

Abstract
Nanoseconds long molecular dynamics (MD) trajectories of differently active complexes of human cyclin-dependent kinase 2 (inactive CDK2/ATP, semiactive CDK2/Cyclin A/ATP, fully active pT160-CDK2/Cyclin A/ATP, inhibited pT14-; pY15-; and pT14,pY15,pT160-CDK2/Cyclin A/ATP) were compared. The MD simulations results of CDK2 inhibition by phosphorylation at T14 and/or Y15 sites provide insight into the structural aspects of CDK2 deactivation. The inhibitory sites are localized in the glycine-rich loop (G-loop) positioned opposite the activation T-loop. Phosphorylation of T14 and both inhibitory sites T14 and Y15 together causes ATP misalignment for phosphorylation and G-loop conformational change. This conformational change leads to the opening of the CDK2 substrate binding box. The phosphorylated Y15 residue negatively affects substrate binding or its correct alignment for ATP terminal phospho-group transfer to the CDK2 substrate. The MD simulations of the CDK2 activation process provide results in agreement with previous X-ray data.
Keywords: cell cycle, CDK regulation, phosphorylated tyrosine, threonine
Cyclin-dependent kinases (CDK) play a central role in the control of the eukaryotic cell division cycle. The function of CDK is to catalyze the phosphoryl transfer of the adenosine-5′-triphosphate (ATP) γ-phosphate to serine or threonine hydroxyl in the protein substrate. The CDK activity is stringently controlled by cyclin binding, phosphorylation, and binding of protein inhibitors. The CDK activation is a two-step process that requires cyclin binding and phosphorylation in the activation loop (known also as the “T-loop”; Morgan 1997; Johnson and Lewis 2001; Lew 2003). The critical CDK/cyclin complexes for the cell division are CDK2/cyclin E, driving a cell across the G1/S-phase border; CDK2/cyclin A, mediating DNA replication; and CDK1/cyclin B, controlling the entry into mitosis (Lew 2003). The CDKs are very intensively studied enzymes, mainly as targets for medical and molecular biological applications (Knockaert et al. 2002), and also as exemplary biochemical models of nonautoinhibitory regulation (Lew 2003).
A detailed description of the activation pathway of p34cdc2 (CDC2, CDK1) came from studies of Xenopus egg extracts, and it is believed that the mechanism is conserved over all eukaryotic organisms (Pagano 1998). The CDK2 activation diagram (Scheme 1 ▶) can be extrapolated from the best-understood CDK1 regulation model (Morgan 1996, 1997). Monomeric CDK2 (Scheme 1, I ▶) is inactive, and for its activation requires binding to a cyclin (cyclin E at the G1/S transition, cyclin A during the S phase). The CDK2/cyclin complex (Scheme 1, IIa and IIb ▶) is recognized by multiple protein kinases, and it results in phosphorylations on T14, Y15, and T160 (in CDK2). The amino acid residue Y15 and to a lesser extent T14 are phosphorylated by human Wee1Hu (Watanabe et al. 1995). This inhibitory phosphorylation is independent of previous cyclin binding (Coulonval et al. 2003). Inhibitory phosphorylation likely precedes the activating T160 phosphorylation by CAK (CDK7/Cyclin H) because activatory phosphorylation requires cyclin binding. The overphosphorylated complex (Scheme 1, III ▶) is inactive and subsequent dephosphorylation of T14 and Y15 by CDC25 (Sebastian et al. 1993; Rudolph et al. 2001) results in activation. Recently, the phosphorylation mechanisms of the cell were revisited with the finding that pY15–CDK2 dephosphorylation by CDC25 is an important regulation mechanism of correct cell cycle timing (Coulonval et al. 2003). The importance of inhibitory sites was also probed by site-directed mutagenesis of T14 (T14A) and Y15 (Y15F). Such mutations stimulate kinase activity (Gu et al. 1992) but the expression of mutated protein (T14A,Y15F)CDK2 is cytotoxic (Chow et al. 2003). The fully active CDK2/cyclin complex (Scheme 1, IV ▶) is phosphorylated only at T160. Feedback from the active form of the pT160–CDK2/cyclin complex stimulates CDC25 activity and inhibits Wee1 activity. Such an “autocatalytic” activation loop leads to a rapid activation of CDK2. Two phosphatases, KAP (Poon and Hunter 1995) and PP2C (Cheng et al. 1999, 2000) were found to be dephosphorylating monomeric CDK2 rather then CDK2/cyclin complex.
Scheme 1.

Scheme of CDK2 regulation. Inactive form CDK2/ATP (I) binds to Cyclin and may be phosphorylated at Y15 by WEE1 kinase. Inhibited complex pY15-CDK2/Cyclin/ATP (II) is phosphorylated by CAK at T160 and pY15,pT160–CDK2/Cyclin/ATP complex (III) is activated at the pY15 site by dephosphorylation by CDC25. The fully active complex pT160–CDK2/Cyclin/ATP (IV) after Cyclin is lost is dephosphorylated by PP2C or KAP at pT160.
CDK2 has the typical bilobal kinase fold (Fig. 1 ▶). The active site is positioned between two lobes—the smaller N-terminal, and the bigger C-terminal. The smaller lobe is primarily composed of β-sheet with one α-helix, the C-helix, whose correct orientation is important for catalysis. The helix includes the conserved PSTAIRE motif (residues 45–51; this helix is also denoted as PSTAIRE helix) important for cyclin binding. The CDK2 activation site of the T-loop is located at T160. Close to the activation segment is a functionally opposite segment, the inhibitory loop (residues 11–18), named the glycine-rich loop (G-loop) because its primary sequence includes three highly conserved glycine residues (CDK2: 11-GEGTYG; Hanks and Quinn 1991). The G-loop includes two possible inhibitory sites, T14 and Y15. The phosphorylation of any of these residues leads to the loss of kinase activity.
Figure 1.

View of CDK2/ATP (1HCK coordinates taken from PDB database) complex is shown in tube representation. The T160 (shown in gray-colored licorice representation) activation site is located on the T-loop. The G-loop (black-colored tube representation) includes two possible inhibitory sites, T14 (gray-colored licorice representation) and Y15 (black-colored licorice representation).
Two recent articles studying the process of CDK2/Cyclin A complex formation and T160 phosphorylation (Morris et al. 2002; Stevenson et al. 2002) have concluded that the CDK2/Cyclin A complex formation a is two-step process; the first step is a CDK2/Cyclin A associate formation followed by CDK2/Cyclin A complex activation by conformational change of the T-loop. The association of unphosphorylated CDK2 with Cyclin A serves to configure the active site for ground-state binding of both ATP and a protein substrate and aligns ATP in the transition state for phosphoryl transfer. The optimization of the mentioned ATP alignment and stabilization of substrate binding is the principal role of phosphorylation at T160. Both articles broaden the knowledge gained from previous X-ray studies on inactive, semiactive, and fully active CDK2 forms (De Bondt et al. 1993; Jeffrey et al. 1995; Schulze-Gahmen et al. 1995; Russo et al. 1996). Cook et al. (2002) deduce that in CDK2, the shift in the T-loop upon phosphorylation creates a pocket that can accept the substrate prolyl side chain, and the substrate basic group (at the P + 3 position) to make contact with the pT160 (phosphothreonine 160) phosphate (Brown et al. 1999; Holmes and Solomon 2001). The serine of the peptide substrate is hydrogen bonded to the ATP γ-phosphate oxygen, to the catalytic aspartate D127, and to the conserved lysine K129 (Cook et al. 2002).
The mechanism of inhibition by phosphorylation is not yet well understood from the structural point of view. It has been suggested that pY15 perturbs the binding of protein substrate at the catalytic site through sterical hindrance (Endicott et al. 1999; Johnson and Lewis 2001). However, to our best knowledge this fact has not yet been clearly confirmed. The aim of this article is a detailed study on CDK2 inhibition and activation by phosphorylation using molecular dynamics simulations.
Results
All the trajectories analyzed below (for a brief summary, see Table 1) were stable during the whole production part of the molecular dynamics (MD) simulation. The trajectory stabilities were confirmed by analysis of secondary structure elements, radius of gyration, root-mean-square deviation to the crystal structure, and total energy (Table 1).
Table 1.
Summary of trajectories production part characteristics
| System | Used PDB structure | E 105 kcal • mole−1 | Rg (MD) Å | Rg (X-ray) Å | RMSD Å |
| CDK2/ATPb | 1HCK | −0.93 ± 0.01 | 20.14 ± 0.07 | 19.85 | 1.72 ± 0.11 |
| CDK2/Cyclin A/ATPc | 1FIN | −1.50 ± 0.01 | 25.54 ± 0.08 | 25.37 | 1.27 ± 0.09 |
| pT160–CDK2/Cyclin A/ATPd | 1JST | −1.43 ± 0.01 | 25.84 ± 0.10 | 25.31 | 1.33 ± 0.07 |
| pY15,pT160–CDK2/Cyclin A/ATPe | 1JSTa | −1.39 ± 0.01 | 25.94 ± 0.07 | — | 1.57 ± 0.15 |
| pT14,pT160–CDK2/Cyclin A/ATPe | 1JSTa | −1.39 ± 0.01 | 26.20 ± 0.13 | — | 1.50 ± 0.09 |
| pT14,pY15,pT160–CDK2/Cyclin A/ATPe | 1JSTa | −1.40 ± 0.01 | 25.99 ± 0.09 | — | 1.53 ± 0.10 |
E, mean total energy; Rg (MD), mean radius of gyration; Rg (X-ray), radius of gyration of the X-ray structure; RMSD, root-mean-square deviation of backbone atoms compared with initial X-ray crystal structures.
a The following were modified by in silico phosphorylation of either Y15 or T14, and both T14,Y15 residues:
b Inactive CDK2.
c Semiactive CDK2.
d Fully active CDK2.
e CDK2 inhibited by phosphorylation.
Enhanced thermal movements observed on the CDK2/ATP complex are localized on the C-helix (PSTAIRE helix, residues 46–56), on residues around T26, E73, L96, Y179, and residues between P228 and L255 (Fig. 2A ▶). The B-factor curves for the CDK2/Cyclin A/ATP system display similar sharp peaks corresponding to the same residues (Fig. 2B ▶). Only the high peak corresponding to the enhanced mobility of the PSTAIRE helix disappeared due to the presence of the bound cyclin that interacts with the PSTAIRE helix and stabilizes its secondary structure fold. The phosphorylation of T160 residue in the CDK2/Cyclin A/ATP complex increases the mobility of the G-loop (Fig. 2B,C ▶), in agreement with experimental X-ray data (Jeffrey et al. 1995; Russo et al. 1996). Phosphorylations in the G-loop lead to an additional increase in the G-loop thermal fluctuations (19, 28, 29, and 34 are the mean B-factors of G-loop residues for pT160–CDK2/Cyclin A/ATP, pT14,pT160–CDK2/Cyclin A/ATP, pY15,pT160–CDK2/Cyclin A/ATP, and pT14,pY15,pT160–CDK2/Cyclin A/ATP complexes, respectively; Fig. 2C–F ▶). The experimental B-factor values are in all cases (Fig. 2A–C ▶) larger than those B-factors calculated from MD simulations. This observation agrees with results described by Philippopoulos and Lim (1995).
Figure 2.
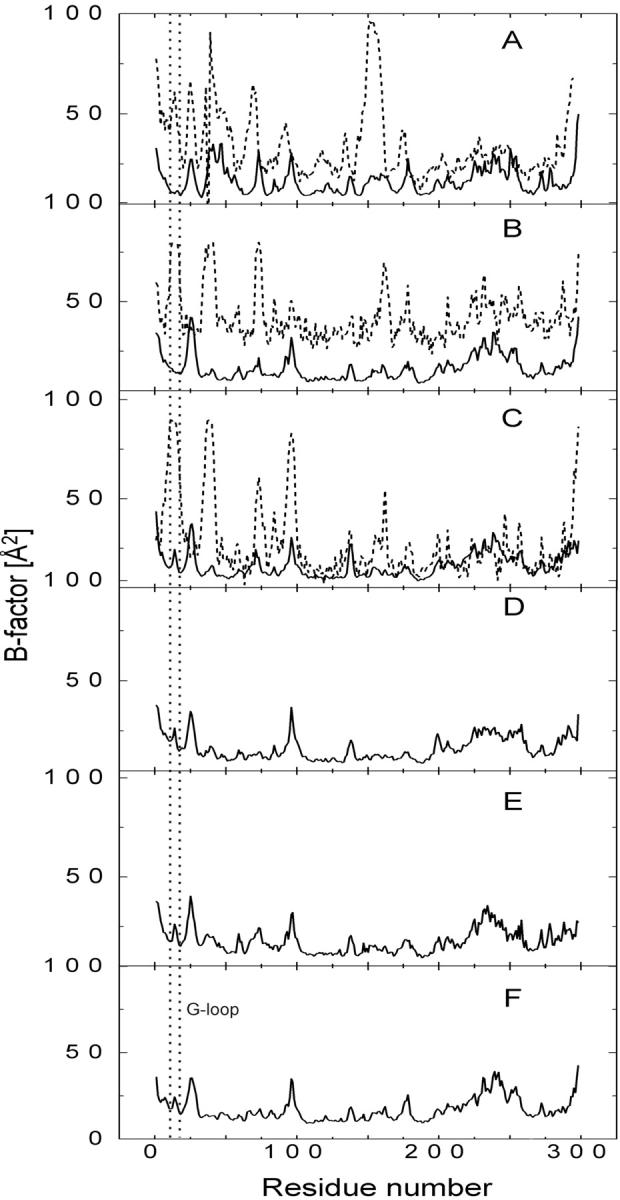
Comparison of temperature B-factors calculated from the last 500 psec of MD simulations (solid line); free CDK2/ATP (A); CDK2/Cyclin A/ATP (B); pT160–CDK2/Cyclin A/ATP (C); pY15,pT160–CDK2/Cyclin A/ATP (D); pT14,pT160–CDK2/Cyclin A/ATP (E); and pT14,pY15,pT160–CDK2/Cyclin A /ATP complex (F). B-factor values of CDK2/ATP (A), CDK2/Cyclin A/ATP (B), and pT160–CDK2/Cyclin A/ATP (C) complexes determined by X-ray experiments are added to the plots (dotted line).
Remarkable differences in secondary structure behavior were noticed between monomeric CDK2 and complexes of CDK2/Cyclin A. The PSTAIRE helix fold is unstable during CDK2/ATP simulation corresponding to our previous results obtained by simulations of CDK2/purine inhibitor complexes. The PSTAIRE helix fold is destabilized by ligand presence in the CDK2 active site (Otyepka et al. 2002). All simulations of CDK2/Cyclin A complexes reveal enhanced stability of the PSTAIRE helix and surrounding secondary structure elements (Fig. 3 ▶). These were also stable during all simulations with systems inhibited by phosphorylation (data not present). Hence, the Cyclin binding to CDK2 remarkably stabilizes the PSTAIRE helix fold in agreement with crystalographic experiments (Jeffrey et al. 1995).
Figure 3.

Secondary structure elements (α-helix and β-sheet in blue and in red, respectively) determined during the whole production part of MD simulations of CDK2/ATP, CDK2/Cyclin A/ATP, and pT160–CDK2/Cyclin A/ATP complexes. Stabilization of the PSTAIRE helix (denoted as α1-helix) after Cyclin A binding to CDK2 is clearly seen.
Analysis of the ATP dihedral angle C8-N9-C1*-C2* (Scheme 2 ▶) between the adenine and ribose moieties reveals that the angle changed during the early stage of CDK2/ATP simulation in comparison with its conformation in the crystal structure (Fig. 4A ▶). This torsion angle was stable for the other studied systems and comparable with its value found in the crystal structures. The conformation of the ATP phosphate moiety was stable during simulation of inactive CDK2 but it changed during the early stage of semi-(CDK2/Cyclin A/ATP) and fully active (pT160–CDK2/Cyclin A/ATP) complex simulations (Fig. 4B,C ▶).
Scheme 2.

Structural formula of adenosine 5′-triphosphate (ATP) molecule with atom labeling used in this article.
Figure 4.
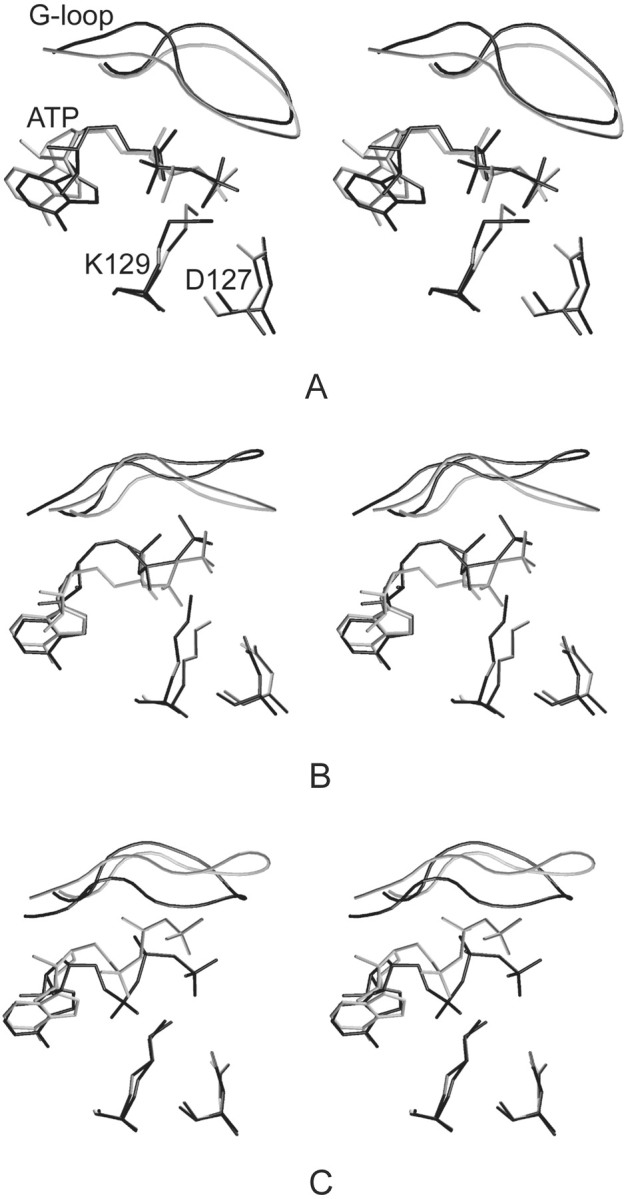
Stereoview of selected residues (D127, K129), ATP and G-loop conformation; comparison of structures determined by MD simulations (gray) and X-ray experiments (black); CDK2/ATP (A), CDK2/Cyclin A/ATP (B), and pT160–CDK2/Cyclin A/ATP (C).
Dramatic changes were observed in ATP conformation and/or position after inhibitory phosphorylations at T14 and/or Y15. The shift of the ATP Pγ group after T14 phosphorylation (Fig. 5B,C ▶) towards the adenine base is especially remarkable. This ATP reorientation also causes a shift in the Mg2+ ion position and consequently affects the Mg2+ ion coordination sphere (Table 2; Fig. 5 ▶).
Figure 5.
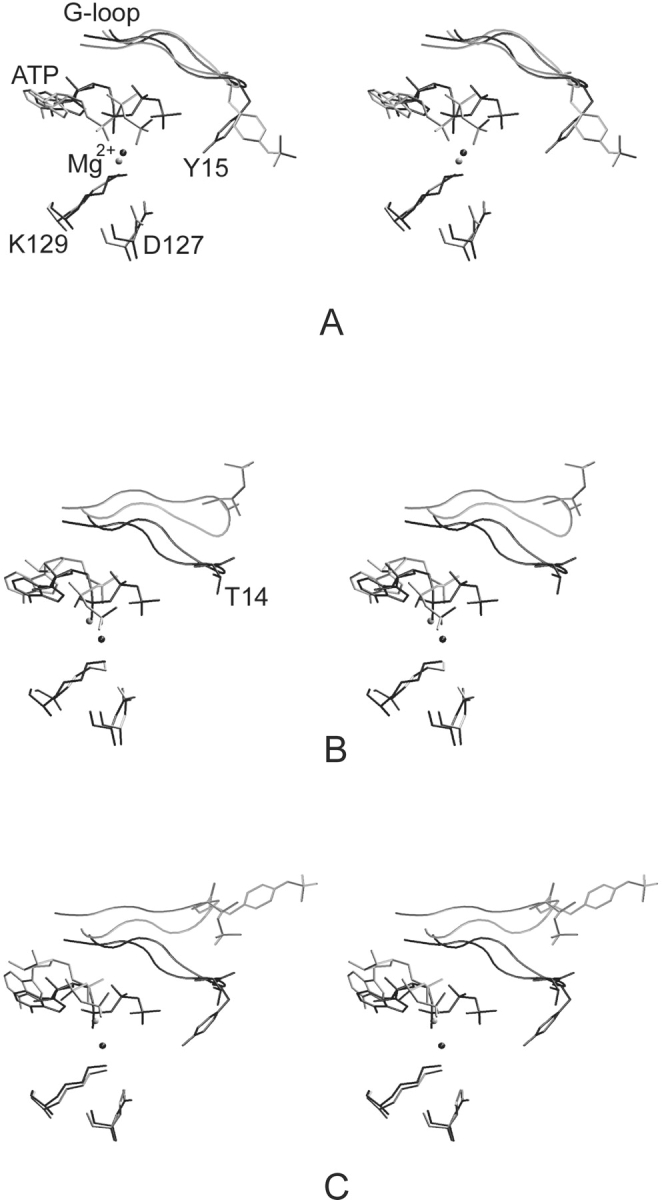
Stereoview of the superimposition of pT160–CDK2/Cyclin A/ATP (black) and complexes phosphorylated in the G-loop (gray); at Y15 residue (A), at T14 residue (B), and at T14/Y15 residues (C). ATP, Mg2+ ion, and D127 and K129 residues are shown in licorice representation. Inhibitory sites at T14 and Y15 residues are depicted in licorice representation, and G-loop in tube representation.
Table 2.
Coordination of the Mg2+ion was monitored during the entire simulation period of all trajectories: the results show the magnesium ion was hexacoordinated during all simulations
| System | Atoms coordinating Mg2+ | |||||
| CDK2/ATP | ATP (Oα1) | ATP (Oβ2) | ATP (Oγ1) | N132 (Oδ1) | D145 (Oδ2) | WAT314 (O) |
| CDK2/Cyclin A/ATP | ATP (Oα2) | ATP (Oβ1) | N132 (Oδ1) | D145 (Oδ2) | WAT4896 (O) | WAT8293 (O) |
| pT160–CDK2/Cyclin A/ATP | ATP (Oα1) | N132 (Oδ1) | D145 (Oδ2) | WAT7781 (O) | WAT7803 (O) | WAT7831 (O) |
| pY15,pT160–CDK2/Cyclin A/ATP | ATP (Oα1) | N132 (Oδ1) | D145 (Oδ2) | WAT1707 (O) | WAT2232 (O) | WAT5368 (O) |
| pT14,pT160–CDK2/Cyclin A/ATP | ATP (Oβ1) | ATP (Oγ3) | D145 (Oδ2) | WAT2413 (O) | WAT2560 (O) | WAT8065 (O) |
| pT14,pY15,pT160–CDK2/Cyclin A/ATP | ATP (Oα2) | ATP (Oγ3) | D145 (Oδ2) | WAT7647 (O) | WAT7693 (O) | WAT8053 (O) |
The G-loop conformation was changed during the early stage of inactive (CDK2/ATP), partly active (CDK2/Cyclin A/ATP), and fully active (pT160–CDK2/Cyclin A/ATP) CDK2 simulations in comparison with its conformation as found in the crystal structures (Figs. 4 ▶,6 ▶). The G-loop moves away from the ATP phosphate moiety binding site after the interaction of CDK2 with Cyclin A and again after CDK2/Cyclin A/ATP complex phosphorylation at the T160 site (Fig. 6A ▶). The shift of the G-loop is equal to 3.5 Å (CDK2/Cyclin A/ATP) and 8.6 Å (pT160–CDK2/Cyclin A/ATP) in comparison with the G-loop position found in the CDK2/ATP system. The G-loop conformation remains stable (low thermal fluctuations) after the shift described above (Fig. 7A ▶). The G-loop backbone possesses similar conformation in the pT160–CDK2/Cyclin A/ATP and pY15,pT160–CDK2/Cyclin A/ATP complex simulations (Fig. 5A ▶) but the pY15 residue reorients its side chain towards a bulk solvent due to hydration of the phosphogroup. The Y15 phosphorylation causes an increase in the G-loop thermal fluctuations (Fig. 7B ▶).
Figure 6.

Stereoview of G-loop and ATP conformations in CDK2/ATP (thin black), CDK2/Cyclin A/ATP (thick gray), and pT160–CDK2/Cyclin A/ATP (thick black) as determined by MD simulations (A) and X-ray experiments (B).
Figure 7.
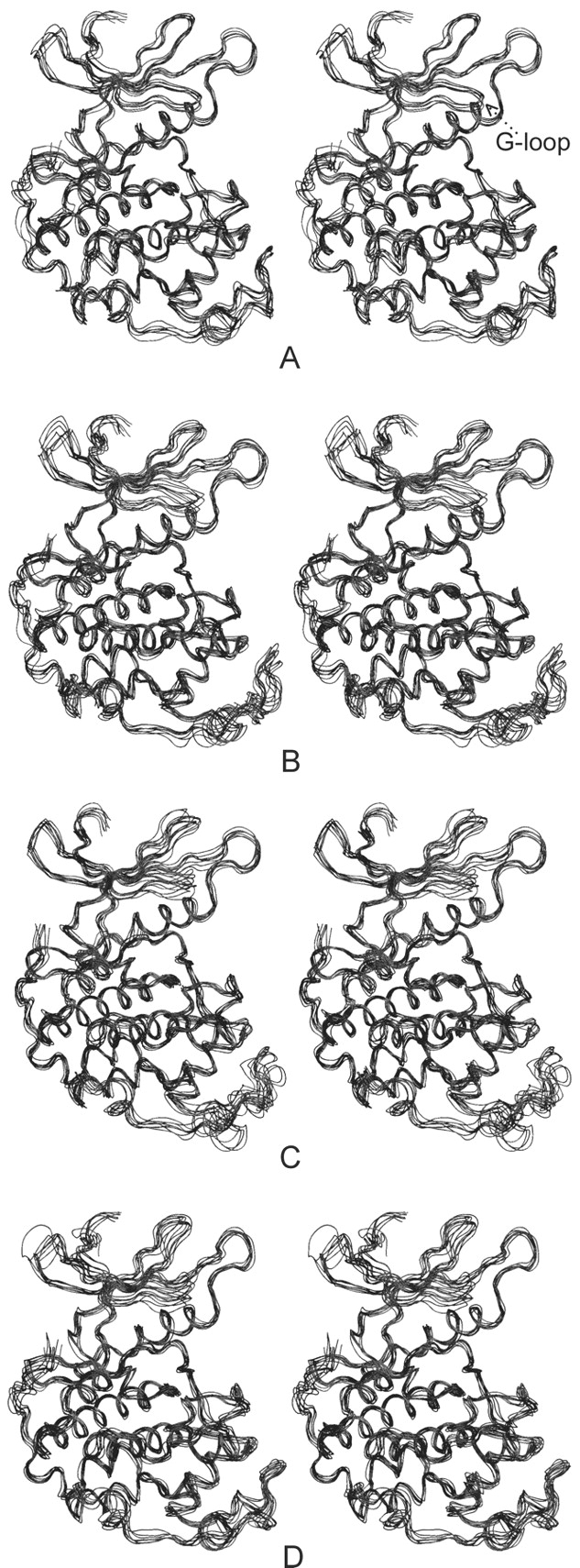
Stereoview of the superimposition of the snapshots taken every 200 psec from the production parts of molecular dynamics simulations of pT160–CDK2/Cyclin A/ATP (A) and complexes phosphorylated in the inhibition segment (G-loop) at Y15 (B), T14 (C), and T14/Y15 together (D).
The G-loop conformation changes remarkably during the simulations of pT14,pT160–CDK2/Cyclin A/ATP and pT14,pY15,pT160–CDK2/Cyclin A/ATP complexes. In these simulations the G-loop moves away from the ATP phosphate binding pocket. Displacement of the G-loop is equal to 4.41 Å (pT14,pT160–CDK2/Cyclin A/ATP) and 5.94 Å (pT14,pY15,pT160–CDK2/Cyclin A/ATP) compared to its position in the fully active CDK2 (Fig. 5B,C ▶). In general, the G-loop phosphorylation increases its thermal fluctuations (Fig. 7A–D ▶) due to the insertion of the bulky hydrophilic group.
Discussion
The Cyclin A binding stabilizes the PSTAIRE helix remarkably and decreases thermal movements in this region by direct interaction of the α1-helix with Cyclin A. This stabilization is also well known from previous crystallographic studies (Jeffrey et al. 1995), demonstrating that molecular dynamics simulations results are in agreement with experimental data. The X-ray studies (Russo et al. 1996) as well as the kinetic experiments (Hagopian et al. 2001) conclude that the phosphorylation of T160 residue causes the correct orientation of the T-loop, the creation of a substrate binding box, and also the correct alignment of ATP for phosphogroup transfer. Moreover, it was suggested that the phosphorylation of T160 leads to substrate stabilization due to the interaction of pT160 with substrate basic residue at the P + 3 position (Holmes and Solomon 1996; Brown et al. 1999; Holmes and Solomon 2001). The MD simulations emphasize the known fact that CDK2/Cyclin A association stabilizes the three-dimensional structure of CDK2 and the activating phosphorylation of T160 stabilizes T-loop conformation for substrate binding and phosphoryl transfer. The molecular mechanism of inhibition by phosphorylation remains until now unclear. Our simulations shed light on the mechanism and suggest a model for how the process of inhibition by phosphorylation may work.
It can be deduced from the simulations and from alignments of X-ray crystal structures or averaged MD structures to X-ray structures of fully active CDK2 with peptide substrate (1QMZ, 1GY3) that the inhibitory sites utilize different mechanisms of action. T14 inhibitory phosphorylation leads to G-loop shift and ATP misalignment for phosphogroup transfer due to the shift of the terminal ATP phosphogroup towards the ATP adenine base moiety in the pT14,pT160–CDK2/Cyclin A/ATP complex (3.8 Å) and also in the pT14,pY15,pT160–CDK2/Cyclin A/ATP structure (3.8 Å). The G-loop shift causes dramatic changes in substrate binding box conformation. Such changes might decrease CDK2 affinity to its substrate. The G-loop shift and substrate box opening is much more remarkable in the pT14,pY15,pT160–CDK2/Cyclin A/ATP complex.
The Y15 residue is buried in an active pT160–CDK2/Cyclin A/ATP complex. After phosphorylation or substrate binding the conformation of Y15 changes, leading to the exposure of the pY15 side chain to solvent (Fig. 5A ▶) and to an increase in G-loop flexibility (Fig. 7B ▶). Conformation of the Y15 residue in the crystal structure of fully active CDK2 with peptide substrate (1QMZ; Brown et al. 1999) is very similar to the conformation of pY15 in the pY15,pT160–CDK2/Cyclin A/ATP complex. The distance between the pY15 phosphogroup and the substrate arginine (P + 2 substrate position) terminal groups is equal to 4.5 Å (Fig. 8 ▶). One can deduce that Y15 phosphorylation affects substrate binding or its correct alignment for phosphorylation, because pY15 can directly interact with the substrate and also decreases substrate interaction with the pT160 residue due to competition. These findings correspond with previously published articles (Endicott et al. 1999; Johnson and Lewis 2001) where the authors deduced from the not-yet published crystal structure of pY15,pT160–CDK2/Cyclin A/ATP complex that the phosphorylation of Y15 does not significantly alter the overall structure of the complex nor does it prevent ATP binding. The authors also suggest that phosphorylation of Y15 may perturb the protein substrate binding at the catalytic site through a steric hindrance.
Figure 8.

Stereoview of G-loop and pY15 residue conformations in the pY15,pT160–CDK2/Cyclin A/ATP complex (gray) and crystal structure of fully active CDK2 with peptide substrate (black; 1QMZ; Brown et al. 1999). Only P–P + 3 peptide substrate residues are depicted for clarity.
The glycine-rich loop (G-loop) enables protein kinase to adopt a wide range of backbone conformations. The significance of this domain is demonstrated by the fact that substitution of the glycine residues in the glycine-rich loop, particularly the first and the second glycine (GxGxxG) with either alanine or serine results in a dramatic decrease in cAMP-dependent protein kinase catalytic (cAPK) activity. The functional importance of the glycine-rich loop has been described in detail for cAPK (Hemmer et al. 1997; Tsigelny et al. 1999; Aimes et al. 2000; Johnson et al. 2001), but its importance for CDK regulation has not been yet discussed. It is believed that the glycine-rich loop catalytic function—that is, correct ATP binding and alignment—is the same as its function in cAPK, but it exhibits a new inhibitory function for CDK.
The functionally important flexibility of the G-loop is assured by its primary sequence where the inhibitory sites T14 and/or Y15 are bordered by conserved glycine residues 11-GEGTYG (conservation profiles for all protein kinases catalytic domains show the G-G–G motif as one of the most conserved protein kinase motifs [Hanks and Quinn 1991], also known as the nucleotide binding loop or P-loop). This sequence has been found in human CDK1 (CDC2), CDK2, CDK3, and CDK5, and also in human CDK10. A very similar sequence has also been found in CDK8 (GRGTYG). The remaining cyclin-dependent kinases have mutated inhibitory sites. The CDK4 (GVGAYG) and CDK6 (GEGAYG) have mutated threonine to alanine, CDK9 (GQGTFG) has changed tyrosine to phenylalanine and CDK7 (GEGQFA) has mutated both sites, and one of the very conserved glycines is also mutated to alanine. The GEGTYG motif and all its aforementioned variations occur only in the CMGC group of protein kinases (classification according to Hanks and Quinn 1991). Important is the fact that the function of these sites can differ among cyclin-dependent kinases. Although T and/or Y residues serve as inhibitory sites in CDK1 homology kinases (CDK1, CDK2, and probably in CDK3), Y serves as an activatory site for CDK5/p35 (Zukerberg et al. 2000). In contrast, Y17 phosphorylation of CDK4 (and perhaps CDK6) is specifically used to initiate control cell cycle start from quiescence but not for G1 traverse (Jinno et al. 1999). Hence, Y17 phosphorylation can also be considered as inhibitory because phosphorylation of CDK4 Y17 residue occurs during entry into quiescence and dephosphorylation must occur sometime during cell cycle start. These findings emphasize the fact that the G-loop plays a very important biological role in CDKs.
Materials and methods
Molecular dynamics simulations of six systems (CDK2/ATP, CDK2/Cyclin A/ATP, pT160–CDK2/Cyclin A/ATP, pT14,pT160–CDK2/Cyclin A/ATP, pY15,pT160–CDK2/Cyclin A/ATP, and pT14,pY15,pT160–CDK2/Cyclin A/ATP; see also Table 1) were carried out using the SANDER module of the AMBER 6.0 software package (Case et al. 1999) with the parm99 force field (Wang et al. 2000). The starting geometries for simulation were prepared using X-ray structures obtained from the Brookhaven Protein Data Bank (http://www.pdb.org). MD simulations of free CDK2 and CDK2/purine inhibitor complexes (Otyepka et al. 2002) were used for comparison in the discussion part.
Starting structures for the molecular dynamics simulations were prepared according to standard procedures. At first, the protonation states of histidines were checked by WHATIF (Vriend 1997) and then all hydrogens were added using Xleap from the AMBER 6.0 package. The structures were neutralized by adding 11, 16, 15, 13, 13, and 11 chloride counter ions for inactive, semi-, and fully active CDK2, and fully active CDK2 phosphorylated at Y15, T14, and T14/Y15 residues, respectively. Such systems were inserted in a rectangular water box where the layer of water molecules was equal to 10 Å. All systems were minimized prior to the production part of molecular dynamics in this way. The protein was frozen, and the solvent molecules with counterions were allowed to move during a 1000-step minimization and a 2 psec-long molecular dynamics run. Then, the side chains were relaxed by several following minimizations with a decreasing force constant applied to the backbone atoms. After the relaxation, the system was heated to 250 K in 10 psec and then to 298.15 K in 40 psec. The production part of CDK2/ATP took 1.2 nsec, of CDK2/Cyclin A/ATP 2 nsec, of pT160–CDK2/Cyclin A/ATP 2.5 nsec, of all inhibited systems 3 nsec. The length of the simulations was chosen as compromise between the quality of configuration space sampling and the size of the studied systems (~60,000 atoms). The 2-fsec time integration step and particle-mesh Ewald (PME) methods for treating electrostatic interaction were used. All simulations were run under periodic boundary condition in the NpT ensemble at 298.16 K and a constant pressure of 1 atm. The SHAKE algorithm with a tolerance of 10−5, was applied to fix all bonds containing hydrogen atoms. The 8.0 Å cutoff was applied to treat nonbonding interactions. Coordinates were stored every 2 psec.
All analyses of MD simulations were carried out by CARNAL and PTRAJ modules of AMBER-6.0 (Case et al. 1999), by GROMACS (Spoel et al. 1991–2002), and by gOpenMol (Laaksonen 1992) program packages. Parametrization of the phosphorylated tyrosine residue (Table 3) was done according to the standard Cornell et al. (1995) scheme. The Gaussian98 program package (Frisch et al. 1998) was used for all necessary ab initio calculations at HF/6-31G(d) level. Partial atomic charges for ATP-Mg2+ and for phosphorylated threonine and tyrosine residues were prepared using the restrained electrostatic potential (RESP) procedure (Cornell et al. 1995).
Table 3.
Torsion angle parameters used in this study for phosphorylated tyrosine residue (pY15)
| Dihedral angle | IDIVF | PK(Vn/2) [kcal • mole−1] | Phase (°) | PN |
| O2-P-OS-CA | 3 | 4.7 | 162.2 | 3 |
| P-OS-CA-CA | 2 | 11.6 | 0.0 | −2 |
| P-OS-CA-CA | 2 | 6.4 | 180.0 | 4 |
These parameters were determined using the standard Cornell et al. scheme (1995). IDIVF, total number of torsion angles about a single bond that the potential applies to; PK, one-half of the barrier magnitude; Phase, phase shift of the torsion function; PN, periodicity of the torsion function.
Acknowledgments
We thank the MetaCenter (http://meta.cesnet.cz) for computer time. This work was supported by the Ministry of Education of the Czech Republic (I.B., Z.K., J.K.; Grant LN00A016) and the Grant Agency of the Czech Republic (M.O.; 301/02/0475). Their financial support is gratefully acknowledged. The authors thank P. Banáš for the parametrization of phosphotyrosine. Our thanks are also addressed to Dr. Jiřǐ Damborský(Brno) for valuable discussions on the paper. L.H. Jones (UK) and R. Turland (UK) are gratefully acknowledged for English corrections.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Dedicated to Professor Milan Kratochvíl on the occasion of his 80th birthday.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03578504.
References
- Aimes, R.T., Hemmer, W., and Taylor, S.S. 2000. Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: Role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry 39 8325–8332. [DOI] [PubMed] [Google Scholar]
- Brown, N.R., Noble, M.E.M., Endicott, J.A., and Johnson, L.N. 1999. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1 438–443. [DOI] [PubMed] [Google Scholar]
- Case, D.A., Pearlman, D.A., Caldwell, J.W., Cheatham III, T.E., Ross, W.S., Simmerling, C.L., Darden, T.A., Merz, K.M., Stanton, R.V., Cheng, A.L., et al. 1999. AMBER 6. University of California, San Francisco, CA.
- Cheng, A., Ross, K.E., Kaldis, P., and Solomon, M.J. 1999. Dephosphorylation of cyclin-dependent kinases by type 2C protein phosphatases. Genes Dev. 13 2946–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, A., Kaldis, P., and Solomon, M.J. 2000. Dephosphorylation of human cyclin-dependent kinases by protein phosphatase type 2C α and β 2 isoforms. J. Biol. Chem. 275 34744–34749. [DOI] [PubMed] [Google Scholar]
- Chow, J.P., Siu, W.Y., Ho, H.T., Ma, K.H., Ho, C.C., and Poon, R.Y.C. 2003. Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. J. Biol. Chem. 278 40815–40828. [DOI] [PubMed] [Google Scholar]
- Cook, A., Lowe, E.D., Chrysina, E.D., Skamnaki, V.T., Oikonomakos, N.G., and Johnson, L.N. 2002. Structural studies on phospho-CDK2/Cyclin A bound to nitrate, a transition state analogue: Implications for the protein kinase mechanism. Biochemistry 41 7301–7311. [DOI] [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, J.K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A 2nd generation force-field for simulation of proteins, nucleic-acids and organic-molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Coulonval, K., Bockstaele, L., Paternot, S., and Roger, P.P. 2003. Phosphorylations of cyclin-dependent kinase 2 revisited using two-dimensional gel electrophoresis. J. Biol. Chem. 278 52052–52060. [DOI] [PubMed] [Google Scholar]
- De Bondt, H.L., Rosenblatt, J., Jancarik, J., Jones, H.D., Morgan, D.O., and Kim, S.H. 1993. Crystal structure of cyclin-dependent kinase 2. Nature 363 595–602. [DOI] [PubMed] [Google Scholar]
- Endicott, J.A., Noble, M.E.M., and Tucker, J.A. 1999. Cyclin-dependent kinases: Inhibition and substrate recognition. Curr. Opin. Struct. Biol. 9 738–744. [DOI] [PubMed] [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Zakrzewski, V.G., Montgomery, J.A., Stratmann, R.E., Burant, J.C., et al. 1998. Gaussian98. Gaussian, Inc., Pittsburgh, PA.
- Gu, Y., Rosenblatt, J., and Morgan, D.O. 1992. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 11 3995–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagopian, J.C., Kirtley, M.P., and Stevenson, L.M. 2001. Kinetic basis for activation of CDK2/Cyclin A by phosphorylation. J. Biol. Chem. 276 275–280. [DOI] [PubMed] [Google Scholar]
- Hanks, S. and Quinn, A.M. 1991. Protein kinase catalytic domain sequence database: Identification of conserved features of primary structure and classification of family members. Methods Enzymol. 200 38–62. [DOI] [PubMed] [Google Scholar]
- Hemmer, W., McGlone, M., Tsigelny, I., and Taylor, S.S. 1997. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J. Biol. Chem. 272 16946–16954. [DOI] [PubMed] [Google Scholar]
- Holmes, J.K. and Solomon, M.J. 1996. A predictive scale for evaluation cyclin-dependent kinase substrates. A comparison of p34cdc2 and p33cdk2. J. Biol. Chem. 271 25240–25246. [DOI] [PubMed] [Google Scholar]
- ———. 2001. The role of Thr160 phosphorylation of Cdk2 in substrate recognition. Eur. J. Biochem. 268 4647–4652. [DOI] [PubMed] [Google Scholar]
- Jeffrey, P.D., Russo, A.A., Polyak, K., Gibbs, E., Hurwitz, J., Massague, J., and Pavletich, N.P. 1995. Mechanism of cdk activation revealed by the structure of a cyclin A–cdk2 complex. Nature 376 313–320. [DOI] [PubMed] [Google Scholar]
- Jinno, S., Hung, S.C., and Okayama, H. 1999. Cell cycle start from quiescence controlled by tyrosine phosphorylation of Cdk4. Oncogene 18 565–571. [DOI] [PubMed] [Google Scholar]
- Johnson, L.N. and Lewis, R.J. 2001. Structural basis for control by phosphorylation. Chem. Rev. 101 2209–2242. [DOI] [PubMed] [Google Scholar]
- Johnson, D.A., Akamine, P., Radzio-Andzelm, E., Madhusudan, and Taylor, S.S. 2001. Dynamics of cAMP-dependent protein kinase. Chem. Rev. 101 2243–2270. [DOI] [PubMed] [Google Scholar]
- Knockaert, M., Greengard, P., and Meijer, L. 2002. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol. Sci. 23 417–425. [DOI] [PubMed] [Google Scholar]
- Laaksonen, L. 1992. A graphics program for the analysis and display of molecular dynamics trajectories. J. Mol. Graph. 10 33–34. [DOI] [PubMed] [Google Scholar]
- Lew, J. 2003. MAP kinases and CDKs: Kinetic basis for catalytic activation. Biochemistry 42 849–856. [DOI] [PubMed] [Google Scholar]
- Morgan, D.O. 1996. The dynamics of cyclin dependent kinase structure. Curr. Opin. Cell Biol. 8 767–772. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13 261–291. [DOI] [PubMed] [Google Scholar]
- Morris, M.C., Gondeau, C., Tainer, J.A., and Divita, G. 2002. Kinetic mechanism of activation of the Cdk2/Cyclin A complex. J. Biol. Chem. 277 23847–23853. [DOI] [PubMed] [Google Scholar]
- Otyepka, M., Kříž, Z., and Koča, J. 2002. Dynamics and binding modes of free cdk2 and its two complexes with inhibitors studied by computer simulations. J. Biomol. Struct. Dynam. 20 141–154. [DOI] [PubMed] [Google Scholar]
- Pagano, M. 1998. Cell cycle control. New York University School of Medicine, New York.
- Philippopoulos, M. and Lim, C. 1995. Molecular dynamics simulation of E. coli ribonuclease H1 in solution: Correlation with NMR and X-ray data and insights into biological function. J. Mol. Biol. 254 771–792. [DOI] [PubMed] [Google Scholar]
- Poon, R.Y.C. and Hunter, T. 1995. Dephosphorylation of Cdk2 Thr160 by the cyclin-dependent kinase-interacting phosphatase KAP in the absence of cyclin. Science 270 90–93. [DOI] [PubMed] [Google Scholar]
- Rudolph, J., Epstein, D.M., Parker, L., and Eckstein, J. 2001. Specifity of natural and artificial substrates for human CDC25A. Anal. Biochem. 289 43–51. [DOI] [PubMed] [Google Scholar]
- Russo, A.A., Jeffrey, P.D., and Pavletich, N.P. 1996. Structural basis of cyclin-dependent kinase 2 activation by phosphorylation. Nat. Struct. Biol. 3 696–700. [DOI] [PubMed] [Google Scholar]
- Schulze-Gahmen, U., Brandsen, J., Jones, H.D., Morgan, D., Meijer, L., Veselý, J., and Kim, S.-H. 1995. Multiple modes of ligand recognition: Crystal structures of cyclin-dependent protein kinase 2 in complex with ATP and two inhibitors, olomoucine and isopenthenyladenine. Proteins 22 378–391. [DOI] [PubMed] [Google Scholar]
- Sebastian, B., Kakizuka, A., and Hunter, T. 1993. Cdc25M2 activation of cyclin-dependent kinases by dephosphorylation of threonine-14 and tyrosine-15. Proc. Natl. Acad. Sci. 90 3521–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoel, D.V.D., Buuren, A.R.V., Apol, E., Meulenhoff, P.J., Tieleman, P.D., Sijbers, A.L.T.M., Hess, B., Feenstra, K.A., Lindhal, E., Drunen, R.V., et al. 1991–2002. GROMACS. University of Groningen, Groningen, The Netherlands.
- Stevenson, L.M., Deal, M.S., Hagopian, J.C., and Lew, J. 2002. Activation mechanism of CDK2: Role of cyclin binding versus phosphorylation. Biochemistry 41 8528–8534. [DOI] [PubMed] [Google Scholar]
- Tsigelny, I., Greenberg, J.P., Cox, S., Nichols, W.L., Taylor, S.S., and Ten Eyck, L.F. 1999. 600 ps molecular dynamics reveals stable substructures and flexible hinge points in cAMP dependent protein kinase. Biopolymers 50 513–524. [DOI] [PubMed] [Google Scholar]
- Vriend, G. 1997. WHAT IF, 5.0 ed. EMBL, Heidelberg, Germany.
- Wang, J.M., Cieplak, P., and Kollman, P.A. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21 1049–1074. [Google Scholar]
- Watanabe, N., Broome, M., and Hunter, T. 1995. Regulation of the human Wee1Hu Cdk tyrosine 15-kinase during the cell cycle. EMBO J. 14 1878–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukerberg, L.R., Patrick, G.N., Nikolic, M., Humbert, S., Wu, C.L., Lanier, L.M., Gertler, F.B., Vidal, M., Van Etten, R.A., and Tsai, L.H. 2000. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 26 543–544. [DOI] [PubMed] [Google Scholar]