

Abstract
7-Azatryptophan (AW), a noncoded isostere of tryptophan (W), possesses interesting spectral properties. In particular, the presence of a nitrogen atom at position 7 in the indolyl nucleus of AW results in a red shift of the absorption maximum and fluorescence emission by 10 and 46 nm, respectively, compared to W. In the present work, we report the chemical synthesis and the conformational and functional characterization of an analog (denoted as Y3AW) of the N-terminal domain 1–47 of hirudin, a highly potent thrombin inhibitor, in which Tyr 3 has been replaced by AW. The results obtained were compared with those of the cooresponding Y3W analog. We found that the replacement W → AW reduces affinity for thrombin by 10-fold, likely because of the lower hydrophobicity of AW compared with that of W. Measurements of the resonance energy transfer effect, which was observed between Tyr13 and the amino acid at position 3 upon disulfide-coupled folding, demonstrate that AW behaves as a better energy acceptor than W for studying protein renaturation. The interaction of Y3AW with thrombin was studied by exciting the sample at 320 nm and recording the change in fluorescence of Y3AW on binding to the enzyme. Our results indicate that the fluorescence of AW of hirudin 1–47 in the Y3AW–thrombin complex is strongly quenched, possibly because of the presence of two structural water molecules at the hirudin–thrombin interface that can promote the nonradiative decay of AW in the excited state. The data herein reported demonstrate that the incorporation of AW can be of broad applicability in the study of protein folding and protein–protein interaction.
Keywords: hirudin, thrombin, 7-azatryptophan, noncoded amino acids, anticoagulants, folding, spectroscopy
In its infancy, the approach of protein engineering was essentially restricted to the possibility of chemically modifying, in a rather unspecific fashion, particular amino acid side chains in proteins (for review, see Freedman 1971). More recently, the advent of recombinant DNA technology allowed the site-specifical alteration of a given polypeptide chain at a glance, thus much expanding the tools available to study the molecular mechanisms of protein folding, stability, and function (Fersht and Winter 1992). From this perspective, the incorporation of noncoded amino acids into proteins (Cornish et al. 1995) represents a further, almost obligatory extension of the studies aimed at elucidating the relationships existing between the structure, stability, and function in proteins (i.e., protein engineering). In fact, the incorporation of noncoded amino acids with tailored side chains allows investigators to more precisely relate the observed changes in protein behavior to the variation of the physicochemical properties at the mutation site. In view of this, several different strategies have been pursued, including chemical synthesis (Kent 1988; De Filippis et al. 1998a, 2002), enzyme-catalyzed semisynthesis (Wallace 1993; De Filippis et al. 1998b), biosynthetic incorporation via auxotrophic bacterial strain expression (Schlesinger 1968; Wong and Eftink 1997; Budisa et al. 1998), and nonsense suppression methodologies in cell-free (Cornish et al. 1995) or whole-cell (Dougherty 2000) expression systems.
A major application of protein engineering with non-coded amino acids regards the introduction into proteins of biophysical probes possessing physicochemical properties (e.g., side-chain volume, hydrophobicity) similar to those of the corresponding natural amino acids but with spectral features distinct from those of the natural counterparts and highly sensitive to the chemical environment in which the probe is located (Cornish et al. 1994). Hence, by the use of the so-called “spectrally enhanced proteins” (Wong and Eftink 1998), it should be possible to effectively monitor the local structure and dynamics of the mutated protein during key events, such as protein folding and denaturation or ligand binding, without significantly perturbing the kinetics and equilibrium properties of the process under investigation. With respect to this, in a recent study Cohen and coworkers (Cohen et al. 2002) were able to site specifically introduce 6-dimethylamino-2-acyl-naphthylalanine (Aladan) into the B1 domain of staphylococcal protein G to obtain estimates of the local dielectric constant of the protein at different sites.
Traditionally, W (Trp) has been widely used as an intrinsic fluorescent probe to study protein dynamics and ligand binding in solution (Eftink 1997). However, its complex photophysics often impairs universal interpretation of fluorescence data (Lakowicz 1999). Moreover, proteins usually contain more than one Trp residue, making it difficult (if not impossible) to assign the change in fluorescence signal to a particular site and interpret the spectral changes that result from intermolecular protein–protein association (Ross et al. 1997). To overcome these problems, investigators have covalently bound several extrinsic fluorophores to protein functional groups (Wu and Brand 1994; Buckler et al. 1995). This approach, however, is limited by possible labeling heterogeneity and structural alteration resulting from the labeling per se.
Among the noncoded W analogs studied so far (i.e., 5-hydroxy- and 5-metoxy-Trp, benzo[b]thiophenylalanine and the spectrally silent fluorotryptophans), AW displays interesting absorption and fluorescence properties (for a recent review, see Twine and Szabo 2003), and its incorporation into proteins has been exploited to study protein structure (Schlesinger 1968; Cornish et al. 1994; Scott et al. 1999), stability (Soumillon et al. 1995; Wong and Eftink 1997, 1998), macromolecular recognition (Mohammadi et al. 2001), and protein conformational change (Blouse et al. 2002). Unlike W, free AW displays single exponential fluorescence decay (Chen et al. 1994), and the presence of a nitrogen atom at position 7 in the indolyl nucleus results in a redshift of 10 nm in the absorption and 46 nm in the emission of AW compared with Trp (Ross et al. 1997). Furthermore, the fluorescence λmax and quantum yield of 7AI are strongly influenced by the polarity of the chemical environment. In particular, going from cyclohexane to water, the emission fluorescence of 7AI is shifted from 325 to 400 nm and the quantum yield is decreased by 10-fold. The quantum yield of AW increases from 0.01 in aqueous solution (pH 7) to 0.25 in acetonitrile (Ross et al. 1997). Hence, it should be possible to selectively excite the fluorescence of AW at the red edge of its absorption (between 310 and 320 nm), where Tyr does not absorb and the contribution of Trp is negligible (see following), and thus investigate the ligand-binding process through variation of AW fluorescence signal. On this basis, AW has great potential as a probe of protein structure, especially in the study of protein–protein interactions in which one of the proteins may contain several Ws. With respect to this, it has been reported that only when the W:AW ratio is as great as 10:1 does the tryptophyl emission become appreciable, and even when this ratio is 40:1, the contribution of AW can be easily detected (Smirnov et al. 1997).
Thrombin is a key enzyme in blood coagulation (Davie et al. 1991) and plays an important role at the interface between coagulation, inflammation, and cell differentiation (Esmon et al. 1999). Under physiological conditions, thrombin exists in equilibrium between a procoagulant (fast) form and an anticoagulant (slow) form, and the slow ⇔ fast transition is triggered by Na+ binding (for review, see Di Cera et al. 1997). Hirudin, a 64-amino-acid polypeptide isolated from the salivary secretions of medicinal leeches, is the most potent inhibitor of thrombin known so far (Markwardt 1994). It is composed of a compact N-terminal domain (residues 1–47) stabilized by three disulfides and a negatively charged C-terminal tail (Haruyama and Wutrich 1989). The high-resolution crystallographic structure of the hirudin–thrombin complex (Rydel et al. 1991) reveals that the C-terminal tail binds to exosite I on thrombin, whereas the N-terminal domain extensively penetrates into the recognition sites, mainly through its first three amino acids, and covers the active site pocket of the enzyme. In particular, the first three amino acids make about half of the total contacts observed for the binding of the N-terminal core domain to thrombin (Rydel et al. 1991) and account for ~30% of the total free energy of binding of intact hirudin (Betz et al. 1992). Hence, fragment 1–47, previously obtained in our laboratory by tryptic cleavage of hirudin (Vindigni et al. 1994), can be a suitable probe for studying the nature of hirudin–thrombin interaction.
In the present work, we report the chemical synthesis and characterization of an analog (hereafter denoted as Y3AW) of the N-terminal domain 1–47 of hirudin in which Tyr 3 has been replaced by AW (Fig. 1 ▶). The conformational and functional properties of Y3AW were compared with those of the Trp 3 analog of hirudin, previously reported (De Filippis et al. 1995). The disulfide-coupled folding reaction of Y3AW was investigated by measuring the resonance energy transfer between the single tyrosine at position 13 and AW at position 3 in the fully reduced and disulfide-oxidized form, and the interaction of Y3AW with thrombin was studied by the change in the fluorescence of Y3AW on binding to the enzyme. The results obtained were explained in the light of the crystallographic structure of the hirudin–thrombin complex. Overall, our findings demonstrate that incorporation of AW into proteins can be of broad applicability in the study of protein folding and protein–protein recognition.
Figure 1.

Amino acid sequence (A; Scacheri et al. 1993) and schematic representation (B) of the solution structure (Nicastro et al. 1997) of the N-terminal fragment 1–47 of hirudin HM2 from the leech Hirudinaria manillensis. The side chains of Tyr 3 and Tyr 13 and disulfide bonds are also shown. The ribbon drawing was generated using WebLab ViewerPro 4.0 (Molecular Simulations Inc. 2000). (C) Chemical structure of L-tryptophan (W) and L-7-azatryptophan (AW).
Results and Discussion
Synthesis and characterization of Y3AW
Prior to chemical synthesis of Y3AW analog, the resolution of the commercially available enantiomeric mixture of AW was carried out as schematized in Figure 2A ▶. The racemic mixture of D,L-AW was acetylated with acetic anhydride in neat acetic acid, as described (Greenstein 1957), to yield D,L-Nα-acetyl-AW (Fig. 2B ▶). Mass spectrometric analysis of the purified species by ESI-TOF mass spectrometry led to unambiguous identification of all of the reaction products (see Fig. 2 ▶ legend). Thereafter, D,L-Nα-acetyl-AW mixture was reacted with Eupergit-C immobilized Asperigillus oryzae acylase-I, an enzyme that is able to enantioselectively hydrolyze L-Nα-acetyl-derivatives of numerous natural and nonnatural amino acids, without reacting with the corresponding D-isomer or with amino acid oligomers (Chenault et al. 1989; Lecointe et al. 1998). As shown in Figure 2C ▶, after a 24-h reaction, the enantioselective deacylation of D,L-Nα-acetyl-AW was quantitative and led to free L-AW enantiomer, which was subsequently purified by preparative RP-HPLC. The enantiomeric purity of L-AW was established by chiral affinity chromatography on an albuminsepharose 4B column (Fig. 2D ▶). This technique takes advantage of the peculiar ability of albumin to bind L-and D-enantiomers of aromatic amino acids with different strength (Stewart and Doherty 1973). As shown in Figure 2D ▶, L-AW elutes after the D-enantiomer, in agreement with the results obtained with D,L-W (Allenmark and Bomgren 1982). Moreover, within the limitations of the detection method used, the chromatographic analysis reported in Figure 2D ▶ provides evidence that our preparation contains only the L-AW enantiomer. The optical purity of L-AW was further verified by using the FLEC-derivatization procedure (not shown) previously reported by Josefsson et al. (1987). Purified L-AW was then reacted with 9-fluorenylmethoxy-carbonyl chloride (Fmoc-Cl), to obtain the resulting Fmoc-derivative with 99% yield, as estimated by RP-HPLC analysis (not shown). The chemical identity of Fmoc-AW was established by ESI-TOF mass spectrometry, IR, and 1H-NMR techniques (not shown). At elevated temperatures, Fmoc-AW decomposes, with a Tm value of 214°C, much higher than that of the corresponding Fmoc-tryptophanyl derivative (Tm = 168°C). Similarly, the melting temperature of 7AI (105°–107°C) is higher than that of indole (52°–53°C).
Figure 2.

(A) Scheme of the enzyme-catalyzed resolution of D,L-AW racemate. (B) RP-HPLC analysis of the acetylation reaction of D,L-AW with acetic anhydride. The material eluted in correspondence of the minor chromatographic peak (denoted as D,L-acetyl-(AW)n) contains several by-products, including a reactive aceticaminoacyl mixed anhydride (Nα,O-diacetyl-AW; Mw, 290.12 a.m.u.), Nα-acetyl dimers (Mw, 435.19 a.m.u.) and trimers (Mw, 620.46 a.m.u.), and the corresponding Nα,O-diacetyl-derivative of AW-dimer (Mw, 477.20 a.m.u.). (C) RP-HPLC analysis of the stereoselective deacetylation of Nα-acetyl-L-AW by acylase-I, to yield free L-AW. (D) Enantioselective separation on the sepharose-BSA chiral column of commercial D,L-AW racemate (lower trace) and analysis of purified L-AW (upper trace) as obtained after acylase-I treatment of Nα-acetyl-D,L-AW (panels A–C).
Incorporation of AW into fragment 1–47 was carried out by standard Fmoc-chemistry, using the HBTU/HOBt activation method (Atherton and Sheppard 1989), and following the automated/manual synthetic procedure previously established (De Filippis et al. 1995, 1998a). After resin cleavage and ether precipitation, the crude peptide with the six Cys residues in the reduced state was allowed to fold (0.5 mg/mL) under air-oxidizing conditions in bicarbonate buffer (pH 8.3) in the presence of 100 μM β-ME (Chatrenet and Chang 1992; De Filippis et al. 1995) to reconstitute the native disulfide topology of hirudin (see Fig. 1A ▶). The RP-HPLC analysis of the synthetic peptide in the reduced (lower curve) and disulfide oxidized (upper curve) state is reported in Figure 3A ▶. The folded species was characterized for its N-terminal amino acid sequence and molecular mass value, obtaining experimental values in agreement with the expected amino acid composition. Renaturated Y3AW was purified by preparative RP-HPLC, lyophilized, and used for subsequent conformational and functional characterization. RP-HPLC analyses (Fig. 3B ▶) of the purified, disulfide-oxidized Y3AW and Y3W analogs, carried out under elution conditions (5 mM sodium phosphate buffer at pH 6.4) at which the N7 atom of AW is not protonated (pKa = 4.5, Negrerie et al. 1991), indicate that Y3AW is more hydrophilic than the corresponding Trp 3 derivative.
Figure 3.
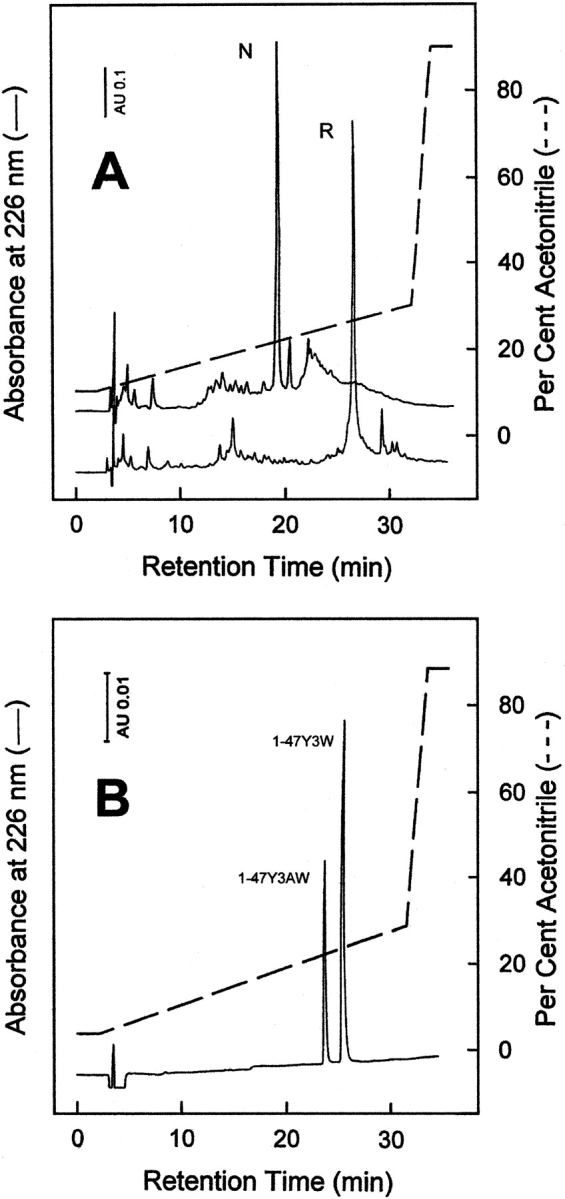
(A) RP-HPLC analysis of the crude synthetic analog Y3AW in the fully reduced, R (lower trace), and disulfide oxidized native, N, state (upper trace). Chromatographic separations were conducted on a Vydac C18 analytical column (4.6 × 250 mm, 5 μm) equilibrated with 0.05% aqueous TFA and eluted with a linear acetonitrile-0,05% TFA (dashed line) gradient at a flow rate of 0.8 mL/min. (B) RP-HPLC analysis of an equimolar mixture of purified, disulfide folded Y3AW and Y3W analogs. The RP-HPLC column was equilibrated with 5 mM sodium phosphate buffer (pH 6.4) and eluted with a linear gradient of neat acetonitrile (dashed line). All other chromatographic conditions were the same as those reported in panel A.
Overall, our results provide evidence that standard Fmoc-based solid-phase synthesis is a convenient approach to site specifically incorporate the fluorescent probe AW into even long polypeptide chains. To the best of our knowledge, Y3AW is the longest peptide chain containing AW obtained so far by chemical methods. With respect to this, only short-chain synthetic peptides have been reported, including bio-tinylated AW (Rich et al. 1995) and the chymotrypsin inhibitor Nα-acetyl-Pro-AW-Asn-NH2 (Rich et al. 1993).
Spectroscopic properties of Y3AW
The spectral properties of Y3AW analog were investigated by UV absorption, fluorescence, and CD, and compared with those of the corresponding Trp 3 derivative (Y3W; Figs 4 ▶, 5 ▶).
Figure 4.
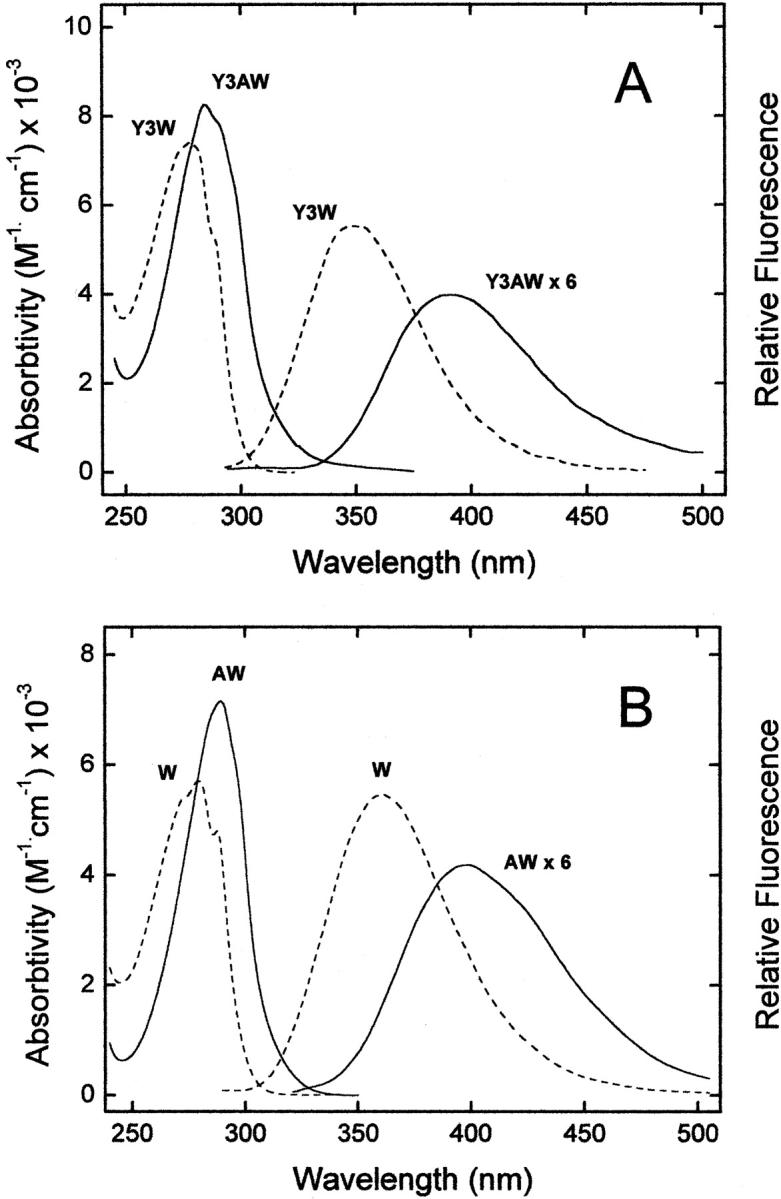
(A) UV absorption and emission fluorescence spectra of Y3AW (solid line) and Y3W (dashed line) analogs of hirudin fragment 1–47. (B) Absorption and fluorescence spectra of the free amino acids W and AW in the zwitterionic form. The fluorescence spectra of free AW and Y3AW analogs were multiplied sixfold. All measurements were carried out at 25°C in 5 mM Tris-HCl buffer (pH 8.0) containing 0.2 M NaCl and 0.1% PEG-8000, at a protein or amino acid concentration of 2 μM. Sample excitation was at 280 nm, using an excitation/emission slit of 5 and 10 nm, respectively.
Figure 5.

Near-UV CD spectra of Y3AW (solid line) and Y3W (dotted line) analogs of hirudin fragment 1–47. For comparison, the CD spectra of the 1–47 analog containing the nonabsorbing amino acid Cha at position 3 is also reported (dashed line). All measurements were carried out at 25°C in 10 mM sodium phosphate buffer (pH 7.0) at a protein concentration of 150 μM. (Inset) Difference spectra obtained by subtracting the spectrum of Y3Cha from that of Y3AW (solid line) or Y3W (dotted line).
The λmax values in the absorption and fluorescence spectra of Y3AW are red shifted by ~10 and 40 nm, respectively, compared with those of Y3W (Fig. 4A ▶). A similar behavior is observed for the free amino acids W and AW in aqueous solution (Fig. 4B ▶). As in the case of W (Lakowicz 1999), the fluorescence of AW is strongly dependent on the polarity of the environment in which this fluorophore is located. In particular, the λmax value of 7AI is red shifted by ~75 nm, from 325 to 400 nm, in going from cyclohexane to water (Chapman and Maroncelli 1992). Hence, the fluorescence maximum of Y3AW at 390 nm indicates that AW at position 3 of hirudin is almost fully exposed to the aqueous solvent, as also expected from the analysis of the tridimensional structure of hirudin fragment 1–47 shown in Figure 1B ▶ (Szyperski et al. 1992; Nicastro et al. 1997).
Near-UV CD is a very sensitive probe of protein tertiary structure and it is usually taken as a fingerprint of the chemical environment in which aromatic amino acids and disulfide bonds are located within protein structure (Strickland 1974; Kahn 1979). The near-UV CD signal of the natural fragment 1–47 is dominated by a broad, prominent band centered at 260 nm, corresponding to the absorbance of the three disulfide bonds. The fine structure observed in the 280–300 nm range is almost exclusively due to Tyr 3, whereas the contribution of Tyr 13 (the only other aromatic amino acid present in the 1–47 sequence) is negligible (De Filippis et al. 1995). The appearance of Tyr 3 fine structure is in agreement with the NMR solution structure of the N-terminal domain of hirudin, showing that the first two amino acids are disordered, whereas Tyr 3 is in a rather fixed conformation (Szyperski et al. 1992; Nicastro et al. 1997). In Figure 5 ▶ the CD spectrum of Y3AW in the near-UV region is reported in comparison with that of the corresponding Trp 3 derivative and with that of the Y3Cha analog, containing the nonabsorbing amino acid Cha. The spectra of Y3W and Y3AW analogs are very similar in both shape and intensity, indicating that the overall tridimensional structure of hirudin fragment 1–47 is unchanged on W →AW exchange. Moreover, the difference spectra, obtained by subtracting the spectrum of Y3Cha from that of Y3AW and Y3W, provide unprecedented information on the chiroptical properties of AW in proteins (Fig. 5 ▶, inset). The 1Lb bands at 288 and 281 nm in the spectrum Y3W are shifted to 311 and 292 nm in the CD spectrum of Y3AW, reflecting the redshifted UV absorption of AW compared with that of W (see also Fig. 4 ▶).
Hirudin folding monitored by tyrosine → 7-azatryptophan energy transfer
Fluorescence energy transfer is a suitable method for investigating the structure and dynamics of macromolecular systems in solution, and provides global geometrical information complementary to that obtainable with other site-specific techniques like NMR (Wu and Brand 1994; Lakowicz 1999). Resonance energy transfer in proteins occurs between two different fluorophores, the donor (usually tyro-sine) and acceptor (usually W), which interact via electromagnetic dipoles transferring the excitation energy of the donor to the acceptor. A prerequisite for this process to occur is that the emission spectrum of the donor significantly overlaps to the excitation spectrum of the acceptor, and that the two counterparts are in close proximity and in a rather fixed (nonorthogonal) orientation. In these conditions, the fluorescence of the donor is quenched and the fluorescence of the acceptor is enhanced. In globular proteins, the energy transfer efficiency approaches zero in the unfolded state, when the donor (tyrosine) and acceptor (W) amino acids are highly flexible and spatially distant, whereas in the more compact native state the donor and acceptor are in close proximity, leading to quenching of Tyr fluorescence. Therefore, tyrosine → W energy transfer can be used as a spectroscopic ruler for long-range distance determination during protein folding. However, because of the intrinsic spectroscopic limitations of the Tyr–Trp donor–acceptor pair (e.g., significant overlapping of Tyr and Trp fluorescence bands, and shielding of the Tyr signal by the more intense Trp emission), only qualitative information can be obtained in most cases (Wu and Brand 1994; Eftink and Shastry 1997). To overcome these problems, investigators covalently bound several extrinsic labeling fluorophores to protein functional groups (Buckler et al. 1995; Ittah and Haas 1995). Alternatively, chemical modifications at specific amino acid side chains (e.g., tyrosine nitration to 3-nitrotyrosine or W oxidation to kynurenine) were incorporated into proteins and used in ligand binding (Yamashita et al. 1996) and protein folding studies (Rischel and Poulsen 1995; Tcherkasskaya and Ptitsyn 1999). These approaches, however, are limited by possible labeling heterogeneity, or nonquantitative modification, and structural alteration resulting from the labeling per se (Wu and Brand 1994; Lakowicz 1999).
In the present study, we have exploited the incorporation of AW to investigate the disulfide-coupled folding of hirudin fragment 1–47 (Fig. 6 ▶). As reported in Figure 6A ▶, the redshifted absorption of AW, compared with that of W, improves the overlapping of the emission band of the donor (Tyr) with the absorption band of the acceptor (AW or W). The fluorescence spectra of Y3AW with the six Cys residues in the fully reduced or correctly folded state are shown in Figure 6B ▶ and compared with those of the corresponding Y3W analog (Fig. 6C ▶). The emission spectrum of the reduced, unfolded Y3AW reveals the presence of two distinct, well-resolved bands at 305 and 397 nm, assigned to the contribution of Tyr 13 and AW at position 3, respectively. In the folded state, the Tyr band disappears, and the fluorescence of AW is blue shifted to 390 nm and enhanced by about 20%. Our results can be rationalized by considering that in the reduced state, hirudin fragment 1–47 is in a random coil conformation (V. De Filippis, unpubl.), with the donor (Tyr 13) and acceptor (AW3) amino acids far apart in space. In the native state, the chain folding brings Tyr 13 in close proximity to AW3 (see Fig. 1B ▶) and allows the energy absorbed by Tyr to be efficiently transferred to AW. In the case of Y3W, a single band at 350 nm is observed in the native state, whereas in the reduced state the contribution of Tyr 13 appears as a very weak shoulder at 303 nm, overwhelmed by the stronger emission of Trp 3 at 355 nm (Fig. 6C ▶). This actually makes it difficult (if not impossible) to follow the folding process of hirudin by Tyr →Trp energy transfer measurements. Therefore, the redshifted emission of AW makes this amino acid a suitable, nonnatural isostere of W in the study of protein folding.
Figure 6.

Disulfide oxidative folding of the Y3AW analog monitored by fluorescence spectroscopy. (A) UV-absorption spectra of free W (dashed line) and AW (solid line), acting as energy acceptors, and fluorescence spectrum of tyrosine (Y, dotted line), acting as an energy donor. Emission fluorescence spectra of Y3AW (B) and Y3W (C) in the reduced (solid line) and disulfide oxidized native state (dashed line). All spectra were taken at 25°C by exciting the samples (5 μM) at 280 nm in 0.1 M NaHCO3 (pH 8.3), except for that of the fully reduced form, which was recorded in 0.1 M MES buffer (pH 6.0). This pH value is sufficiently low to impair disulfide formation for at least 4 h (not shown) and, concomitantly, avoid protonation of the N7-atom of the azaindole nucleus, which has a pKa value of 4.5 (Négrerie et al. 1991).
Thrombin inhibitory properties of Y3AW
The synthetic Y3AW analog was assayed for its thrombin inhibitory potency with respect to Y3W and wild-type fragment 1–47 (Table 1). Antithrombin activity was determined by measuring at 405 nm the release of p-nitroaniline from the synthetic substrate D-Phe-Pro-Arg-p-NA, under experimental conditions that favor the procoagulant, fast form (0.2 M NaCl) or the anticoagulant, slow form (0.2 M ChCl). From these experiments, the values of the dissociation constant, Kd, of thrombin–inhibitor complexes have been determined, and the difference in the free energy change of binding, ΔΔGb, relative to the wild-type fragment 1–47 has been calculated. The data reported in Table 1 indicate that replacement of W with the isosteric AW leads to a significant reduction (~10-fold) in the affinity for the fast form of thrombin, whereas the affinity for the slow form is reduced by only threefold. These results can be explained by the reduced hydrophobicity of AW compared with that of W, as given by the values of octanol →water partition coefficient (logP) of indole (logPindole = 2.33) and 7AI (logPazaindole = 1.72), determined experimentally (see Materials and Methods). The more polar character of Y3AW compared with that of Y3W is also confirmed by the fact that at pH 6.4 (a pH value at which the N7 atom of AW is not protonated), Y3AW is eluted from a C18 column at a shorter retention time (see Fig. 3B ▶). Our findings demonstrate that replacement of even a single atom (C → N) can substantially affect binding to thrombin and that the effect of increasing hydrophobicity at position 3 has different consequences on the affinity of fragment 1–47 for the two allosteric forms of the enzyme.
Table 1.
Thermodynamic data for the binding of synthetic analogs 1–47 to the fast and slow form of thrombin
| Fast form | Slow form | ||||
| Synthetic analogs 1–47 | Kd (nM) | ΔΔ Gba (kcal/mole) | Kd (nM) | ΔΔ Gba (kcal/mole) | Δ Gcb (kcal/mole) |
| Natural | 41 ± 2 | — | 1500 ± 100 | — | −2.1 |
| Y3W | 6.6 ± 0.7 | −1.08 | 550 ± 40 | −0.60 | −2.6 |
| Y3AW | 60 ± 2 | 0.22 | 1550 ± 24 | 0.02 | −1.9 |
a All measurements were carried out at 25°C in 5 mM Tris (pH 8.0) containing 0.1% PEG in the presence of 0.2 M NaCl for the fast form or 0.2 M ChCl when the slow form was being studied.
ΔΔGb is the difference in the free energy change of binding to thrombin between the synthetic analog (ΔGb*) and the natural fragment (ΔGbwt): ΔΔGb = ΔGb* − ΔGbwt. A negative value of ΔΔGb indicates that the mutated species binds more tightly to thrombin than the natural fragment. Errors are ±0.1 kcal/mole or less.
b ΔGc is the free energy of coupling to thrombin, measured as ΔGc = ΔGb, fast− Δ Gb, slow (Ayala and Di Cera 1994). The value of ΔGc is negative if the inhibitor binds to the fast form with higher affinity than to the slow form.
Hirudin–thrombin interaction probed by fluorescence spectroscopy
A central issue in structural biology and modern drug discovery regards the elucidation of the local physicochemical properties at protein–ligand and protein–protein interfaces (Lo Conte et al. 1999; Hu et al. 2000), including amino acid side-chain dynamics (Kay 1998), local dielectric constant (Cohen et al. 2002), and water penetration and escape (Desinov et al. 1996; Garcia and Hummer 2000). In particular, it has been well documented that structural water molecules at the interface can play a key role in modulating binding strength (Huang et al. 1995; Shaltiel et al. 1998).
In the present study, we have exploited the strong dependence of the fluorescence properties of AW on solvent polarity (Taylor et al. 1969; Chapman and Maroncelli 1992) to investigate the binding of the Y3AW analog to thrombin (Fig. 7 ▶). To minimize the contribution of the nine Trp residues present in the thrombin sequence, we excited AW at the red edge of its absorption range (320 nm), where the contribution of Trp is expected to be negligible (Ross et al. 1997). The emission spectrum of the isolated Y3AW analog is dominated by the fluorescence of AW at 390 nm (Fig. 7A ▶), in agreement with the surface localization AW at position 3 in the free inhibitor (see Fig. 1B ▶). The fluorescence λmax of free thrombin is at ~342 nm, reflecting the fact that in the tridimensional structure of thrombin, six of nine Ws are exposed to water (Bode et al. 1992). The spectrum of thrombin complexed to Y3AW is similar to that of the isolated enzyme. The only difference is in the 380–440 nm range, where a slight increase in the fluorescence signal of the complex is observed. Interestingly, the theoretical spectrum obtained by summing those of the free enzyme and the Y3AW analog is remarkably more intense than that of the thrombin–Y3AW complex, obtained experimentally (Fig. 7A ▶). The difference spectrum, obtained by subtracting the spectrum of free thrombin from that of the enzyme–inhibitor complex, displays a λmax value centered at 400 nm and an intensity that is about 25% of that of the free inhibitor. Interestingly, this spectrum would account for the contribution of Y3AW in the hirudin–thrombin complex. However, with a dissociation constant (Kd) for the thrombin–Y3AW complex of 60 nM and a concentration of enzyme and inhibitor of 2 μM, it is expected that >80% of the total amount of Y3AW is bound to thrombin, whereas ~20% remains in the free form. For comparison, the spectrum of a model compound solution of W and AW, isolated or mixed in the same molar ratio (9:1) as that present in the thrombin–inhibitor complex, is reported in Figure 7B ▶. Clearly, the emission of W, although less intense than that of AW, is still significant even after excitation at 320 nm. As expected, the theoretical sum spectrum (not shown) is almost superimposable to the experimental spectrum of the 9W:1AW model solution. Taken together, these results indicate that the fluorescence of Y3AW is almost fully quenched on binding to thrombin recognition sites and that the slight changes in the fluorescence spectrum of the thrombin–Y3AW complex mainly arise from the fraction of free Y3AW in equilibrium with the thrombin-bound form. With respect to this, titration of Y3AW with increasing concentrations of thrombin would yield a direct estimate of the dissociation constant of the enzyme–inhibitor complex.
Figure 7.
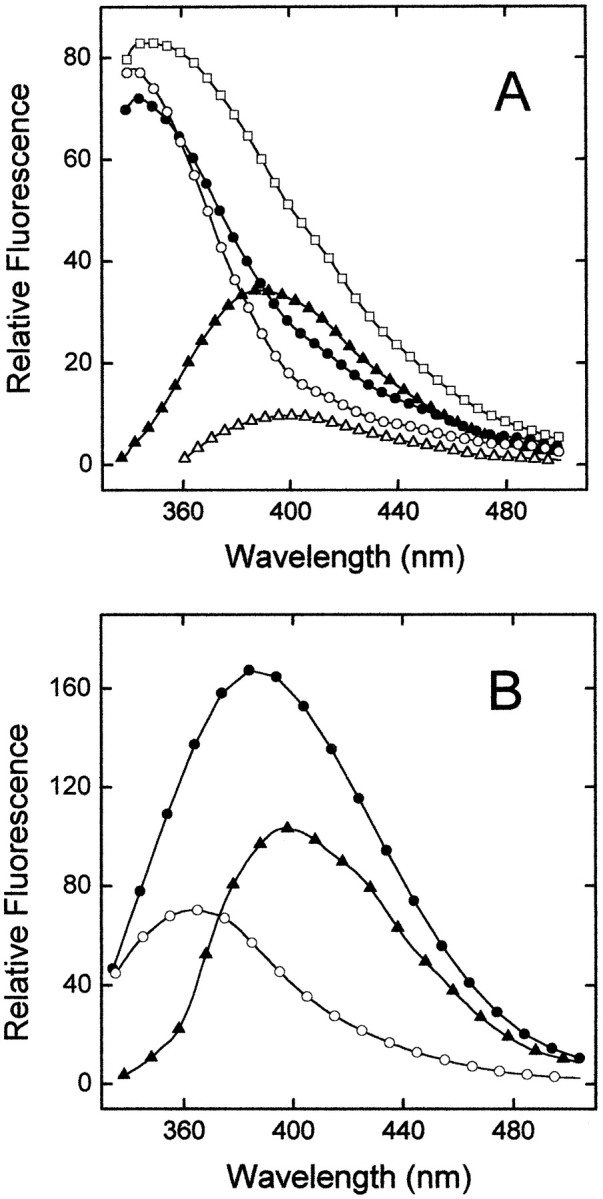
(A) Hirudin–thrombin interaction probed by fluorescence spectroscopy. Fluorescence emission spectra of isolated thrombin (open circles) and Y3AW analog (filled triangles), and equimolar mixture of the enzyme and inhibitor (filled circles). All spectra were recorded at 25°C in 5 mM Tris-HCl buffer (pH 8.0) containing 0.2 M NaCl and 0.1% PEG-8000, at a protein concentration of 2 μM, and by exciting the samples at 320 nm. The theoretical sum spectrum (open squares) was obtained by adding the spectrum of free thrombin to that of free Y3AW analog. The contribution of Y3AW in the thrombin-bound form is estimated by the difference spectrum (open triangles), obtained by subtracting the spectrum of isolated thrombin from that of the enzyme-inhibitor complex. (B) Fluorescence spectra of the model compounds Nα-acetyl-tryptophanamide (open circles, 18μM) and AW (filled triangles, 2 μM), and of a solution obtained by mixing Nα-acetyl-tryptophanamide (Ac-Trp-NH2) and AW in the same molar ratio (9:1) as that present in the thrombin/Y3AW complex (filled circles). Experimental conditions are as in panel A.
Given the known solvent-dependent emission of AW and the apolar character of the S2/S3 binding sites of thrombin, this result is rather surprising. In fact, AW is a water-quenched fluorophore, and in going from water to cyclohexane, the emission λmax is shifted from 400 to 325 nm and the fluorescence quantum yield is enhanced by 10-fold (Chapman and Maroncelli 1992). The model structure of the N-terminal tripeptide of the hirudin analog Y3AW interacting with thrombin recognition sites (Fig. 8 ▶) shows that the -NH group of AW points toward the S3 site, traditionally referred to as the “apolar binding site” (Berliner and Shen 1977) and formed by Trp 215, Leu 99, and Ile 174, whereas the six-membered ring points toward Tyr 60a and Trp 60d in the S2 site (Rydel et al. 1991). Thus, we would have expected that binding of Y3AW to thrombin be accompanied by a blueshifted emission of AW and a substantial increase in its fluorescence intensity. Conversely, we observe a strong quenching of AW fluorescence on binding to thrombin.
Figure 8.

Schematic representation of the interaction of the N-terminal tripeptide of hirudin analog Y3AW with thrombin. The inhibitor is color coded (carbon, gray; nitrogen, blue; oxygen, red); Tyr 60a and Trp 60d in the S2 site and Trp 215, Leu 99, and Ile 174, forming the apolar S3 site on thrombin, are shown in magenta. Water molecules at the hirudin–thrombin interface in the S2–S3 sites are indicated by green spheres. The model structure of Y3AW was obtained by keeping the position of all atoms unchanged and using the same dihedral angles that Tyr 3 has in the X-ray structure of wild-type hirudin complexed to thrombin (χ1 = −61°; χ2 = −56°; PDB code, 4HTC). Water molecule w432 (B-factor 36 Å2, occupancy 1.0) is in the same plane of the azaindole ring of AW at position 3, and N7 occupies the same position of the -OH group in Tyr 3, which is linked to Tyr 60a through a water bridge involving w606 (B-factor 22 Å2, occupancy 0.52).
At this point, a more detailed understanding of the environment-dependent fluorescence emission of 7AI (which is the fluorophore of AW) is necessary to interpret our results. The spectroscopic properties of 7AI were originally investigated as a model for studying the interactions in DNA base pairs (Taylor et al. 1969) and, since that time, the photo-physics of 7AI in different solvents has been the subject of numerous investigations (Chapman and Maroncelli 1992; Chou et al. 1992; Smirnov et al. 1997; Mente and Maroncelli 1998). The sensitivity of 7AI to its environment depends on either bulk or specific interactions with solvent (Smirnov et al. 1997). The bulk interactions are those typical of most indoles and find their origin in the large change in the electric dipole moment of the low-lying excited states with respect to the ground state. On the other hand, specific interactions of 7AI with the solvent are dominated by the hydrogen bond capability of the -NH group. In Figure 9A ▶, we have reported the fluorescence spectra of 7AI in different solvents, namely, diethyl ether, acetonitrile, n-propanol, water, and water-saturated diethyl ether. The emission of 7AI is shifted from 325 nm in cyclohexane (not shown) to 345 nm in diethyl ether, and to 362 nm in acetonitrile. In protic solvents, the signal intensity is remarkably reduced and the λmax value is shifted to longer wavelengths. In n-propanol the fluorescence spectrum of 7AI displays a prominent band at 367 nm, and a second band at 520 nm. In bulk water, the longer wavelength band disappears and the λmax is shifted to 400 nm. In water-saturated diethyl ether ([H2O]: 0.487 M) the λmax value is shifted to 355 nm, the intensity is reduced to about 16% of that measured in neat diethyl ether (Chapman and Maroncelli 1992), and an additional emission is observed at ~520 nm (Fig. 9A ▶, inset; Chou et al. 1992). Notably, the intensity of this band accounts for ~1%–2% of that measured at the λmax in water-saturated ether or in bulk water. A widely accepted interpretation of the peculiar spectroscopic properties of 7AI is schematically reported in Figure 9B ▶ and can be summarized as follows (Chou et al. 1992; Gordon 1996; Nakajima et al. 1997; Ross et al. 1997; Smirnov et al. 1997; Mente and Maroncelli 1998):
Figure 9.

(A) Fluorescence emission spectra of 7AI in different solvents: acetonitrile (open squares), diethyl ether (filled triangles), n-propanol (open circles), water-saturated diethyl ether (open triangles), and deionized water (filled circles). The emission of 7AI in n-propanol is multiplied 20-fold. (Inset) Fluorescence spectra of 7AI in diethyl ether (filled triangles) and water-saturated diethyl ether (open triangles) expanded by a factor of 80 to show the growth of the second band at ~520 nm in organic-water solution. All spectra were recorded at 25°C and 2 μM 7AI concentration, by exciting the samples at 280 nm. (B) Consensus scheme explaining the emission properties of 7AI in alcohol (R = alkyl) or water solution (R = −H). The polyhydrated species in water are more populated, and emit radiation at 400 nm. In water-restricted environments (e.g., water-saturated diethyl ether) the concentration of the “normal” monohydrated species increases and, on excitation, it is converted into the “tautomer” form, which is responsible for the longer wavelength emission at ~520 nm (Inset to panel A).
In protic solvents 7AI exists in equilibrium between monohydrate and polyhydrate species, the latter being more stable and thus highly represented in solution. This is due to the fact that in the (cyclically bonded) monohydrate species, a single water or alcohol molecule can make only weak, nonlinear hydrogen bonds to 7AI, whereas in bulk solvent, eight-membered ring or neighbor-bonded structures prevail simply because strong hydrogen bonds can be formed (see Fig. 9B ▶).
The formation of a 1:1 cyclically bonded 7AI/R-OH (R = H, alkyl) is a prerequisite for the formation of a “tautomer” species in the excited state, through double proton transfer reaction between N1-H and N7. In its ground electronic state, the normal structure of 7AI is predicted to be 10 kcal/mole more stable than the higher energy tautomer, whereas in the lowest excited state, the relative stabilities of the tautomers is reversed. Hence, the extent of tautomer formation in the excited state depends on the molar fraction of 7AI existing in solution as 1:1 7AI:H2O complex (monohydrate form). With respect to this, the energy barrier for tautomerization (~60 kcal/mole) is significantly reduced (20 kcal/mole) when the 1:1 complex is formed.
The “tautomer” form is responsible for the longer wavelength emission of 7AI observed at ~530 nm in alcohols and water-saturated ether. The absence of this longer wavelength band in bulk water simply reflects the low concentration of this “reactive” cyclically bonded species compared with that present in alcohols. In water-restricted environments, like water-saturated ether, the formation of a 1:1 7AI–water complex is less hampered, giving rise to a detectable 530-nm band (see inset to Fig. 9B ▶; Chou et al. 1992).
In the light of these considerations, the strong quenching effect on the fluorescence of 7AW, observed on binding of Y3AW to thrombin, can be explained on the basis of the tridimensional model structure of Y3AW bound to thrombin. As shown in Figure 8 ▶, the -NH group of AW interacts with water molecule w432 (B-factor: 36 Å2; occupancy: 1.0), whereas N7 may be linked to Tyr 60a through a water bridge involving the structural water molecule w606 (B-factor: 22 Å2; occupancy: 0.52; Rydel et al. 1991). Hence, the rigid, structural water molecules at the hirudin–thrombin interface can have a crucial role in quenching the fluorescence of 7AW, because they can promote the nonradiative decay of AW in the excited state more effectively than the labile water molecules solvating the 7AW in the free Y3AW analog. This interpretation relies on the quite reasonable assumption that the binding mode of Y3AW to thrombin is unchanged, compared with that of the wild-type fragment 1–47. In fact, the mutation introduced is too “conservative” to produce an extensive rearrangement at the enzyme–inhibitor interface. With respect to this, Tyr 3 →AW exchange leads to an increase in the side-chain volume of only 15%. In addition, the conformation of the specificity sites of thrombin in the crystal structure of the hirudin–thrombin complex (Rydel et al. 1991; PDB code: 4HTC) is closely similar to those they acquire in the hirugen–thrombin complex, where either the active site or the specificity sites of the enzyme are unoccupied (Vijayalakshmi et al. 1994; PDB code 1HAH), suggesting that in our case the structural plasticity of the enzyme is low. Further experimental support of our interpretation of the fluorescence quenching of Y3AW would come from the comparison of the fluorescence lifetime values of AW in the Y3AW–thrombin complex with those of the model compound 7AI in different solvent conditions.
In conclusion, the data herein reported demonstrate that solid-phase synthesis is a convenient method to introduce AW into even long polypeptide chains, and that the incorporation of this Trp analog can be of broad applicability in structure–activity relationship studies, where a Trp isostere is required, or as a spectroscopic probe in the study of protein folding and protein–protein recognition.
Materials and methods
Materials
Nα-Fmoc-protected amino acids, solvents, and reagents for peptide synthesis were purchased from Applied Biosystems or from Advanced Chemtech. D,L-AW, indole and 7AI, cyanogen bromide, and BSA were from Sigma. TFA, β-ME, acetic anhydride, neat acetic acid, PEG-8000, EDT, DTT, octanol, and eupergit-C immobilized acylase-I from Asperigillus oryzae were from Fluka. Human α-thrombin was from Calbiochem. Sepharose-4B was purchased from Merck. L-arginine-p-nitroanilide was from Bachem AG. All other reagents, buffers, and organic solvents were of analytical grade and obtained from Fluka. Water used for preparing all of the solutions was prepared with the coupled Milli-Rho/Milli-Q water production system from Millipore.
Methods
Preparation of L-7-azatryptophan
D,L-AW racemate was resolved by enantioselective hydrolysis of Nα-acetyl-derivative of D,L-AW by eupergit-C immobilized acyl-ase-I from Asperigillus oryzae (Chenault et al. 1989). To a solution of 205 mg of D,L-AW (1 mmole) in 2.56 mL (45 mmole) neat acetic acid, 114 μL of acetic anhydride (1.2 mmole) was added under vigorous stirring. The reaction was allowed to proceed at room temperature under stirring and monitored by RP-HPLC. At regular time intervals, aliquots (10 μL) of the reaction mixture were loaded onto a Waters C-18 Pico-Tag (3.9 × 150 mm, 5-μm particle size, 80–100 Å porosity) analytical column eluted with a linear acetonitrile-0.1% TFA gradient from 1% to 30% in 10 min at a flow rate of 0.8 mL/min, recording the absorbance of the effluent at 280 nm. After a 2-h reaction, 114 μL of acetic anhydride was added, and the reaction was terminated after 5 h by adding 0.5 mL of ice cold acetone. The reaction mixture was vacuum dried in a Speedvac concentrator (Savant) and the resulting microcrystalline solid of Nα-acetyl-D,L-AW was dissolved in 20 mL of 5 mM sodium phosphate buffer (pH 7.7). The pH value was kept constant by adding 1 M NaOH. To this solution, 112 mg of eupergit-C immobilized acylase-I was added. The time-dependent deacylation reaction of Nα-acetyl-AW was monitored by RP-HPLC, using the chromatographic conditions reported earlier. The reaction was stopped after 48 h. The mixture was centrifuged at 14,000 r.p.m. to separate the eupergit-bound enzyme, and the solution phase (containing L-AW) was purified by semipreparative RP-HPLC on a Vydac (The Separation Group) C-18 (1 × 10 cm, 10-μm granulometry) column, eluted with a linear acetonitrile-0.1% TFA gradient from 1% to 10% in 15 min at a flow rate of 2.5 mL/min. After purification and vacuum drying on P2O5, 95 mg of lyophilized L-AW was obtained, for a final yield of ~90%, calculated on the amount of the L-enantiomer present in the racemic mixture.
The optical purity of L-AW was established by chiral chromatography on an albumin-sepharose column, prepared by BrCN-activation of sepharose-4B gel and subsequent covalent binding to BSA (Allenmark and Bomgren 1982). To a suspension of swollen sepharose-4B gel (50 mL) in 2 M sodium carbonate buffer (pH 12) was added 7.5 mL of an acetonitrile solution containing 11 g of BrCN. The activation reaction was conducted for about 2 min under gentle stirring at room temperature. Thereafter, the resin was filtered and washed fivefold with 200 mL of cold 0.1 M sodium bicarbonate buffer (pH 8.3). The activated sepharose-4B was reacted with 40 mL of a BSA solution (50 mg/mL) in the same buffer at room temperature for 6 h under gentle stirring and at 4°C for 15 h without stirring. The gel was extensively washed with 50 mM sodium phosphate buffer (pH 7.0), and the BSA-sepharose stationary phase was used to pack a 1 × 20-cm column. The en-antiomeric purity of our L-AW preparation was established by loading 5 μg of the purified L-AW onto the albumin-sepharose column connected to an FPLC instrument (Pharmacia) and eluted at a constant flow rate of 0.8 mL/min with 20 mM sodium phosphate buffer (pH 8.0). The absorbance of the effluent was recorded at 280 nm. The optical purity of L-AW was further checked by the method of Josefsson et al. (1987), using FLEC as a chiral precolumn derivatization reagent.
Synthesis of Nα-Fmoc-(L)-7-azatryptophan
The corresponding Nα-Fmoc-AW was obtained by using Fmoc-ONSu as an acylating agent, according to the method of Ten-Kortenaar et al. (1986). Briefly, 158 mg (467 mmole) of Fmoc-ONSu in acetonitrile (1.14 mL) was added to an aqueous solution (1.5 mL) of L-AW (104 mg, ~0.5 mmole) containing 0.75 mmole (67 μL) of TEA. The reaction was allowed to proceed for 45 min, keeping the pH value constant at 8.5. After that, acetonitrile was eliminated under vacuum. To the remaining solution was added 20 mL of 5% sodium bicarbonate (pH 8.0), and the excess Fmoc-ONSu was extracted with cold diethyl ether. The aqueous phase was acidified to pH 3.0 with 1 M KHSO4, and the Fmoc-protected amino acid extracted with ethyl acetate. The organic phase was extensively washed with water, treated with anhydrous sodium sulfate, filtered, and evaporated under vacuum, yielding 86 mg of Nα-Fmoc-(L)-AW. Time-course kinetics of derivatization reaction was monitored by RP-HPLC on a Waters Pico-Tag C18 column eluted with a linear acetonitrile-0.1% TFA gradient from 1% to 50 % in 30 min, at a flow rate of 0.8 mL/min, recording the absorbance of the effluent at 280 nm. The homogeneity and chemical identity of Nα-Fmoc-AW was confirmed by RP-HPLC analysis, high-resolution mass spectrometry on a Mariner ESI-TOF spectrometer (Perseptive Biosystems), by 1H-NMR on a model AMX-300 instrument from Bruker AG, and by FT-IR on a model 1760 instrument (Perkin-Elmer). Melting point determinations were performed with a Gallenkamp apparatus, according to the manifacturer’s procedures.
Synthesis and characterization of the 7-azatryptophan analog of hirudin fragment 1–47
The Y3AW analog was synthesized by the solid-phase Fmoc-method (Atherton and Sheppard 1989) in two sequential steps on a Lys-derivatized (0.67 mmole/g of resin) p-alkoxybenzylester polystyrene resin as previously described (De Filippis et al. 1995, 1998a). In the first step, the peptide chain corresponding to the sequence 6–47 was assembled stepwise using a model 431 automated peptide synthesizer (Applied Biosystems). After the assembly of the peptide chain 6–47 was completed, the coupling of the remaining five amino acids, including Fmoc-azaTrp, was accomplished with 100 mg (10 μmole) of the side-chain-protected pep-tidyl 6–47 resin by manual chemical synthesis using the HBTU/HOBt activation procedure and a fourfold molar excess of protected amino acids. Cleavage of the peptide from the resin and side-chain deprotection was simultaneously achieved by treatment for 90 min at 0°C with a mixture of TFA/H2O/EDT (95:2.5:2.5, v/v). The crude reduced peptides, obtained after precipitation with ice-cold diethylether, were dissolved (2 mg/mL) in 0.1 M NaHCO3 buffer (pH 8.3) and allowed to fold for 24 h under air-oxidation conditions in the presence of 100 μM β-ME (Chatrenet and Chang 1992; De Filippis et al. 1995). The refolded disulfide-oxidized peptides were purified by preparative RP-HPLC on a Vydac C18 column (1 × 25 cm, 10-μm particle size), eluted with a linear acetonitrile-0.05% TFA gradient, and their chemical identity established by high-resolution mass spectrometry, which gave mass values in agreement with the expected amino acid composition within 10 ppm mass accuracy. The correctness of disulfide pairing was established by enzymatic fingerprint analysis, as previously described (De Filippis et al. 1995).
Determination of protein concentration
The concentration of the synthetic analogs Y3W and Y3AW was determined by UV absorption at 280 nm on a double beam model Lambda-2 spectrophotometer from Perkin-Elmer. Molar absorptivity at 280 nm for the Y3W and Y3AW analogs was calculated as 7330 and 7610 M−1•cm−1, respectively, using a molar absorption coefficient of 1280 M−1•cm−1 for tyrosine, 5690 for W, 120 M−1•cm−1 for cysteine (Gill and von Hippel 1989), and 5970 M−1•cm−1 for AW (Wong and Eftink 1997). The concentration of thrombin was determined by using a molar absorptivity at 280 nm of 66,795 M−1•cm−1 (Fenton II et al. 1977).
Circular dichroism
CD spectra were recorded on a Jasco model J-710 spectropolarimeter equipped with a thermostated cell holder and a NesLab model RTE-110 water circulating bath. Far- and near-UV CD spectra were recorded at 25°C in 10 mM phosphate buffer (pH 7.0) at a peptide concentration ranging from 50 to 150 μM, using 1- or 5-mm pathlength quartz cells in the far- and near-UV region, respectively.
Fluorescence
Fluorescence emission spectra were recorded at 25°C on a Perkin-Elmer spectrofluorimeter model LS-50B, equipped with a thermostated cell holder, at a scan rate of 240 nm/min, and using an excitation/emission slit of 5 and 10 nm, respectively. Unless otherwise specified, all spectra were recorded at a protein concentration of 2 μM in 5 mM Tris-HCl buffer (pH 8.0), containing 0.1 % PEG-8000 and resulting from the average of four accumulations after baseline subtraction.
The interaction of Y3AW with thrombin was monitored by recording the fluorescence spectra of the enzyme and inhibitor before and after complex formation. Typically, the emission spectrum of the isolated Y3AW analog was obtained by adding 7 μL of a 62-μM stock solution of Y3AW in water to 177 μL of 5 mM Tris-HCl buffer (pH 8.0), containing 0.1% PEG-8000, together with 16 μL of the same buffer, to get a final concentration of 2 μM in 0.2 mL. The emission fluorescence spectrum of thrombin was obtained by adding 16 μL of a 25-μM stock solution of thrombin in 20 mM Tris-HCl/0.1 M NaCl (pH 7.4) to 177 μL of 5 mM Tris-HCl buffer (pH 8.0) containing 0.1% PEG-8000. Thereafter, 7 μL of the synthetic 1–47 analog was added to the thrombin solution and left to incubate for 5 min. The spectra were recorded by exciting the samples at 320 nm and collecting the fluorescence signal from 330 to 550 nm.
Thrombin inhibitory activity
Binding of hirudin N-terminal fragments was quantified from analysis of the competitive inhibition for the hydrolysis of D-Phe-Pro-Arg-pNA (FPR) substrate (Ayala and Di Cera 1994), following the procedure already described (De Filippis et al. 1998a). The chromogenic substrate was synthesized in our laboratory, as previously described (De Filippis et al. 2002) Measurements were carried out at 25°C in 5 mM Tris (pH 8.0), containing 0.1% (w/w) PEG-8000, and 0.2 M NaCl. The active thrombin concentration was 100 pM, the substrate concentration was fixed at 20 μM, and the inhibitor concentration was in the 0.4- to 7-μM range.
Determination of logP values
Octanol-water partition coefficients (P) for indole and 7AI were determined according to the shake-flask method (Leo et al. 1971). The aqueous phase (5 mM Tris-HCl buffer at pH 8.0) was saturated with octanol by gently shaking for 5 min at room temperature (24°–25°C) equal volumes (10 mL) of Tris-buffer and octanol in a 50-mL polypropylene tube. Indole or 7AI (104 μmole, ~12 mg) was dissolved in 2 mL of buffer-saturated octanol. Thereafter, 8 mL of Tris-buffer was added, and the aqueous/organic mixture was gently shaken in a 10-mL tube as described earlier. The concentration of indole and 7AI was determined from the absorbance of the corresponding aqueous phase at 280, by using an extinction coefficient, measured in water at pH 7.0, of 6770 and 7050 M−1•cm−1 for indole and 7AI, respectively (Chen et al. 1993).
Computational methods
The structure of the synthetic analogs of fragment 1–47 of hirudin HM2 in the free state was modeled on the NMR solution structure of natural fragment 1–47 (Nicastro et al. 1997), using the Insight-II software run on a Silicon Graphics O2 workstation. The best representative model in the NMR ensemble was selected using the program OLDERADO (Kelley and Sutcliffe 1997) available online at the site http://neon.chem.le.ac.uk/. The structure of the synthetic analogs in the thrombin-bound state was modeled on the 2.1-Å resolution crystallographic structure of the hirudin–thrombin complex (Rydel et al. 1991; PDB entry code, 4htc).
Acknowledgments
The authors gratefully thank Professor G. Zagotto for performing NMR spectra of Fmoc-AW. This work was supported in part by a Grant (PRIN-2000) from the Italian Ministry of University and Scientific Research to V.D.F. Part of this work was presented at the 45th National Conference of the Italian Society of Biochemistry and Molecular Biology, SIB 2000, Naples (Italy) September 20–23, 2000. Commun. A92.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
7AI, 7-azaindole
a.m.u., atomic mass units
AW, (L)-7-azatryptophan
W, (L)-tryptophan
BSA, bovine serum albumin
CD, circular dichroism
Cha, cyclohexylalanine
ChCl, choline chloride
DTT, dithiothreitol
EDT, ethanedithiol
ESI, electrospray ionization
FLEC, (+)-1–(9-fluorenyl)ethylchloroformate
Fmoc, 9-fluorenyl-methyl-oxycarbonyl
Fmoc-ONSu, 9-fluorenyl-methyloxycarbonyl-O-N-hydroxy-succinimide
FPLC, fast protein liquid chromatography
FT-IR, Fourier transformed infrared
HBTU, 2–(1H-benzotriazol-1-yl-)-1,1,3,3-tetrameth-yluronium hexafluorophosphate
HOBt, 1-hydroxybenzotriazole
HPLC, high-pressure liquid chromatography
β-ME, β-mercaptoethanol
MES, morpholinoethanesulfonic acid
NMR, nuclear magnetic resonance
PEG, polyethylene glycol
RP, reverse-phase
TEA, triethylamine
TFA, trifluoroacetic acid
UV, ultraviolet
TOF, time of flight
Y3AW, synthetic analog of the hirudin HM2 fragment 1–47 in which Tyr 3 has been replaced by L-7-azatryptophan
Y3W, synthetic analog of the hirudin HM2 fragment 1–47 in which Tyr 3 has been replaced by tryptophan
HM2, hirudin variant isolated from the leech Hirudinaria manillensis
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03542104.
References
- Allenmark, S. and Bomgren, B. 1982. Resolution of enantiomers by liquid affinity chromatography on albumin-agarose under isocratic conditions. J. Chromatogr. 237 473–477. [Google Scholar]
- Atherton, E. and Sheppard, R.C. 1989. Solid phase peptide synthesis: A practical approach. IRL Press, Oxford, UK.
- Ayala, Y. and Di Cera, E. 1994. Molecular recognition by thrombin. Role of the slow →fast transition, site-specific ion binding energetics and thermodynamic mapping of structural components. J. Mol. Biol. 235 733–746. [DOI] [PubMed] [Google Scholar]
- Berliner, L.J. and Shen, Y.Y.L. 1977. Physical evidence for an apolar binding site near the catholytic center of human α-thrombin. Biochemistry 16 4622–4626. [DOI] [PubMed] [Google Scholar]
- Betz, A., Hofsteenge, J., and Stone, S.R. 1992. Interaction of the N-terminal region of hirudin with the active-site cleft of thrombin. Biochemistry 31 4557–4562. [DOI] [PubMed] [Google Scholar]
- Blouse, G.E., Perron, M.J., Thompson, J.H., Day, D.E., Link, C.A., and Shore, J.D. 2002. A concerted structural transition in the plasminogen activator inhibitor-1 mechanism of inhibition. Biochemistry 41 11997–12009. [DOI] [PubMed] [Google Scholar]
- Bode, W., Turk, D., and Karshikov, A. 1992. The refined 1.9-Å X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human α-thrombin: Structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1 426–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler, D.R., Haas, E., and Scheraga, H.A. 1995. Analysis of the structure of ribonuclease A in native and partially denatured states by time-resolved nonradiative dynamic excitation energy transfer between site-specific extrinsic probes. Biochemistry 34 15965–15978. [DOI] [PubMed] [Google Scholar]
- Budisa, N., Minks, C., Medrano, F.J., Lutz, J., Huber, R., and Moroder, L. 1998. Residue-specific bioincorporation of non-natural, biologically active amino acid into proteins as possible drug carriers: Structure and stability of the per-thiaproline mutant of annexin V. Proc. Natl. Acad. Sci. 95 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, C.F. and Maroncelli, M. 1992. Excited-state tautomerization of 7-azaindole in water. J. Phys. Chem. 96 8430–8441. [Google Scholar]
- Chatrenet, B. and Chang, J.-Y. 1992. The folding of hirudin adopts a mechanism of trial and error. J. Biol. Chem. 267 3038–3043. [PubMed] [Google Scholar]
- Chen, Y., Rich, R.L., Gai, F., and Petrich, J.W. 1993. Fluorescent species of 7-azaindole and 7-azatryptophan in water. J. Phys. Chem. 97 1770–1780. [Google Scholar]
- Chen, Y., Gai, F., and Petrich, J.W. 1994. Single-exponential fluorescence decay of the nonnatural amino acid 7-azatryptophan and the nonexponential decay of tryptophan in water. J. Phys. Chem. 98 2203–2209. [Google Scholar]
- Chenault, H.K., Dahmer, J., and Whitesides, G.M. 1989. Kinetic resolution of unnatural and rarely occurring amino acids: Enantioselective hydrolysis of N-acyl amino acids catalyzed by acylase I. J. Am. Chem. Soc. 111 6354–6364. [Google Scholar]
- Chou, P.-T., Martinez, M.L., Cooper, W.C., McMorrow, D., Collins, S.T., and Kasha, M. 1992. Monohydrate catalysis of excited-state double-proton transfer in 7-azaindole. J. Phys. Chem. 96 5203–5205. [Google Scholar]
- Cohen, B.E., McAnaney, T.B., Park, E.S., Jan, Y.N., Boxer, S.G., and Jan, L.Y. 2002. Probing protein electrostatics with a synthetic fluorescent amino acid. Science 296 1700–1703. [DOI] [PubMed] [Google Scholar]
- Cornish, V.W., Benson, D.R., Altenbach, C.A., Hideg, K., Hubbell, W.L., and Schultz, P.G. 1994. Site-specific incorporation of biophysical probes into proteins. Proc. Natl. Acad. Sci. 91 2910–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish, V.W, Mendel, D., and Schultz, P.G. 1995. Probing protein structure and function with an expanded genetic code. 1995. Angew. Chem. Int. Ed. Engl. 34 621–633. [Google Scholar]
- Davie, E.W., Fujikawa, K., and Kisiel, W. 1991. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 30 10363–10370. [DOI] [PubMed] [Google Scholar]
- De Filippis, V., Vindigni, A., Altichieri, L., and Fontana A. 1995. Core domain of hirudin from leech Hirudinaria manillensis: Chemical synthesis, purification and characterization of a Trp3 analog of fragment 1–47. Biochemistry 34 9552–9564. [DOI] [PubMed] [Google Scholar]
- De Filippis, V., Quarzago, D., Vindigni, A., Di Cera, E., and Fontana, A. 1998a. Synthesis and characterization of more potent analogues of hirudin fragment 1–47 containing non-natural amino acids. Biochemistry 37 13507–13515. [DOI] [PubMed] [Google Scholar]
- De Filippis, V., De Antoni, F., Frigo, M., Polverino de Laureto, P., and Fontana, A. 1998b. Protein stabilization by Ala → Aib replacements. Biochemistry 37 1686–1696. [DOI] [PubMed] [Google Scholar]
- De Filippis, V., Colombo, G., Russo, I., Spadari, B., and Fontana, A. 2002. Probing hirudin-thrombin interaction by incorporation of noncoded amino acids and molecular dynamics simulation. Biochemistry 43 1537–1550. [DOI] [PubMed] [Google Scholar]
- Desinov, V.P., Peters, J., Hörlein, H.D., and Halle, B. 1996. Using buried water molecules to explore the energy landscape of proteins. Nat. Struct. Biol. 3 505–509. [DOI] [PubMed] [Google Scholar]
- Di Cera, E., Dang, Q.D., and Ayala, Y.M. 1997. Molecular mechanisms of thrombin function. Cell. Mol. Life Sci. 53 701–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty, D.A. 2000. Unnatural amino acids as probes of protein structure and function. Curr. Opin. Chem. Biol. 4 645–652. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. 1997. Fluorescence methods for studying equilibrium macromoleculeligand interactions. Methods Enzymol. 278 221–257. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. and Shastry, M.C.R. 1997. Fluorescence methods for studying kinetics of protein folding reactions. Methods Enzymol. 278 258–286. [DOI] [PubMed] [Google Scholar]
- Esmon, C.T., Fukudome, K., Mather, T., Bode, W., Regan, L.M., Stearns-Kurosawa, D.J., and Kurosawa, S. 1999. Inflammation, sepsis, and coagulation. Trends Hematol. 84 254–259. [PubMed] [Google Scholar]
- Fenton II, J.W., Fasco, M.J., and Stackrow, A.B. 1977. Human thrombins: Production, evaluation, and properties of α-thrombin. J. Biol. Chem. 252 3587–3598. [PubMed] [Google Scholar]
- Fersht, A. and Winter, G. 1992. Protein engineering. Trends Biochem. Sci. 17 292–294. [DOI] [PubMed] [Google Scholar]
- Freedman, R.B. 1971. Applications of the chemical reactions of proteins in studies of their structure and function. Q. Rev. Chem. Soc. 25 431–462. [Google Scholar]
- Garcia, A.E. and Hummer, G. 2000. Water penetration and escape in proteins. Proteins 38 261–272. [PubMed] [Google Scholar]
- Gill, S.G. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Gordon, M.S. 1996. Hydrogen transfer in 7-azaindole. J. Phys. Chem. 100 3974–3979. [Google Scholar]
- Greenstein, J.P. 1957. Resolution of DL mixtures of α-amino acids. Methods Enzymol. 3 554–570. [Google Scholar]
- Haruyama, H. and Wüthrich, K. 1989. Conformation of recombinant desulfatohirudin in aqueous solution determined by nuclear magnetic resonance. Biochemistry 28 4301–4312. [DOI] [PubMed] [Google Scholar]
- Hu, Z., Ma, B., Wolfson, H., and Nussinov, R. 2000. Conservation of polar residues as hot spots at protein interfaces. Proteins 39 331–342. [PubMed] [Google Scholar]
- Huang, K., Lu, W., Anderson, S., Laskowski Jr., M., and James, M.N.G. 1995. Water molecules participate in proteinase-inhibitor interactions. Protein Sci. 4 1985–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittah, V. and Haas, E. 1995. Nonlocal interactions stabilize long range loops in the initial folding intermediates of reduced bovine pancreatic trypsin inhibitor. Biochemistry 34 4493–4506. [DOI] [PubMed] [Google Scholar]
- Josefsson, B., Einarsson, S., Moller, P., and Sanchez, D. 1987. Separation of amino acid enantiomers and chiral amines using precolumn derivatization with (+)-1–(9-fluorenyl)ethylchloroformate and reversed-phase liquid chromatography. Anal. Chem. 59 1191–1195. [DOI] [PubMed] [Google Scholar]
- Kahn, P.C. 1979. The interpretation of near-ultraviolet circular dichroism. Methods Enzymol. 61 339–378. [DOI] [PubMed] [Google Scholar]
- Kay, L.E. 1998. Protein dynamics from NMR. Nat. Struct. Biol. Suppl: 513–517. [DOI] [PubMed]
- Kelley, L.A. and Sutcliffe, M.J. 1997. OLDERADO: On-line database of ensemble representatives and domains. Protein Sci. 6 2628–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, S.B. 1988. Chemical synthesis of peptides and proteins. Annu. Rev. Biochem. 57 957–989. [DOI] [PubMed] [Google Scholar]
- Lacowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Kluwer Academic/Plenum, New York.
- Lecointe, L., Rolland-Fulcrand, V., Roumestant, M.L., Viallefont, P., and Martinez, J. 1998. Chemoenzymatic synthesis of the two enantiomers of 7-azatryptophan. Tetrahedron 9 1753–1758. [Google Scholar]
- Leo, A., Hansch, C., and Elkins, D. 1971. Partition coefficients and their uses. Chem. Rev. 71 525–555. [Google Scholar]
- Lo Conte, L., Chothia, C., and Janin, J. 1999. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285 2177–2198. [DOI] [PubMed] [Google Scholar]
- Markwardt, F. 1994. The development of hirudin as an antithrombotic drug. Thromb. Res. 741–23. [DOI] [PubMed] [Google Scholar]
- Mente, S. and Maroncelli, M. 1998. Solvation and the excited-state tautomerization of 7-azaindole and 1-azacarbazole: Computer simulations in water and alcohol solvents. J. Phys. Chem. 102 3860–3876. [Google Scholar]
- Mohammadi, F., Prentice, G.A., and Merrill, A.R. 2001. Protein-protein interaction using tryptophan analogues: Novel spectroscopic probes for toxin-elongation factor-2 interactions. Biochemistry 40 10273–10283. [DOI] [PubMed] [Google Scholar]
- Nakajima, A., Hirano, M., Hasumi, R., Kaya, K., Watanabe, H., Carter, C.C., Williamson, J.M., and Miller, T.A. 1997. High-resolution laser-induced fluorescence spectra of 7-azaindole-water complexes and 7-azaindole dimmer. J. Phys. Chem. A 101 392–398. [Google Scholar]
- Negrerie, M., Gai, F., Bellefeuille, S.M., and Petrich, J.W. 1991. Photophysics of a novel optical probe: 7-Azaindole. J. Phys. Chem. 95 8663–8670. [Google Scholar]
- Nicastro, G., Baumer, L., Bolis, G., and Tatò, M. 1997. NMR solution structure of a novel hirudin variant HM2, N-terminal 1–47 and N64 → V + G mutant. Biopolymers 41 731–749. [DOI] [PubMed] [Google Scholar]
- Rich, R.L., Negrerie, M., Li, J., Elliott, S., Thornburg, R.W., and Petrich, J.W. 1993. The photophysical probe, 7-azatryptophan, in synthetic peptides. Photochem. Photobiol. 58 28–30. [Google Scholar]
- Rich, R.L., Gai, F., Lane, J.W., Petrich, J.W., and Schwabacher, A.W. 1995. Using 7-azatryptophan to probe small molecule-protein interactions on the picosecond time scale: The complex of avidin and biotinylated 7-azatryptophan. J. Am. Chem. Soc. 117 733–739. [Google Scholar]
- Rischel, C. and Poulsen, F.M. 1995. Modification of a specific tyrosine enables tracing of the end-to-end distance during apomyoglobin folding. FEBS Lett. 374 105–109. [DOI] [PubMed] [Google Scholar]
- Ross, A., Szabo, A.G., and Hogue, C.W.V. 1997. Enhancement of protein spectra with tryptophan analogs: Fluorescence spectroscopy of protein-protein and protein-nucleic acid interactions. Methods Enzymol. 278 151–190. [DOI] [PubMed] [Google Scholar]
- Rydel, T.J., Tulinsky, A., Bode, W., and Huber, R. 1991. Refined structure of the hirudin-thrombin complex. J. Mol. Biol. 221 583–601. [DOI] [PubMed] [Google Scholar]
- Scacheri, E., Nitti, G., Valsasina, B., Orsini, G., Visco, C., Ferreia, M., Sawyer, R.T., and Sarmientos, P. 1993. Novel hirudin variants from the leech Hirudinaria manillensis. Amino acid sequence, cDNA cloning and genomic organization. Eur. J. Biochem. 214 295–304. [DOI] [PubMed] [Google Scholar]
- Schlesinger, S. 1968. The effect of amino acid analogues on alkaline phosphatase formation in Escherichia coli K-12. J. Biol. Chem. 243 3877–3883. [PubMed] [Google Scholar]
- Scott, D.J., Leejeerajumnean, S., Brannigan, J.A., Lewis, R.J., Wilkinson, A.J., and Hoggett, J.G. 1999. Quaternary re-arrangement analysed by spectral enhancement: The interaction of sporulation repressor with its antagonist. J. Mol. Biol. 293 997–1004. [DOI] [PubMed] [Google Scholar]
- Shaltiel, A., Cox, S., and Taylor, S.S. 1998. Conserved water molecules contribute to the extensive network of interactions at the active site of protein kinase A. Proc. Natl. Acad. Sci. 95 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov, A.V., English, D.S., Rich, R.L., Lane, J., Teyton, L., Schwabacher, A.W., Luo, S., Thorburg, R.W., and Petrich, J.W. 1997. Photophysics and biological applications of 7-azaindole and its analogs. J. Phys. Chem. B 101 2758–2769. [Google Scholar]
- Soumillon, P., Jespers, L., Vervoort, J., and Fastrez, J. 1995. Biosynthetic incorporation of 7-azatryptophan into the phage lambda lysozyme: Estimation of tryptophan accessibility, effect on enzymatic activity and protein stability. Protein Eng. 8 451–456. [DOI] [PubMed] [Google Scholar]
- Stewart, K.K. and Doherty, R.F. 1973. Resolution of D,L-tryptophan by affinity chromatography on bovine-serum albuminagarose columns. Proc. Natl. Acad. Sci. 70 2850–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland, E.H. 1974. Aromatic contributions to circular dichroism spectra of proteins. CRC Crit. Rev. Biochem. 3 113–175. [DOI] [PubMed] [Google Scholar]
- Szyperski, T., Güntert, P., Stone, S.R., and Wütrich, K. 1992. Nuclear magnetic resonance solution structure of hirudin (1–51) and comparison with corresponding tridimensional structures determined using the complete 65-residue hirudin polypeptide chain. J. Mol. Biol. 228 1193–1205. [DOI] [PubMed] [Google Scholar]
- Taylor, C.A., El-Boyoumi, M.A., and Kasha, M. 1969. Excited-state two proton tautomerism in hydrogen-bonded N-heterocyclic base pairs. Proc. Natl. Acad. Sci. 63 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkasskaya, O. and Ptitsyn, O.B. 1999. Direct energy transfer to study the 3D structure of non-native proteins: AGH complex in the molten globule state of apomyoglobin. Protein Eng. 12 485–490. [DOI] [PubMed] [Google Scholar]
- Ten-Kortenaar, P.B.W., Van Dijk, B.G., Peeters, M.J., Raabe, B.J., Adams, P.J., and Tesser, G.I. 1986. Rapid and efficient method for the preparation of Fmoc-amino acids starting from 9-fluorenylmethanol. Int. J. Pept. Protein Res. 27 398–400. [Google Scholar]
- Twine, S.M. and Szabo, A.G. 2003. Fluorescent amino acid analogs. Methods Enzymol. 360 104–127. [DOI] [PubMed] [Google Scholar]
- Vijayalakshmi, J., Padmanabhan, K.P., Mann, K.G., and Tulinsky, A. 1994. The isomorphous structures of prethrombin2, hirugen-, and PPACK-thrombin: Changes accompanying activation and exosite binding to thrombin. Protein Sci. 3 2254–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindigni, A., De Filippis, V., Zanotti, G., Visco, C., Orsini, G., and Fontana, A. 1994. Probing the structure of hirudin from Hirudinaria manillensis by limited proteolysis: Isolation, characterization and thrombin-inhibitory properties of N-terminal fragments. Eur. J. Biochem. 226 323–333. [DOI] [PubMed] [Google Scholar]
- Wallace, C.J. 1993. Understanding cytochrome c function: Engineering protein structure by semisynthesis. FASEB J. 7 505–515. [DOI] [PubMed] [Google Scholar]
- Wong, C.-Y. and Eftink, M.R. 1997. Biosynthetic incorporation of tryptophan analogues into staphylococcal nuclease: Effect of 5-hydroxytryptophan and 7-azatryptophan on structure and stability. Protein Sci. 6 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1998. Incorporation of tryptophan analogues into staphylococcal nuclease: Stability toward thermal and guanidine-HCl induced unfolding. Biochemistry 37 8947–8953. [DOI] [PubMed] [Google Scholar]
- Wu, P. and Brand, L. 1994. Resonance energy transfer: Methods and applications. Anal. Biochem. 18 1–13. [DOI] [PubMed] [Google Scholar]
- Yamashita, S., Nishimoto, E., Szabo, A.G., and Yamasaki, N. 1996. Steady-state and time-resolved fluorescence studies on the ligand-induced conformational change in an active derivative, Kyn62-lysozyme. Biochemistry 35 531–537. [DOI] [PubMed] [Google Scholar]