

Abstract
Site-directed mutagenesis has frequently been used to replace proline with other amino acids in order to determine if proline isomerization is responsible for a slow phase during refolding. Replacement of Pro 85 with alanine in cellular retinoic acid binding protein I (CRABP-I) abolished the slowest refolding phase, suggesting that this phase is due to proline isomerization in the unfolded state. To further test this assumption, we mutated Pro 85 to valine, which is the conservative replacement in the two most closely related proteins in the family (cellular retinoic acid binding protein II and cellular retinol binding protein I). The mutant protein was about 1 kcal/mole more stable than wild type. Retinoic acid bound equally well to wild type and P85V-CRABP I, confirming the functional integrity of this mutation. The refolding and unfolding kinetics of the wild-type and mutant proteins were characterized by stopped flow fluorescence and circular dichroism. The mutant P85V protein refolded with three kinetic transitions, the same number as wild-type protein. This result conflicts with the P85A mutant, which lost the slowest refolding rate. The P85V mutation also lacked a kinetic unfolding intermediate found for wild-type protein. These data suggest that proline isomerization may not be responsible for the slowest folding phase of CRABP I. As such, the loss of a slow refolding phase upon mutation of a proline residue may not be diagnostic for proline isomerization effects on protein folding.
Keywords: proline isomerization; protein folding; folding intermediates; β-sheet proteins; structural homology; stopped-flow kinetics, iLBP
The folding of the intracellular lipid binding proteins (iLBPs) has been the subject of considerable research (e.g., Burns et al. 1998; Clark et al. 1998; Dalessio and Ropson 2000; Hodsdon and Frieden 2001; Yeh et al. 2001). These small (14–16 kD, 127–136 amino acids) predominantly β-sheet proteins function in the intracellular trafficking of small amphipathic ligands (Banaszak et al. 1994; Zimmerman and Veerkamp 2002). The structures of these proteins are very similar, consisting of 10 antiparallel β-strands and two short α-helices (Banaszak et al. 1994). The topology of the protein is a simple β-meander. The first two strands are connected by a helix–turn–helix motif, and the remaining strands are connected by reverse turns (Fig. 1 ▶). The β-strands are organized into two nearly orthogonal β-sheets surrounding a large internal ligand-binding cavity, which is filled with solvent in the absence of ligand. Only one of these proteins has a disulfide bond and all of the prolines are in the trans configuration in the native structures (Banaszak et al. 1994).
Figure 1.

Structure of CRABP I (1cbi, Kleywegt et al. 1994; Thompson et al. 1995) emphasizing the local structure and important residues near proline 85. The right panel is a 90° rotation of the left panel.
The folding of these proteins is completely reversible with denaturants under most solvent conditions (Clark et al. 1996; Burns et al. 1998; Dalessio and Ropson 2000; Burns and Ropson 2001) and fits a two-state model for the folding process at equilibrium. However, stopped-flow experiments have shown that at least one intermediate was present on the unfolding and refolding pathways for most of these proteins (Clark et al. 1996; Burns et al. 1998; Dalessio and Ropson 2000; Burns and Ropson 2001). The structural properties of the intermediates observed during unfolding fall into two classes. Some of the proteins have intermediates with the spectral characteristics of molten globules (nativelike circular dichroism [CD] spectra but nonnative fluorescence). Other proteins in the family have intermediates that lack secondary structure by CD criteria, but have fluorescence spectral properties that are different from the unfolded state (Burns et al. 1998; Dalessio and Ropson 2000; Burns and Ropson 2001). As such, it appears that the same final structure may be achieved by different mechanisms. There are differences in the number of observed intermediates, the spectral properties of those intermediates, and the rate at which folding and unfolding occurs among these proteins, suggesting that the energetic landscapes for the folding and unfolding of these sequences are quite different, despite their similar final structures.
One member of the family, cellular retinoic acid binding protein I (CRABP-I) both folds and unfolds significantly slower than other family members, resulting in a similar stability in the absence of urea. The refolding of this protein shows four distinct kinetic phases, a burst phase, followed by three additional observed phases (Burns et al. 1998; Clarke et al. 1998; Eyles and Gierasch 2000; Burns and Ropson 2001). The slowest refolding phase has a small amplitude, minimal dependence of the rate on the final denaturant concentration, and is slow enough to be caused by a proline isomerization event in the unfolded state (Nall 1994). There are four prolines in CRABP-I, all of which are in the trans configuration in the native state. Conventional double-jump mixing experiments suggest that proline isomerization in the unfolded state is not an important factor in the folding of this protein (data not shown). However, these experiments are inconclusive for this protein because the rate at which unfolding occurs is similar to that of proline isomerization in the unfolded state (Nall 1994; Eyles and Gierasch 2000). The addition of peptidyl prolyl isomerase during refolding did not alter the folding kinetics (Eyles and Gierasch 2000; data not shown). Replacement of Pro 85 with alanine abolished this slow phase, supporting a role for cistrans isomerization of this residue in the unfolded state as responsible for the slowest refolding phase (Eyles and Gierasch 2000). The two most closely related proteins in this family, cellular retinoic acid binding protein II and cellular retinol binding protein I (CRABP II and CRBP II, respectively), have valines at this sequence location instead of proline and lack this slow refolding phase. As such, the replacement of Pro 85 with the sequence-conserved valine was expected to confirm the role of proline isomerization in this slow phase. However, as described below, this mutant protein does have a slow folding phase that is very similar in rate and amplitude to that of wild-type protein, making the role of proline isomerization in the folding of this protein uncertain.
Results
Spectra of the native and denatured states of P85V-CRABP I
The CD spectra of WT-CRABP I and P85V-CRABP I were virtually identical in shape and intensity, demonstrating that mutation of Pro 85 to Val did not perturb the structure of CRABP I (data not shown). Both proteins displayed the typical β-sheet minimum at 216 nm and a small minimum at 232 nm. The minima at 232 nm were shown to be due to favorable interactions between the π electrons of the aromatic ring of Trp 109 and the positively charged side chain of Arg 111, providing a unique spectral signature for this region of the protein (Fig. 1 ▶; Clark et al. 1996). The presence of these minima in the P85V-CRABP I CD spectra indicated that this specific tertiary interaction was intact in the variant protein. The CD spectra of the denatured proteins were identical, suggesting that the two proteins were equally unfolded in 8 M urea. Similarly, the fluorescence emission spectra of denatured WT-CRABP I and P85V-CRABP I were coincident with a maximum of 354 nm. As such, the three tryptophans of both proteins were equally exposed in the unfolded state (Creighton 1993). However, the environment of the tryptophans in the native state for the wild-type and the P85V mutant were not equivalent. Native P85V-CRABP I was more fluorescent and the emission maximum was redshifted approximately 10 nm in comparison to the wild-type protein. There are three tryptophans in CRABP I (Trp 7, Trp 87, and Trp 109). Cys 95 is a significant quencher of Trp 109 (Clark et al. 1998) and is also within 4.4 Å of Pro 85 in the wild-type protein (Kleywegt et al. 1994; Thompson et al. 1995). Given the enhanced fluorescence of the P85V-CRABP I mutant, it appears that the mutation perturbs the structure near Cys 95, resulting in less efficient quenching of Trp 109. Trp 109 is in an unusually polar environment for the interior of a protein. Polar residues with side-chain atoms within 5 Å of the Trp 109 side chain include Glu 73, Ser 83, Cys 95, Gln 97, and Arg 111. Several water molecules are also within 5 Å of the Trp 109 side chain in both the apo- and holo-structures (Kleywegt et al. 1994; Thompson et al. 1995). The emission spectrum of this tryptophan is redshifted compared to the other tryptophans of the protein (Clark et al. 1998). Reduced quenching of the emission of this tryptophan would cause a redshift in the emission maxima of the entire protein. However, the overall orientation of the aromatic ring of Trp 109 with respect to Arg 111 must remain similar to that of the wild-type protein because the inflection at 232 nm in the CD spectrum was present.
Ligand binding studies
CRABP I has a nanomolar affinity for retinoic acid and binds this ligand with a 1:1 stoichiometry (Norris et al. 1994). The binding of retinoic acid by P85V-CRABP I and WT-CRABP I is shown in Figure 2 ▶. The binding isotherms of the two proteins were nearly coincident (Kd of P85V-CRABP I, 16 ± 3 nM; Kd of WT-CRABP I, 16 ± 1 nM). The binding stoichiometry of the variant protein was 1:1 (inset of Fig. 2 ▶), identical to that of WT-CRABP I. As such, replacement of Pro 85 with valine did not affect the function of the mutant protein.
Figure 2.

Retinoic acid binding to WT-CRABP I (open symbols) and P85V-CRABP I (closed symbols). (Inset) The data for ligand binding stoichiometry for P85V- CRABP I by the method of continuous variation (Huang 1982). A value of 0.5 for the intersection of the two lines in the inset indicates a 1:1 binding stoichiometry.
Equilibrium folding studies
The equilibrium unfolding and refolding of P85V-CRABP I and WT-CRABP I by urea is shown in Figure 3 ▶. The data for WT-CRABP I have been reported previously (Burns et al. 1998). All folding transitions for the variant protein were completely reversible and overlaid well. The data collected by both CD and fluorescence were best fit by a two-state model, suggesting that no significant population of intermediates was present.
Figure 3.

Equilibrium unfolding of WT-CRABP I and P85V-CRABP I with urea. The line through the data is the fit by nonlinear regression to a two-state model for the folding transition at equilibrium.
P85V-CRABP I was somewhat more stable than the wild-type protein as reflected by the right shift in the midpoint of urea denaturation (4.13 vs. 3.76 M, respectively) and the ΔΔGH2O value of 0.77 kcal/mole at 3.76 M urea. Because replacement of proline residues usually destabilizes the native state, the increase in stability of this variant was unexpected. The imino ring of prolines decreases the rotational freedom of this side chain, restricting the degree of randomness in the unfolded state. Proline residues can stabilize the native state by narrowing the entropic energy barrier between the native and unfolded states.
Refolding kinetics
Similar to WT-CRABP I, the folding transitions of P85V-CRABP I by fluorescence were best fit by a tri-exponential equation (Fig. 4 ▶). As such, at least two intermediates were present on the folding path. The three observed folding rates differed by up to three orders of magnitude from each other and displayed a logarithmic dependence on the final urea concentration (Fig. 5 ▶). A significant burst phase was also present. This burst phase intermediate (IB) had a 4.5-fold greater intensity than that expected for the denatured state in 1.1 M urea (Fig. 6 ▶). In comparison, the wild-type burst phase intermediate was twofold greater in intensity than that expected for the denatured state under similar conditions (Burns et al. 1998). Because both the equilibrium spectra of the denatured states and the slope of the denatured state baselines for the mutant and wild-type protein were virtually identical, differences in the extrapolated denatured state fluorescence did not account for the fluorescent intensity of IB.
Figure 4.

A typical refolding transition for P85V-CRABP I. The top trace shows an average of five refolding transitions. Protein in 6.9 M urea was diluted 1:6 with buffer to initiate refolding. The dashed lines indicate the expected intensities of the native and unfolded states at this final concentration of denaturant. (Inset) The first 5 sec of folding, emphasizing the initial burst phase. The residuals of the fits to a biphasic and triphasic model for the transition are shown.
Figure 5.

The dependence of the refolding and unfolding rates of P85V-CRABP I on urea concentration. The rates monitored by fluorescence and CD are shown by open and closed symbols, respectively. Error bars not indicated are within the size of the symbol. The lines on the graph are the rates observed for the wild-type protein.
Figure 6.

Burst phase analysis of P85V-CRABP I. The normalized initial and final amplitudes from both refolding and unfolding stopped-flow fluorescence experiments were plotted versus urea concentration. (Open and closed circles) Final amplitudes for unfolding and refolding, respectively. (Open triangles) Observed fluorescence at t =0 sec for unfolding transitions. (Open diamonds) Observed fluorescence at t =0 sec for refolding transitions. (Dashed lines) Expected fluorescence intensity of the native (N) and unfolded (U) states. The curve through the data is a fit to a two-state equilibrium model for the folding transition.
The folding of P85V-CRABP I by fluorescence can be described as follows,
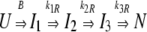 |
where B is the burst rate and k1R, k2R, and k3R are the observed rates for the folding process (Fig. 6 ▶). The amplitudes associated with k1R, k2R, and k3R when folding in 1.1 M urea were 76%, 13%, and 11% of the total amplitude change, respectively (data not shown). Therefore, most of the detectable tertiary changes (76%) occurred during the formation of I2. The proportion of the amplitude change associated with each rate were comparable to those observed for wild type.
The folding rates were independent of protein concentration (5–80 μM), suggesting that the fluorescence changes were not the result of protein association (data not shown). Stopped-flow CD measurements showed a burst phase and a single observed phase corresponding to the fastest observed fluorescence phase (Fig. 5 ▶).
Unfolding kinetics
The rates of unfolding as a function of urea concentration by both fluorescence and CD are shown in Figure 5 ▶. Data collected by both methods were best fit by a mono-exponential equation at all urea concentrations. The entire expected amplitude associated with the rate was present at each final concentration of denaturant (Fig. 6 ▶). Thus, the kinetic model that best fit the unfolding data of P85V-CRABP I was
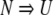 |
where N and U stand for the native and unfolded states, respectively. This behavior was different from that of WT-CRABP I (Burns et al. 1998). The unfolding transition of WT-CRABP I was biphasic by fluorescence but monophasic by CD. The kinetic model that best fit the data is
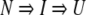 |
where I represents the intermediate state. The rate detected by CD during the unfolding of WT-CRABP I coincided with the slower rate observed by fluorescence. Thus, the intermediate observed during unfolding of WT-CRABP I has nativelike secondary structure associated with it.
Discussion
Models for proline-isomerization-limited protein-folding reactions assume that isomerization occurs in the unfolded state and that refolding is limited by the rate of interconversion between the isomers (Nall 1994; Kiefhaber 1995). A significant population of an incorrect isomer in the unfolded state causes the slow folding phase. According to the model, mutation of one or more proline residues to some other amino acid should abolish the slowest folding phase(s) if proline isomerization were responsible for that phase. In most cases this appears to be a common and predictable result (Nall 1994; Kiefhaber 1995). Generally, the proline residue thought to be responsible for the slow folding step is replaced by alanine. Replacement of Pro 85 with alanine in CRABP I removed the slowest folding phase, supporting the hypothesis that isomerization of Pro 85 causes the slowest folding phase in this protein.
However, the replacement of Pro 85 with valine did not remove this phase. An alternative explanation is that the alanine replacement relaxes a steric constraint on the native structure involving the correct positioning of the imino side chain of proline into the native structure. As such, a slow folding reaction may be caused by a barrier associated with the correct positioning of the proline ring in the final folded structure rather than proline isomerization. The replacement of proline with the β-branched amino acid valine may mimic the steric problems associated with the correct positioning of the proline side chain at this structural location, resulting in the appearance of the slowest phase in folding.
Proline 85 is located in a region of the protein that has been postulated to be an initiating site for folding of these proteins (Ropson and Frieden 1992; Dalessio and Ropson 2000) and is within the strand that spans both the top and bottom β-sheet (Fig. 1 ▶). As such, the constraints on the folding of this region of the protein may be more stringent.
An alternative explanation postulates that the valine substitution may have created a new barrier not present in the wild-type protein that causes a similar folding rate. In this case, the slowest phase in the folding of the WT-CRABP I was indeed due to proline isomerization. It is also possible that the replacement of proline with the smaller alanine residue reduced the observed amplitude of the slowest phase to the point where it could no longer be detected, even though it was still there.
It has long been known that processes other than proline isomerization can cause slow folding phases. In the cases of human lysozyme (Herning et al. 1991) and trp aporepressor (Mann et al. 1995; Gloss et al. 2001) removal of all of the proline residues failed to remove the slowest folding phase. The data shown here indicate that the loss of a particular folding phase in a mutant protein where proline is replaced with alanine is not definitive proof that proline isomerization is responsible for that phase.
Materials and methods
Protein source and purification
Mouse P85V-CRABP I was constructed using the Transformer Site-Directed Mutagenesis Kit (Clontech). The entire coding sequence was confirmed to prove the absence of second site mutations. P85V-CRABP I was purified by the same methods used for wild-type protein (Burns et al. 1998). The extinction coefficient at 280 nm was calculated as 20,970 M−1cm−1 (Pace et al. 1995).
Reagents
Denaturant stock (10 M urea) solutions were prepared and stored at −20°C until the day of the experiment as previously described (Burns et al. 1998; Dalessio and Ropson 1998). Working solutions (8 M urea) were prepared by adding buffer components (final concentrations: 25 mM NaPO4, 75 mM NaCl, 0.1 mM EDTA, 0.1 mM DTT) to the denaturant stock and adjusting the pH to 8.0. Final denaturant concentrations were determined by refractive index measurements (Pace 1986). All buffers were filtered using a 0.2-mm Whatman nylon membrane. All chemicals were reagent grade.
All-trans retinoic acid (Sigma) was reconstituted with 100% ethanol, aliquoted into microfuge tubes in the dark, and stored at −20°C. The concentration of retinoic acid was estimated using an extinction coefficient of 45,000 M−1cm−1 (Horwitz and Heller 1973).
Equilibrium studies
Equilibrium unfolding and refolding transitions as a function of denaturant concentration were monitored using far-UV CD and intrinsic tryptophan fluorescence as previously described (Burns et al. 1998; Dalessio and Ropson 1998). All measurements were made at 25°C after at least 1 h equilibration. Reversibility of unfolding was demonstrated by coincident equilibrium curves for native protein and protein that was previously denatured. The data were corrected for the background signal of the buffer and urea solutions (Burns et al. 1998; Dalessio and Ropson 1998).
Fluorescence kinetic studies
Unfolding and refolding kinetics were monitored using an Applied Photophysics DX-17MV stopped-flow spectrophotometer as previously described (Burns et al. 1998; Dalessio and Ropson 1998). Excitation was at 290 nm (2.3 nm bandpass) using a 0.2-cm path-length. Emission intensity was monitored above 305 nm at 90° through a WG345 Schott glass filter (Oriel) at 25°C. Five parts of denaturant solution were mixed with one part of protein solution (11 μM, final protein concentration). Three to seven kinetic traces were averaged at each final denaturant concentration. The dead time of the instrument was determined to be between 5 and 10 msec (Ropson and Dalessio 1997).
CD kinetic studies
The kinetics of unfolding and refolding were monitored at 218 nm (2 nm bandpass) in a 0.1-cm cell with a Jasco J-710 spectropolarimeter (CD) in conjunction with a RX1000 stopped-flow apparatus (Applied Photophysics) as previously described (Ropson and Dalessio 1997). For all experiments five parts of denaturant were mixed with one part of protein (32 μM protein, final concentration). Five to seven transitions were averaged at each denaturant concentration. The dead time of the instrument was 15–30 msec (Ropson and Dalessio 1997).
Ligand binding studies
The binding of retinoic acid to CRABP I and P85V-CRABP I was monitored by fluorescence using an Aminco-Bowman Series 2 luminescence spectrometer. Inner filter corrections were unnecessary due to the low absorbance of retinoic acid over this concentration and wavelength range (Birdsall et al. 1983). Samples were prepared by mixing retinoic acid (0–1.5 μM, concentration range) and protein (0.7 μM, final concentration). Samples were incubated for 1 h at 25°C in the dark before analysis. The data were corrected for the background signal of the buffer and free retinoic acid and were done in duplicate. The stoichiometry of ligand binding was determined using the continuous variation method (Huang 1982). Samples contained 0–1 mole fraction of ligand and 0–1 mole fraction of protein. The total concentration of protein plus ligand for any given sample was 1 μM. Samples were analyzed by fluorescence as described above. Each sample was corrected for the signal of protein in the absence of ligand. The stoichiometry of ligand binding was determined from two normalized data sets.
Computer fitting of data
Ligand binding isotherms were normalized and fit to the following equations by nonlinear least squares regression with KaleidaGraph (Synergy Software):
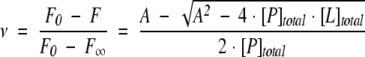 |
where ν is the fractional saturation, F is the normalized and corrected fluorescence, F0 is the fluorescence of the protein in the absence of ligand, F∞ is the fluorescence of the protein at saturation, [P]total and [L]total are the total concentration of protein and ligand, respectively, and A =[P]total + [L]total + Kd.
Ligand binding stoichiometry was determined by the value where the two lines that best fit the data intersected in the continuous variation plot (Huang 1982). The lines were fit by linear regression with KaleidaGraph.
Nonlinear least-square fits of the equilibrium data were determined with KaleidaGraph as previously described (Burns et al. 1998). Midpoints of the transition for the equilibrium data were determined by substituting ΔG H2O/midpoint for mG into the six-term equation as previously described (Dalessio and Ropson 1998).
The program supplied by Applied Photophysics was used to determine the nonlinear least-square fits of the kinetic data to mono-, bi-, and tri-exponential equations using Kaleidagraph (Burns et al. 1998).
The extent of the burst phase reaction occurring in the dead time of the stopped-flow CD and fluorescence experiments was determined by constructing an equilibrium plot using the normalized initial and final intensities obtained from both folding and unfolding transitions over a range of urea concentrations. The intensities were normalized using minimum and maximum values. The expected intensities of the native and denatured states at high and low denaturant concentrations were determined from the native and denatured state baselines in this plot. The extent of the reaction occurring in the dead time was determined by comparing the calculated intensity change with the observed intensity change.
Standard criteria were used to assess the quality of the fit to the various models (Mannervik 1982; Motulsky and Ransnas 1987). When the standard error of a parameter exceeded the value of the fitted parameter, the parameter was eliminated from the equation, and the fit repeated. This process continued until all of the remaining terms were significant.
Acknowledgments
This work was supported by NIH grant GM-57906 to I.J.R.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03317804.
References
- Banaszak, L., Winter, N., Xu, Z., Bernlohr, D.A., Cowan, S., and Jones, A.T. 1994. Lipid-binding proteins: A family of fatty acid and retinoid transport proteins. Adv. Protein Chem. 45 89–151. [DOI] [PubMed] [Google Scholar]
- Birdsall, B., King, R.W., Wheeler, M.R., Lewis, C.A.J., Goode, S.R., Dunlap, R.B., and Roberts, G.C. 1983. Correction for light absorption in fluorescence studies of protein-ligand interactions. Anal. Biochem. 132 353–361. [DOI] [PubMed] [Google Scholar]
- Burns, L.L. and Ropson, I.J. 2001. Folding of intracellular retinol and retinoic acid binding proteins. Proteins 43 292–302. [DOI] [PubMed] [Google Scholar]
- Burns, L.L., Dalessio, P.M., and Ropson, I.J. 1998. Folding mechanism of three structurally similar β-sheet proteins. Proteins 33 107–118. [DOI] [PubMed] [Google Scholar]
- Clark, P.L., Liu, Z.P., Zhang, J., and Gierasch, L.M. 1996. Intrinsic tryptophans of CRABP I as probes of structure and folding. Protein Sci. 5 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, P.L., Weston, B.F., and Gierasch, L.M. 1998. Probing the folding pathway of a β-clam protein with single tryptophan constructs. Fold. Des. 3 401–412. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1993. Proteins: Structures and molecular properties. W.H. Freeman and Company, New York.
- Dalessio, P.M. and Ropson, I.J. 1998. pH dependence of the folding of intestinal fatty acid binding protein. Arch. Biochem. Biophys. 359 199–208. [DOI] [PubMed] [Google Scholar]
- ———. 2000. β-sheet proteins with nearly identical structures have different folding intermediates. Biochemistry 39 860–871. [DOI] [PubMed] [Google Scholar]
- Eyles, S.J. and Gierasch, L.M. 2000. Multiple roles of prolyl residues in structure and folding. J. Mol. Biol. 301 737–747. [DOI] [PubMed] [Google Scholar]
- Gloss, L.M., Simler, B.R., and Matthews, C.R. 2001. Rough energy landscapes in protein folding: Dimeric E. coli Trp repressor folds through three parallel channels. J. Mol. Biol. 312 1121–1134. [DOI] [PubMed] [Google Scholar]
- Herning, T., Yutani, K., Taniyama, Y., and Kikuchi, M. 1991. Effects of proline mutations on the unfolding and refolding of human lysozyme: The slow refolding kinetic phase does not result from proline cistrans isomerization. Biochemistry 30 9882–9891. [DOI] [PubMed] [Google Scholar]
- Hodsdon, M.E. and Frieden, C. 2001. Intestinal fatty acid binding protein: The folding mechanism as determined by NMR studies. Biochemistry 40 732–742. [DOI] [PubMed] [Google Scholar]
- Horwitz, J. and Heller, J. 1973. Interactions of all-trans, 9-, 11-, and 13-cis-retinal, all-trans-retinyl acetate, and retinoic acid with human retinol-binding protein and prealbumin. J. Biol. Chem. 248 6317–6324. [PubMed] [Google Scholar]
- Huang, C.Y. 1982. Determination of binding stoichiometry by the continuous variation method: The Job plot. Methods Enzymol. 87 509–525. [DOI] [PubMed] [Google Scholar]
- Kiefhaber, T. 1995. Protein folding kinetics. In Protein stability and folding (ed. B.A. Shirley), Vol. 40, pp. 313–341. Humana Press, Totowa, NJ. [DOI] [PubMed]
- Kleywegt, G.J., Bergfors, T., Senn, H., Le Motte, P., Gsell, B., Shudo, K., and Jones, T.A. 1994. Crystal structures of cellular retinoic acid binding proteins I and II in complex with all-trans-retinoic acid and a synthetic retinoid. Structure 2 1241–1258. [DOI] [PubMed] [Google Scholar]
- Mann, C.J., Shao, X., and Matthews, C.R. 1995. Characterization of the slow folding reactions of trp aporepressor from Escherichia coli by mutational analysis of prolines and catalysis by a peptidyl-prolyl isomerase. Biochemistry 34 14573–14580. [DOI] [PubMed] [Google Scholar]
- Mannervik, B. 1982. Regression analysis, experimental error, and statistical criteria in the design and analysis of experiments for discrimination between rival kinetic models. Methods Enzymol. 87 370–390. [DOI] [PubMed] [Google Scholar]
- Motulsky, H.J. and Ransnas, L.A. 1987. Fitting curves to data using nonlinear regression: A practical and nonmathematical review. FASEB J. 1 365–374. [PubMed] [Google Scholar]
- Nall, B.T. 1994. Proline isomerization as a rate-limiting step. In Mechanisms of protein folding (ed. R.H. Pain), pp. 80–99. Oxford University Press, New York.
- Norris, A.W, Cheng, L., Gigue′re, V., Rosenberger, M., and Li, E. 1994. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochim. Biophys. Acta 1209 10–18. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropson, I.J. and Dalessio, P.M. 1997. Fluorescence spectral changes during the folding of intestinal fatty acid binding protein. Biochemistry 36 8594–8601. [DOI] [PubMed] [Google Scholar]
- Ropson, I.J. and Frieden, C. 1992. Dynamic NMR spectral analysis and protein folding: Identification of a highly populated folding intermediate of rat intestinal fatty acid-binding protein by 19F NMR. Proc. Natl. Acad. Sci. 89 7222–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.R., Bratt, J.M., and Banaszak, L.J. 1995. Crystal structure of cellular retinoic acid binding protein 1 shows increased access to the binding cavity due to formation of an intermolecular β-sheet. J. Mol. Biol. 252 433–446. [DOI] [PubMed] [Google Scholar]
- Yeh, S.R., Ropson, I.J., and Rousseau, D.L. 2001. Hierarchical folding of intestinal fatty acid binding protein. Biochemistry 40 4205–4210. [DOI] [PubMed] [Google Scholar]
- Zimmerman, A.W. and Veerkamp, J.H. 2002. New insights into the structure and function of fatty-acid binding proteins. Cell. Mol. Life Sci. 59 1096–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]