleaner comparison between our novel methods and the single sequence method by using the same sequence data for parameter optimization.
Figure 1.

The layout of TMHMM. Each box corresponds to a compartment. Arrows correspond to possible intercompartmental transitions.
For predicting the topology of a TM protein, the traditional approach is to find the most probable path for the particular sequence through the HMM, and in each state of this path, emit the label of that state. This sequence of state labels is the topology prediction. Variants of this approach are used by both PRO-TMHMM and S-TMHMM. The method introduced with HMMTOP (Tusnády and Simon 1998,Tusnády and Simon 2001) describes another possible procedure for making a prediction. Here, the input sequence is first used to estimate new model parameters. This model is then used to make the prediction in the same way as described earlier. The objective of this approach is to maximize the divergence of the amino acid distributions of the different regions. A version of this procedure is used in PRODIV-TMHMM.
Both PRO-TMHMM and PRODIV-TMHMM are profile-based HMMs. A straightforward way of including multiple sequence information into TMHMM is to create a multiple sequence alignment based on the query sequence and its homologs and to calculate the geometric mean of the probabilities of the single aligned sequences, using the alignment to force the residues of each particular column to be emitted in the same state. In principle, this is the scoring method developed by Edgar and Sjölander (2003). In practice, the only difference between multiple sequence alignment scoring and the standard HMM single sequence scoring method is the way of calculating the “probability” of a particular alignment column being emitted in a particular state. Strictly, this is not a probability, but the term is used here in analogy with the corresponding term “emission probability” in the single sequence context. For alignments, this is calculated as ∏AJ = 1es(aj)Xi(aj), where A is the alphabet size, es(a) is the emission probability of letter a in state s, and Xi(a) is the fraction of letter a in alignment column Xi. This means that profiles can be used both for predictions and for estimating model parameters. For a detailed description, refer to Materials and Methods.
This method differs from how multiple sequence information is used in HMMTOP. In HMMTOP, a set of sequences that are homologous to the query sequence are used in the prediction phase, so that the divergence-optimizing parameter reestimation is done on the set of homologous sequences rather than on the query sequence alone. The important difference between using single homologous sequences and multiple sequence alignments is that when using alignments, the amino acids of a particular column are forced to be emitted in the same state. This is not necessarily the case for single homologous sequences that may take separate paths through the model.
Results
Prediction performance
In Table 1, a comparison of the performance of two novel methods (PRO-TMHMM and PRODIV-TMHMM) with several well-tested methods and one less well-tested method (HMMTOP_multi) is shown. The performance of the methods can be divided into three groups: methods that neither use multiple sequence information nor are “model-based” predict 32%–39% of the topologies correctly; methods that are either model-based (S-TMHMM, HMMTOP, MEMSAT, TMHMM2.0) or use multiple sequence information (PHD_htm2.1) predict 41%–52% of the topologies correctly; and finally, methods that are both model-based and use multiple sequence information predict 60%–66% of the topologies correctly. The difference between the third group and the previous two is statistically significant, as the error rate is ~4% for all methods. The only difference between S-TMHMM and PRO-TMHMM is that the latter uses multiple sequence alignment data, whereas the former does not. This means that both TMHMM and HMMTOP improve their prediction accuracy when including evolutionary information, from 49% and 52% to 61% and 60%, respectively. The usefulness of evolutionary information is emphasized further by the increased performance of PHD_htm (from 35% to 46%). It is possible that using alignments/profiles is more efficient than using single homologous sequences, because the performance increase for TMHMM is slightly larger than for HMMTOP. Using sequence profiles is often computationally more efficient than using homologous sequences.
Table 1.
Comparison of the prediction performance of different prediction methods on a data set of 147 sequences
| Prediction results for the 3D + 1D data set, 147 seqs | |||||||
| Method | Topo | Model-based | Multi | Over | Under | Q3 | Q2 |
| PRODIV-TMHMM | 97 (66%) | Yes | Yes | 28 | 11 | 82% | 88% |
| PRO-TMHMM | 90 (61%) | Yes | Yes | 21 | 26 | 82% | 87% |
| HMMTOP2.0_multi | 88 (60%) | Yes | Yes | 25 | 26 | 76% | 87% |
| HMMTOP2.0 | 76 (52%) | Yes | No | 33 | 28 | 74% | 87% |
| S-TMHMM | 72 (49%) | Yes | No | 20 | 40 | 79% | 88% |
| MEMSAT1.8 | 71 (48%) | Yes | No | 25 | 40 | 70% | 87% |
| PHD_htm2.1_msa | 67 (46%) | No | Yes | 26 | 48 | 72% | 86% |
| TMHMM2.0 | 61 (41%) | Yes | No | 19 | 49 | 70% | 88% |
| TopPred2.0 | 58 (39%) | No | No | 25 | 49 | 68% | 87% |
| PHD_htm2.1 | 52 (35%) | No | No | 31 | 46 | 70% | 86% |
| THUMBUP | 47 (32%) | No | No | 12 | 74 | 67% | 84% |
| ERROR | 4% | — | — | — | — | 2% | 1% |
HMMTOP2.0 (Tusnády and Simon 1998,Tusnády and Simon 2001) is the single sequence version of HMMTOP. HMMTOP2.0_multi is the multiple sequence version of HMMTPO. The input sequences used were the homologous sequences found in the BLAST search at E-value cutoff 10−5. PHD_htm2.1 (Rost et al. 1996) is the single sequence version of PHD, PHD_htm2.1_msa is the multiple sequence alignment version of PHD. The alignments used were those created by the BLAST search at E-value cutoff 10−5. MEMSAT1.8 (Jones et al. 1994), THUMBUP (Zhou and Zhou 2003), and TopPred2.0 (von Heijne 1992; Claros and von Heijne 1994) were all run with default parameter settings. PRODIV-TMHMM and PRO-TMHMM were run on the same multiple sequence alignments as PHD. The column Topo shows the number of (share of) correctly predicted topologies. The columns Over and Under describe the number of overpredictions and underpredictions that were made by each method. The column Model-based refers to the classification of each method as either residue-based or model-based (see introduction). Multi refers to whether a method includes multiple sequence information or not. Q3 is the per protein average of the share of residues that are correctly predicted using a three-state prediction evaluation (helix, inside, outside). Q2 is the per protein average of the share of residues that are correctly predicted using a two-state prediction evaluation (helix, nonhelix). The results of PRODIV-TMHMM, PRO-TMHMM, and S-TMHMM were all obtained using the cross-validation procedure described in Materials and Methods.
The PRODIV-TMHMM method is an extended version of PRO-TMHMM, which also includes the reestimation procedure for maximum divergence described earlier. This leads to a performance increase of another five percentage units.
Comparing the different methods on the basis of the perresidue scores (Q2 and Q3) shows that there is no significant difference between most of the methods regarding two-state (helix, nonhelix) predictions. However, the results for the three-state per-residue predictions (helix, inside, outside) seem to correlate with the results for the overall topology predictions; that is, methods using model-based approaches and/or multiple sequence information perform better than the simpler hydrophobicity-based methods.
The topology prediction accuracies obtained here agree with the results obtained by Käll and Sonnhammer (2002) and Melén et al. (2003), who both showed that earlier studies had overestimated the accuracy of the methods. In addition, the “completely correct” measure in the study by Chen et al. (2002) agrees very well with the topology prediction results, and the Q2 results agree with their conclusions that there are no significant differences between the best methods using “per residue” measures.
Creation of profiles
The performance of a profile-based HMM method is dependent on the quality of the profile. Our initial hypothesis was that only relatively close homologs should be included in the profiles, because it is important that the aligned sequences share the same topology as the query sequence. However, the sequence relationship should also be distant enough to provide additional evolutionary information. To compare different types of profiles, we created multiple sequence alignments running BLAST (Altschul et al. 1990) and PSI-BLAST (Altschul et al. 1997; for five iterations) with E-value cutoffs between 10−1 and 10−9. HMMs were then created and evaluated using these alignments and the PRO-TMHMM training and scoring method.
As can be seen from Figure 2 ▶, the profiles from BLAST clearly outscore those of PSI-BLAST, and the PSI-BLAST profiles tend to score better with more conservative cutoffs. It is well known that there is a significant risk of high-scoring false-positive hits when using PSI-BLAST and membrane proteins because all membrane regions, in some sense, are similar to each other (Hedman et al. 2002). Here it seems as though even a very restrictive E-value cutoff makes PSI-BLAST profiles drift too much from the query sequence and thereby pick up too remotely related sequences to be optimal for topology prediction purposes. Moreover, the scores for the BLAST profiles peak around a quite conservative E-value cutoff, 10−5.
Figure 2.

Comparison of prediction scores (Y-axis) for sequence alignments created using different E-value cutoffs (X-axis) and different search methods (BLAST, PSI-BLAST).
Specificity
Using a measurement method developed by Melén et al. (2003), it is possible to calculate the reliability of a prediction. Figure 3 ▶ shows that this prediction reliability is also improved using multiple sequence information compared with using single sequences. With PRO-TMHMM, the coverage is ~70% for a prediction accuracy of 75%, compared with using S-TMHMM, where the coverage is ~40% for the same prediction accuracy.
Figure 3.

Relation between test-set cumulative coverage and the fraction of correct topology predictions for S-TMHMM and PRO-TMHMM.
Discussion
Training and using profile information in TMHMM
The results for S-TMHMM and PRO-TMHMM in Table 1 were achieved using the same method both for parameter optimization (training) and evaluation (scoring). A fundamental question is whether it is sufficient to use the multiple sequence alignments only in the evaluation phase (scoring on an HMM trained on single sequences). It turns out that this is not the case. There is a slight improvement (54% correct predictions) using alignment scoring versus single sequence scoring (51%). This means that the extra information gained from the profiles is as important for estimating a good model as it is for the prediction itself. The results when using single sequence scoring on a model trained using the profile method (57%) gives further evidence for the hypothesis that the model estimated using the profiles is indeed better than the one estimated from single sequences.
Analysis of prediction differences between PRO-TMHMM and S-TMHMM
A comparison between the predictions of PRO-TMHMM and S-TMHMM (Table 2 shows an overview of this comparison) shows that 22 sequences are predicted correctly with PRO-TMHMM and predicted wrongly with S-TMHMM, whereas for 4 sequences the prediction results are the opposite. Of the 22 sequences, 11 are predicted correctly by S-TMHMM (the single sequence scoring method) if the model used for scoring is trained using the alignment method. We interpret this type of improvement as being due to the estimated model becoming better when training using alignments for the reasons discussed earlier.
Table 2.
Classification of the data set into different categories to illustrate the effect of including multiple sequence information in the prediction procedure
| Prediction comparison of S-TMHMM vs. PRO-TMHMM, 147 seqs | |
| Sequence category | No. seqs |
| Whole data set | 147 |
| Both correct | 68 |
| Both false | 53 |
| Only PRO_TMHMM correct | 22 |
| False S-THMMM when multioptimized | 11 |
| >50% correct aligned sequences | 6 |
| <30% correct aligned sequences | 5 |
| Correct S-THMMM when multioptimized | 11 |
| Only S_TMHMM correct | 4 |
Indented lines mean that these categories are subsets of the category of the nonindented line directly above. The four outer categories divide the sequences into those that are correctly predicted by both PRO-TMHMM and S-TMHMM, those that are predicted correctly by one method, and those that are wrongly predicted by both methods. The sequences that are predicted correctly only by PRO-TMHMM are divided into two groups: those that are predicted correctly using the S-TMHMM prediction method, the model using multiple sequence alignments, and those having been optimized are predicted falsely using this procedure. Further, the sequences of the last category are divided into two categories: sequences for which more than half of the aligned sequences are predicted to have the correct topology of the query sequence (when predicted as single sequences using the single sequence optimized model) and sequences for which less than 30% of the aligned sequences are predicted to have the correct topology of their respective query sequence.
For the remaining 11 sequences, there is also some information in the alignments themselves, which causes the correct prediction. When studying the properties of these alignments, it is clear that they fall into two categories. The first category, which contains five sequences, consists of proteins for which more than half of the aligned sequences can be correctly predicted, that is, predicted to have the same topology as the query sequence by S-TMHMM. In particular, many of the sequences whose sequence identity with the query sequence is low (<35%) are predicted correctly by S-TMHMM. Here, it seems reasonable to believe that it is the averaging property of the multiple sequence alignments that is the cause of the prediction becoming correct using PRO-TMHMM; that is, the profile representations of the sequences include more of the typical statistical properties for a sequence with this particular topology than the query sequence alone. The remaining six sequences make up the second category. Here, it is not clear that the average of the sequences should have more of the typical properties for the topology, because <30% of all sequences can be predicted correctly by S-TMHMM. For these sequences, the difference in prediction is usually caused by something subtler, such as small changes in the sizes of the hydrophobic regions, which lead to avoided false splits or false merges. The four additional mispredictions that appear when including multiple sequence information are also caused mainly by this type of subtle change.
Data set composition: Single-spanning versus multispanning TM proteins
Two recent studies (Käll and Sonnhammer 2002; Melén et al. 2003) point out that the data sets used in many earlier studies are not representative for TM proteins taken across whole genomes and that this has led to a general overestimation of prediction accuracies. For this reason, we wanted to use a data set with an internal diversity as large as possible. When studying Table 1, there are two striking observations. First, the prediction results of all methods are rather poor in comparison with earlier studies, in agreement with the estimations made by Melén et al. (2003) and Käll and Sonnhammer (2002). Second, all methods but HMMTOP and PRODIV-TMHMM have a tendency for underpredictions.
We believe that these observations can be explained largely by the composition of the data set we used. First, our data set was homology-reduced at 30% sequence similarity to make its internal diversity as large as possible. Second, it contained a fairly large amount of eukaryotic TM proteins (35%), for which the positive inside rule is generally regarded as being less prominent. Third, our data set contained no single-spanning TM proteins, that is, proteins with only one TM helix. This is something that in particular affects the balance between overpredictions and underpredictions. There are two reasons for this. First, in single-spanning TM proteins, the TM regions are generally more hydrophobic than in multispanning TM proteins, which will make these regions easier to find; that is, the risk for underpredictions is generally greater in multispanning TM proteins. Second, single-spanning TM proteins often have large globular domains, something that increases the risk for overpredictions; that is, overpredictions are generally more common for TM proteins with few TM helices and large globular domains. The fact that HMMTOP and PRODIV-TMHMM are the only exceptions to the underprediction consensus is also reasonable, because their prediction methods are based on maximal divergence in a way, which makes them more insensitive to absolute differences in hydrophobicity level.
Consequently, PRODIV-TMHMM is not optimal for distinguishing membrane proteins from nonmembrane proteins, as it will try to fit any sequence to a TM protein HMM architecture to maximize the likelihood of the sequence being produced by the model. This weakness can be seen most clearly when comparing the prediction results for the different methods on a set of 1087 globular proteins (data not shown). For 856 (79%) of these, PRODIV-TMHMM predicted at least one TM helix. The corresponding number for PRO-TMHMM was 11 (1%). Together with PHD_msa (14) and TMHMM2.0 (13), PRO-TMHMM had the lowest number of false positives of all methods in our comparison. Therefore, PRODIV-TMHMM should be combined with PRO-TMHMM if used for large-scale detection and topology prediction of membrane proteins.
For comparison, we also ran all prediction methods on a smaller data set with different properties than our original set (Zhou and Zhou 2003. Parameter estimation for all TMHMM-methods was done using the original data set). This data set contained only TM proteins for which a 3D structure has been determined and consisted of single-spanning TM proteins to a degree of ~50%. On this data set, prediction results are generally better than for our original data set (Table 3), but the ordering of the methods is exactly the same, except for THUMBUP (Zhou and Zhou 2003), which has been optimized using this data set. Further, there is a better balance between overpredictions and underpredictions using the smaller data set, indicating that overpredictions are more common on single-spanning TM proteins. For instance, 8 of the 12 overpredictions made by PRO-TMHMM were made on single-spanning proteins.
Table 3.
Comparison of different prediction methods on a “high-resolution” 3D data set of 73 sequences
| Prediction results for the 3D data set, 73 sequences | |||||
| Method | Topo | Over | Under | Q3 | Q2 |
| PRODIV-TMHMM | 56 (77%) | 13 | 1 | 82% | 87% |
| THUMBUP | 55 (75%) | 1 | 7 | 72% | 83% |
| PRO-TMHMM | 53 (73%) | 12 | 7 | 79% | 85% |
| HMMTOP2.0_multi | 50 (68%) | 8 | 3 | 78% | 88% |
| HMMTOP2.0 | 44 (60%) | 14 | 5 | 75% | 86% |
| S-TMHMM | 44 (60%) | 13 | 10 | 74% | 85% |
| MEMSAT1.8 | 44 (60%) | 9 | 5 | 71% | 86% |
| PHD_htm2.1_msa | 38 (52%) | 8 | 14 | 71% | 85% |
| TMHMM2.0 | 42 (58%) | 10 | 12 | 76% | 86% |
| TopPred2.0 | 38 (52%) | 15 | 8 | 69% | 86% |
| PHD_htm2.1 | 35 (48%) | 12 | 14 | 70% | 85% |
| ERROR | 5% | — | — | 3% | 1% |
All methods were run as described in Table 1.
Because of the apparent difference in prediction on multispanning versus single-spanning TM proteins, a conclusion is that the prediction accuracy of PRODIV-TMHMM may be increased further by adding a separate “path” for single-spanning proteins to the TMHMM architecture.
Conclusions
In this study, we show that topology predictors that use both HMMs and multiple sequence information perform superiorly to other methods. In particular, we introduce a new method, PRODIV-TMHMM, which is based on the best features of the earlier HMM predictors TMHMM and HMMTOP and adds the ability to use sequence profiles. PRODIV-TMHMM correctly identifies 66% of all topologies, whereas the best methods that do not use both HMMs and multiple sequence information (HMMTOP and TMHMM) only identify 52%. We show that the best results are obtained using quite conservative profiles; otherwise, it is likely that proteins with different topologies are included in the profile.
Materials and methods
Data sets
The larger 3D + topology data set of 147 sequences was provided by Johan Nilsson and created from proteins with experimentally determined topologies. This data set is homology-reduced at 30% sequence identity using the Hobohm 2 algorithm (Hobohm et al. 1992) and includes only multispanning TM proteins.
The smaller 3D data set of 73 sequences was created by Zhou and Zhou (2003). It contains only TM proteins with experimentally determined 3D structures. This data set contains 39 single-spanning proteins and 34 multispanning proteins and is not reduced for homology.
The set of globular proteins used for the discrimination test consists of 1087 sequences extracted from SWISSPROT (Käll et al. 2004).
Multiple sequence alignments were created running BLAST/ PSI-BLAST on the combined nrdb90 and SCOP1.57 database. PSI-BLAST was run for five iterations with E-value cutoffs set regarding both final inclusion and inclusion after each iteration.
HMM training and scoring
All HMM training and scoring was performed using the program modhmm, which is available from the authors. The scoring method described in the article was implemented using the N-best algorithm (Krogh 1997). Multiple sequence alignment scoring increases the time complexity of this algorithm with a factor the size of the alphabet compared with using single sequences. When the score of an alignment is calculated, only the columns corresponding to the query sequence residues are used.
It is possible to derive the alignment scoring method from the standard single sequence scoring method in the following way: Given an HMM with model parameters θ, the joint probability of a set of sequences (x1, . . ., xn) being emitted using a particular state path p is the product of the probabilities of the single sequences,
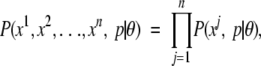 |
which can also be
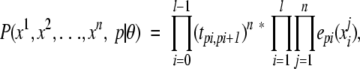 |
where tk,k′ is the transition probability from state k to state k′, ek(a) is the emission probability for letter a in state k, and l is the sequence length. This is the same as calculating the joint probability for the sequences in a multiple sequence alignment. To normalize for the size of the alignment, we raise this expression to the power of 1/n. This means that the expression can be written
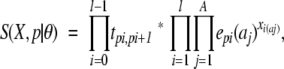 |
where A is the size of the alphabet and Xi is the profile vector calculated from column i of the multiple sequence alignment. Note that S(X,p|θ) is no longer a probability because of the normalization operation, hence the notational switch from P to S. The only practical difference between doing calculations using this method and the standard single sequence method is the emission “probability” (or score) of a column of a sequence profile being calculated as
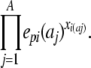 |
Except for the normalization part, this scoring method follows the same basic principle as the scoring method developed by Edgar and Sjölander (2003). Informally, this method calculates the geometric mean of the probabilities of the sequences in the alignment, which means that it will tend to prefer paths where all sequences have a fairly high probability to paths where some sequences have high and others have low probabilities; therefore, we refer to this scoring method as GM-score (Geometric Mean).
Note that this derivation is only completely accurate when there are no gaps in the alignment. Two approaches are available for handling gaps. The first is to add “GAP” as a letter of the alphabet. The second is to artificially set each gap letter of a column to a pseudoletter, which is interpreted as consisting of all of the original letters of the alphabet to a share corresponding to the distribution of the particular profile column.
For parameter optimization, the Baum-Welch training algorithm (Durbin et al. 1998) was used, with the only addition being the calculation of the emission score, as described earlier, for the forward and backward algorithms and the calculation of the expected number of times each emission is used. For the profile method, this was calculated as
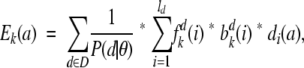 |
where D is the set of profiles, ld is the length of profile d, and di(a) is the share of letter a in column i of profile d.
The procedure used for training a model was the same regardless of the scoring method used and in most aspects identical to the procedure used by Krogh et al. (2001) when creating TMHMM2.0; that is, training was done in five steps using labeled sequences (Krogh 1994,Krogh 1997). First, the labels of the sequences were made flexible around the region borders so that the 12 residues around a border were allowed to match any state. Model estimation was then done using the Baum-Welch algorithm with noise injection as described by Hughey and Krogh (1996). In the third step, the label boundaries were reestimated using the model from step two. Next, a new model estimation was done using the relabeled sequences and the Baum-Welch training algorithm (without noise injection). In the fifth and final step, the model was once again reestimated using conditional maximum likelihood training as described by Krogh (1994). For a more thorough description of the training procedure used, refer to the original TMHMM articles (Sonnhammer et al. 1998; Krogh et al. 2001).
For the comparison between the different E-value cutoffs, the noise injection step was skipped to achieve nonrandom comparable models.
As an alternative to the geometric mean, an approximation of the arithmetic mean of the joint probabilities can be calculated by using the correlation coefficient between the two profiles. This and several other comparison methods were also tried (data not shown), but the best performance was obtained using the geometric mean. For computational efficiency, the alignments are represented as frequency profiles.
During model estimation, a nine-component Dirichlet mixture density created by Sjölander et al. (1996) was used to avoid over-fitting the emission probabilities. We also tried adding prior information to the profiles during both model estimation and prediction, but this did not seem to improve the prediction performance, nor did including gap information in the multiple sequence alignments (data not shown).
Cross-validation
For the results obtained for S-TMHMM, PRO-TMHMM, and PRODIV-TMHMMM, 10-fold cross validation was used. For this, the training set was divided into 10 equally sized subsets. A model was then optimized for each of the 10 subsets using the other 9/10 of the data set and evaluated using the remaining 1/10.
This procedure was not used on the models created for E-value cutoff comparison. Here, both training and scoring was done using the original data set.
Evaluation methodology
The requirement used for a correct topology prediction is that the correct number of TM helices is predicted and that the positioning of the predicted helices overlaps the experimentally determined positioning with at least five residues. Furthermore, the prediction of the orientation of the N and C termini must be correct.
Because we evaluate prediction performance on sequence basis, we have defined the categories of prediction errors to be as follows:
Overprediction: when too many TM helices are predicted.
Underprediction: when too few TM helices are predicted.
Other: The number of TM helices is predicted correctly, but at least one TM helix is predicted in the wrong location, or the orientation of the sequence relative to the membrane is inverted.
Compared with evaluating predictions (or rather the type of misprediction) on a helix basis, an overprediction usually corresponds to one or more false-positive helix predictions. These can either be predictions of helices where none is present, or a so-called false split, which means that one helix is incorrectly split into two. Conversely, an underprediction corresponds to one or more false-negative helix predictions, that is, either simply missing a true TM helix or incorrectly joining together two adjoining TM helices, a so-called false merge.
The standard error for the topology predictions was estimated using the following bootstrapping approach:
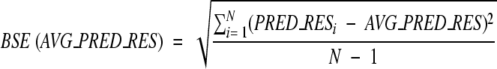 |
Here PRED_RESi is the share of correct predictions for a random subset of the original data set of size n/2, where n is the size of the original data set. AVG_PRED_RES is the average of the N random predictions. In our test, N was set to 100.
For the predictions of the aligned sequences in the multiple sequence alignments (when predicted as single sequences), the correct topology was set to be the same as for the query sequence of the alignment and the overlap criterion was skipped because insertions and deletions make it difficult to know exactly where the TM regions should be.
Q2 and Q3 are per-residue-based scores (i.e., the share of correctly predicted residues), calculated as the mean of the Q2 and Q3 scores for each protein. Q2 uses two states: helix and nonhelix. Q3 uses three states: helix, inside, and outside. The standard error for the Q2 and Q3 predictions were calculated as 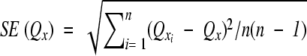 ,that is, Qx is the mean of the predictions for all sequences, and SE(Qx) is the standard error of this sample mean.
,that is, Qx is the mean of the predictions for all sequences, and SE(Qx) is the standard error of this sample mean.
Availability
The final versions of the topology predictors PRODIV-TMHMM, PRO-TMHMM, and S-TMHMM are available at http://www.sbc-.su.se/PRODIV-TMHMM/. All training and scoring was done using the modhmm package, which is available from http://www.s-bc.su.se/modhmm/. modhmm is a module-based HMM package developed to aid creating, training, and scoring HMMs, and which implements all scoring methods described in this paper as well as the standard HMM algorithms.
Acknowledgments
We thank Johan Nilsson and Lukas Käll for supplying data sets used in this study, and Gunnar von Heijne, Johan Nilsson, and Karin Melén for helpful comments and discussions regarding this project. We also thank Anders Krogh for providing us with the architecture and parameters of TMHMM2.0. This work was supported by grants from the Swedish Research Council and the Foundation for Strategic Research to A.E.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
HMM, hidden Markov model
TM, transmembrane
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04625404.
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., and Weissig, H. 2000. The protein data bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P.C., Kernytsky, A., and Rost, B. 2002. Transmembrane helix predictions revisited. Protein Sci. 11 2774–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claros, M.G. and von Heijne, G. 1994. Toppred II: An improved software for membrane protein structure prediction. Comput. Appl. Biosci. 10 685–686. [DOI] [PubMed] [Google Scholar]
- Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. 1998. Biological sequence analysis: Probabilistic models of proteins and amino acids. Cambridge University Press, Cambridge, UK.
- Edgar, R.C. and Sjölander, K. 2003. SATCHMO: Sequence alignment and tree construction using hidden Markov models. Bioinformatics 19 1404–1411. [DOI] [PubMed] [Google Scholar]
- Hedman, M., Deloof, H., von Heijne, G., and Elofsson, A. 2002. Improved detection of homologous membrane proteins by inclusion of information from topology prediction. Protein Sci. 11 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobohm, U., Scharf, M., Schneider, R., and Sander, C. 1992. Selection of representative protein data sets. Protein Sci. 1 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughey, R. and Krogh, A. 1996. Hidden Markov models for sequence analysis: Extension and analysis of the basic method. Comput. Appl. Biosci. 12 95–107. [DOI] [PubMed] [Google Scholar]
- Ikeda, M., Arai, M., Lao, D.M., and Shimizu, T. 2002. Transmembrane topology prediction methods: A re-assessment and improvement by a consensus method using a dataset of experimentally characterized transmembrane topologies. In Silico Biol. 2 19–33. [PubMed] [Google Scholar]
- Jayasinghe, S., Hristova, K., and White, S.H. 2001. MPtopo: A database of membrane protein topology. Protein Sci. 10 455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1994. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33 3038–3049. [DOI] [PubMed] [Google Scholar]
- Käll, L. and Sonnhammer, E.L.L. 2002. Reliability of transmembrane predictions in whole-genome data. FEBS Lett. 532 415–418. [DOI] [PubMed] [Google Scholar]
- Käll, L., Krogh, A., and Sonnhammer, E.L. 2004. A combined transmembrane topology and signal Peptide prediction method. J. Mol. Biol. 338 1027–1036. [DOI] [PubMed] [Google Scholar]
- Kim, H., Melén, K., and von Heijne, G. 2003. Topology models for 37 Saccharomyces cerevisiae membrane proteins based on C-terminal reporter fusions and predictions. J. Biol. Chem. 278 10208–10213. [DOI] [PubMed] [Google Scholar]
- Krogh, A. 1994. Hidden Markov models for labeled sequences. In Proceedings of the 12th IAPR International Conference on Pattern Recognition, pp. 140–144. IEEE Computer Society Press, Los Alamitos, CA.
- ———. 1997. Two methods for improving performance of a HMM and their application for gene finding. In Proceedings of the Fifth International Conference on Intelligent Systems for Molecular Biology (eds. T. Gaasterland et al.), pp. 179–186. AAAI Press, Menlo Park, CA. [PubMed]
- Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E.L.L. 2001. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305 567–580. [DOI] [PubMed] [Google Scholar]
- Martelli, P.L., Fariselli, P., Krogh, A., and Casadio, R. 2002. A sequence-profile-based HMM for predicting and discriminating β-barrel membrane proteins. Bioinformatics 18 46–53. [DOI] [PubMed] [Google Scholar]
- Melén, K., Krogh, A., and von Heijne, G. 2003. Reliability measures for membrane protein topology prediction algorithms. J. Mol. Biol. 327 735–744. [DOI] [PubMed] [Google Scholar]
- Möller, S., Croning, M.D.R., and Apweiler, R. 2001. Evaluation of methods for the prediction of membrane proteins. Bioinformatics 17 646–653. [DOI] [PubMed] [Google Scholar]
- Persson, B. and Argos, P. 1994. Prediction of transmembrane segments in proteins utilising multiple sequence alignments. J. Mol. Biol. 237 182–192. [DOI] [PubMed] [Google Scholar]
- Rost, B. and Sander, C. 1993. Improved prediction of protein secondary structure by use of sequence profiles and neural networks. Proc. Natl. Acad. Sci. 90 7558–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, B., Fariselli, P., and Casadio, R. 1996. Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci. 5 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sääf, A., Johansson, M., Wallin, E., and von Heijne, G. 1999. Divergent evolution of membrane protein topology: The Escherichia coli RnfA and RnfE homologues. Proc. Natl. Acad. Sci. 96 8540–8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjölander, K., Karplus, K., Brown, M., Hughey, R., Krogh, A., Mian, I.S., and Haussler, D. 1996. Dirichlet mixtures: A method for improved detection of weak but significant protein sequence homology. Comput. Appl. Biosci. 12 327–345. [DOI] [PubMed] [Google Scholar]
- Sonnhammer, E.L.L., von Heijne, G., and Krogh, A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. In Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology (eds. J. Glasgow et al.), pp. 175–182. AAAI Press, Menlo Park, CA. [PubMed]
- Tusnády, G.E. and Simon, I. 1998. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Biol. 283 489–506. [DOI] [PubMed] [Google Scholar]
- ———. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17 849–850. [DOI] [PubMed] [Google Scholar]
- von Heijne, G. 1986. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the transmembrane topology. EMBO J. 5: 3021–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225 487–494. [DOI] [PubMed] [Google Scholar]
- ———. 1994. Membrane proteins: From sequence to structure. Annu. Rev. Biophys. Biomol. Struct. 23 167–192. [DOI] [PubMed] [Google Scholar]
- Zhou, H. and Zhou, Y. 2003. Predicting the topology of transmembrane helical proteins using mean burial propensity and a hidden-Markov-model-based method. Protein Sci. 12 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]