

Abstract
Penicillin G acylase (PGA) is a heterodimeric enzyme synthesized as a single-polypeptide precursor that undergoes an autocatalytic processing to remove an internal spacer peptide to produce the active enzyme. We constructed a single-chain PGA not dependent on autoproteolytic processing. The mature sequence of the β-domain was expressed as the N terminus of a new polypeptide, connected by a random tetra-peptide to the α-domain, to afford a permuted protein. We found several active enzymes among variants differing in their linker peptides. Protein expression analysis showed that the functional single-chain variants were produced when using a Sec-dependent leader peptide, or when expressed inside the bacterial cytoplasm. Active-site titration experiments showed that the single-chain proteins displayed similar kcat values to the ones obtained with the wild-type enzyme. Interestingly, the single-chain proteins also displayed close to 100% of functional active sites compared to 40% to 70% functional yield usually obtained with the heterodimeric protein.
Keywords: penicillin acylase, circular permutation, posttranslational processing, Tat secretion, Sec secretion
PGA is an industrially important enzyme used to produce 6-aminopenicillanic acid (6-APA) from penicillin G during the manufacturing of semisynthetic antibiotics. PGA from Escherichia coli is a periplasmic heterodimeric enzyme composed of two subunits (α- and β-subunits of 23.8 and 62.2 kD, respectively). The mature periplasmic protein is synthesized as a single-polypeptide cytoplasmic precursor containing a 26-amino acid signal peptide and a 54-amino acid connector peptide that joins the α- and β-chains. The PGA precursor is autocatalytically processed (Kasche et al. 1999) inside the bacterial periplasm to remove the spacer peptide and produce the mature enzyme. Because PGA expression in E. coli is limited by precursor secretion and maturation, much work has been reported on strategies aimed to improve the expression level of heterodimeric PGA (Lin et al. 2001).
Crystal structures of PGA are known for the dimeric, precursor, and substrate-bound forms of the enzyme (Duggleby et al. 1995; Done et al. 1998; Alkema et al. 2000; Hewitt et al. 2000; McVey et al. 2001). The crystal structure of the dimeric enzyme (Duggleby et al. 1995) shows a catalytic center that includes the N-terminal serine residue of the β-subunit. This particularity made PGA the founding member of the Ntn (N-terminal nucleophile) hydrolase superfamily (Brannigan et al. 1995). Structural analysis of different enzyme–substrate complexes (Done et al. 1998; Alkema et al. 2000; McVey et al. 2001), show ligand-induced conformational changes in the enzyme-binding pocket. In particular, residue αF146 is restricting the active-site entry and needs to move away to allow access to bulky substrates, like penicillin G.
In vitro refolding studies of PGA subunits have suggested a key role for the β-subunit during heterodimer folding: The 3-subunit is unable to fold properly in the absence of the α-subunit (Lindsay and Pain 1991). Furthermore, the successful production of functional enzyme when the protein subunits are separately expressed inside the cytoplasm (Burtscher and Schumacher 1992) indicates that the presence of the α-subunit in the same polypeptide precursor is not necessary for the successful assistance in the folding of the β-subunit.
The structure of dimeric PGA (Duggleby et al. 1995) shows that the C terminus of the β-subunit is nearby the N terminus of the β-subunit. Thus, we decided to attempt the construction of a single-chain PGA by joining the α- and β-subunits with a short peptide and generating new N and C termini (a permutation). If functional, this polypeptide arrangement will no longer depend on autoproteolytic processing.
Results
Single polypeptide, permuted PGA design
The first four amino acids from the α-subunit (1EQSS4) and the last two amino acids from the β-subunit (556QR557) were removed to leave new ends of the antiparallel β-strand elements (6EIKIVRD12 from the α-subunit and 547KESQEVLH554 from the β-subunit), which will be now less than 5 Å apart (Fig. 1 ▶). We chose to connect the β-strand elements with four amino acids to favor the formation of a potential β-turn. To increase the probability of having a functional single-chain enzyme, we connected the β- and α-subunits with a random tetra-peptide linker. The single-chain PGA was fused to a β-lactamase signal peptide previously selected to export a β-lactamase protein with a serine residue at position +1 of the mature protein (Palzkill et al. 1994). The final construct was ligated to plasmid pT4Bla (Osuna et al. 2002) and electroporated into bacterial cells (see Supplementary Fig. 1 and Materials and Methods section for details about the construction strategy). The final library size was around 1.2 × 106 variants (with a complexity of 324 = 1.05 × 106).
Figure 1.
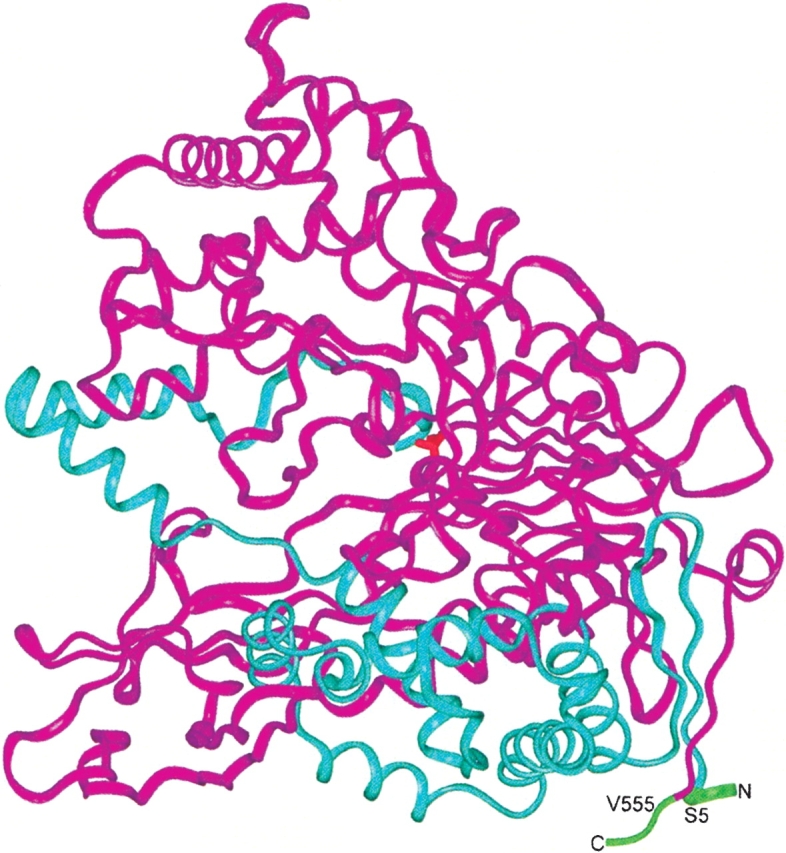
The PGA dimeric structure. The β-subunit is shown in magenta, and the α-subunit, in blue ribbons. The polypeptide regions trimmed from the N terminus of the α-subunit and from the C terminus of the β-subunit are indicated in green. The amino acid residues to be connected with the four amino acids linker are labeled. In red, at the center of the molecule, the catalytic serine residue is indicated.
Selection of single-chain PGA active variants
The single-chain library described above was selected for penicillin G resistance to already 6-APA resistant E. coli hosts (HB101 mutant [Chou et al. 1999] generously provided by Dr. C. Perry Chou, or E. coli cells containing a β-lactamase with a better specificity to 6-APA compared to penicillin G [Osuna et al. 1995]). Previous attempts in our laboratory to construct a linker-specific, circularly permuted single-chain PGA enzyme resulted in expressed protein products showing detrimental effects on bacterial viability (data not shown). Thus, we also decided to construct a library of single-chain PGA variants in a cloning vector with tight expression control (pThioC; Invitrogen; see Supplementary Fig. 1 and Materials and Methods section). Colony counts in repressed and induced states indicate that around 40% of the library is not able to grow in the induced state (data not shown). The preference for glycine residues, in particular at position i+1 of the linker peptide (Supplementary Table 1), may be due to a G nucleotide bias during the oligonucleotide synthesis, because we observed the same tendency when the library was produced in a cloning vector unable to produce a complete single-chain protein (see Supplementary Table 1).
Specific activity characterization of single-chain variants
Specific activity data (Table 1) using total protein extracts from pT4Bla and pThioC expressed clones, showed good activity for some single-chain proteins compared with the wild-type enzyme. Western analysis of the same total protein extracts shown in Table 1 indicated that lower amounts of single-chain proteins are being produced compared to the heterodimeric enzyme (data not shown).
Table 1.
Linker sequences of functional single-chain penicillin acylases
| Enzyme | Specific activitya % |
| WTb | 100 |
| NEGMb | 22 |
| PGLR | 15 |
| GGEA | 14 |
| DPAGb | 35 |
| GGLK | 8 |
| EPQR | 5 |
| GGG | 18 |
| RGAG | 27 |
| PGVG | 18 |
| GDAL | 16 |
| QGAG | 13 |
| GGSG | 7 |
| RAAV | 3 |
| AESSc | 29 |
| GARDc | 39 |
a Measured using an XL1-Blue total crude soluble-protein extract by the PDAB method. The PGA wild-type value (28.9 μg per min of 6-APA formed using 100 μg of soluble protein) was taken as 100%.
b Those clones were expressed in both pT4Bla and pThioC expression vectors.
c Functional clones obtained from the library constructed with the pThioC expression vector.
Secretion signal effects in the production of functional and therefore, correctly processed, single-chain protein
Because we required a precise design of the signal peptide, so as to afford the proper processing that generates the active site residue, and because the natural PGA precursor is secreted by the unusual twin-arginine translocation (TAT) pathway (Ignatova et al. 2002), we decided to explore the effects of different signal peptides on the production yield of the expressed single-chain proteins. Inspection of Table 1 indicates that variants with connecting peptides NEGM, DPAG, RGAG, AESS, and GARD show the best specific activities compared to the wild-type values. Therefore, we decided to fuse the NEGM, DPAG, and GARD variants to different secretion signals (Fig. 2 ▶ and Supplementary Fig. 1B; see Materials and Methods section for details). For activity, the single-chain penicillin acylases must be transported to the periplasm and processed to liberate the serine at the N-terminal position of the mature protein. Thus, we expect that all active variants are located in the periplasmic space. If single-chain variants are expressed in the cytoplasm, the N-terminal methionine should be removed to produce an active protein. We assume that the inactive variants were not transported and retained the leader peptide, or that they were processed in an incorrect way during export to the periplasm.
Figure 2.

Western blot of single-chain variants expressed with or without a leader peptide. (A) Variant NEGM was fused to different leader peptides or expressed without a leader peptide. (B) The three indicated variants were expressed without a leader peptide or (C) with a modified β-lactamase leader peptide. The PGA wild-type protein was expressed using its natural leader peptide. All the cultures were OD600 normalized. T means a total protein sample, and S means a soluble protein sample. The upper band in the PGA-WT lane corresponds to the unprocessed polypeptide (precursor protein) and the lower band corresponds to the β-subunit. The percentages indicated below the soluble protein bands in parts B and C correspond to the relative specific activities measured using 20 times the protein amount shown in the figure; 11.66 μg per min of produced 6-APA using 100 μg of soluble protein of the wild-type clone was taken as 100%.
We made no attempts to fractionate the samples other than separation of insoluble and soluble material, but Western analysis of protein expression showed that variants containing the PGA wild-type signal peptide were found mainly as insoluble aggregates (more than 95% of the produced NEGM variant; Fig. 2A ▶). Variants containing the TorA leader peptide were produced in good soluble yield (around 30% of the NEGM total protein; Fig. 2A ▶). Proteins using the β-lactamase signal peptide were produced in lower total yields than that obtained using the TAT-dependent leader peptides, but were significantly found at the soluble protein fraction (around 40% of the produced protein; Fig. 2C ▶). The single-chain variants expressed without a signal-peptide were mostly found at the soluble protein fraction (around 40% of the produced protein) and were produced in good yields (Fig. 2B ▶). Functional proteins were only obtained when the single-chain variants were targeted to the bacterial periplasmic space using the β-lactamase leader (Fig. 2C ▶) or when produced inside the bacterial cytoplasm (Fig. 2B ▶). The absence of function of the different variants when attempted to be secreted with TAT-specific signal peptides indicates incompatibility of the single-chain protein with the TAT secretion pathway or that an incorrect processing of the signal peptide could be taking place. The similar activities shown by the cytoplasmically or periplasmically expressed variants (Fig. 2 ▶), despite better expression yields obtained with the former, indicate that a poor removal of the N terminus methionine could be taking place in the bacterial cytoplasm.
Catalytic parameter measurements for two single-chain variants
Active-site titration of penicillin acylase with PMSF has been used (Siewinsky et al. 1984; del Rio et al. 1995; Janssen et al. 2002; van Langen et al. 2002) to quantify the amount of catalytically competent enzyme present in crude bacterial extracts, whole-cell preparations, or immobilized enzyme preparations. The active site titration experiments provide the necessary information for the quantification of catalytic parameters such as kcat and help to minimize the known presence of a catalytically incompetent protein fraction in heterodimeric enzyme preparations. The kcat values, obtained from PMSF inhibition studies, were similar for all the assayed proteins (Table 2). Analysis of the PMSF inhibition course, for the three enzymes tested, showed a linear inhibition behavior in all cases (Fig. 3 ▶). This means that the intersection with the abscissa in the graph should correspond to the active site molar concentration and the slope to kcat. The active site titration experiments (Fig. 3 ▶; Table 2) also indicate close to 100% of active conformation for the single-chain proteins compared to around 60% of active conformation for our semipure wild-type enzyme. Western experiments using normalized amounts of the purified proteins (Supplementary Fig. 2) did not show any proteolysis of the single-chain proteins during the experimental course.
Table 2.
Active-site titration experiments for some variants and the heterodimeric protein
| kcata | Active site concentration μM PMSF | ||
| Enzyme | sec−1 | Experiment 1 | Experiment 2 |
| WT | 25.1 ± 1.4 | 0.28 (0.55)b | 0.56 (1.1) |
| NEGM | 36.8 ± 2.7 | 0.13 (0.14) | 0.22 (0.28) |
| DPAG | 15.5 ± 0.8 | 0.20 (0.21) | 0.54 (0.62) |
a Determined by PMSF inhibition studies (Fig. 3 ▶).
b The data in parentheses indicate the protein molar concentration (in μM) used for the inhibition experiments. The different inhibition experiments consisted in modifying the amount of initial protein before measuring the active-site molar concentration.
Figure 3.

Plot of residual PGA activity vs. PMSF inhibitor concentration. The open and closed symbols indicate different concentration of protein used during the inhibition assays (enzyme concentration values are shown in Table 2). The slope of the parallel lines was taken as the kcat and the line intersection with the abscissa indicates the active site molar concentration. Circles, diamonds, and triangles correspond to PGA-WT, DPAG, and NEGM data, respectively.
Discussion
We have constructed an active, single-polypeptide, permuted penicillin G acylase. The key for obtaining active single-chain PGA variants was to randomize the linker that fused the β- and α-protein domains and then use a selection approach to identify the active variants. The importance of the selection step was made evident when a high proportion of putative variants with a strong detrimental effect to bacterial viability were detected. In other words, a lot of different linker sequences joining the β- and α-subunits are not allowed due to expression impairment of bacterial viability.
Recently, it was proposed that the wild-type E. coli PGA signal peptide directs the protein to the twin-arginine translocation (TAT) pathway (Ignatova et al. 2002). Along with the wild-type PGA signal peptide, we tested a classical TAT signal peptide (the TorA signal peptide). None of the TAT signal peptides tested produced functional single-chain PGA.
The fact that the different single-chain proteins were successfully produced when expressed without a leader peptide indicates that the new domain arrangement in the polypeptide chain is not impairing the folding process. However, adding the PGA wild-type signal peptide to the single-chain proteins resulted in aggregated protein products. There can be several reasons that could explain the observed behavior of the TAT routed single-chain proteins. Because the proteins must fold first before being transported by the TAT pathway (Palmer and Berks 2003), the simplest explanation would be an impairment of the folding capabilities of the β-subunit as a result of the extension in the N terminus of the protein with the wild-type leader peptide. Additionally, the new domain arrangement in the polypeptide could make the proteins not compatible with the TAT secretion pathway as reported for other proteins (Stanley et al. 2002).
Single-chain proteins using the Sec-dependent β-lactamase signal peptide were correctly processed, as demonstrated by the production of functional protein. The success of the β-lactamase leader to correctly secrete the single-chain PGA proteins could be explained by the suggested capabilities of this leader peptide to collect a set of protein chaperones that help in the translocation process (Zahn et al. 1994; Wild et al. 1996; Kebir and Kendall 2002). It is also possible that our modifications of the β-lactamase leader (see Results section) could be more permissive to the correct processing of the single-chain proteins. Finally, the fact that a known Sec-dependent signal peptide is important to produce functional (and therefore, fully processed) protein is not a proof that the single-chain proteins are following the Sec secretion pathway. Repeating the expression experiments using the reported TAT-deficient bacterial strains could be the next step to help clarify this point.
The successful production of functional PGA when the subunits are separately expressed inside the cytoplasm was recently reported as bacterial strain dependent (Burtscher and Schumacher 1992). The authors reported a wild-type PGA expression level of about 10% that of the subunits using the same permissive bacterial strain. Nonetheless, the wild-type PGA showed a better specific activity than the one obtained with PGA formed from separately expressed subunits. In our work, the single-chain variants were expressed in lower yields than the ones obtained with wild-type PGA. However, the single-chain PGA specific activity values were similar to the ones showed by the wild-type PGA.
The better performance of single-chain PGA as compared with PGA formed from separately expressed subunits could be explained by a more successful production of functional enzyme with the former as shown by the 100% yield of functional active sites displayed by the single-chain variants compared with 40% to 70% functional yield obtained with the wild-type enzyme, and also reported during in vitro enzyme refolding (Lindsay and Pain 1991; Shamolina and Svedas 2000). Joining the β- and α-subunits in a single polypeptide should improve the interdomain contacts, and might help the calcium ion to define the structure of the binding site (McVey et al. 2001).
We believe our results open new, alternative avenues for PGA production. A natural next step would be to improve the single-chain PGA production yield by using the selection method described in this work. Directed evolution experiments could be planned to increase the amount of soluble single-chain protein in the periplasm of bacteria by looking for mutations that eliminate the detected aggregation tendencies of the single-chain penicillin acylases here described.
Materials and methods
All chemicals were from Sigma. Restriction enzymes and T4 DNA ligase were from New England Biolabs. Taq Gold polimerase was from PE Applied Biosystems. PCR and Plasmid DNA purification kits were from Qiagen. Antimouse secondary alkaline phosphatase-conjugated antibody and alkaline phosphatase substrates were from ICN. Oligonucleotides were synthesized in an in-house facility (Instituto de Biotecnología). PCR reactions were performed in an iCycler thermal cycler from Bio-Rad.
Single-chain PGA gene construction
A single-chain PGA gene containing a stop codon in the connector peptide was used as a DNA template. The α-domain coding DNA was amplified with the oligonucleotides ACLINK (5′ CGCAG GAAGTGTTGCACGTTNNG/CNNG/CNNG/CNNG/CAGTGAG ATAAAGATTGTT 3′ [Nucleotides in bold code for the random connector; underlined nucleotides code for the first amino acids of the α domain; and nucleotides in italics code for the last amino acids of the β-domain.]) and ACTAP (5′ AGCCGTGGATCGT GGGTCGACTCATTATGCTGTTTGCGA 3′ [Underlined nucleotides are complementary to the part of the gene that codes for the last amino acids of the α-domain including a new stop codon; nucleotides in italics correspond to the SalI restriction site.]). The PCR product was used as a megaprimer, and extended to reach a MluI restriction site upstream in the α-domain coding DNA using the backward oligonucleotide BMLU (5′ GGTGTACCGCAGGC CGCA 3′). The final product was digested with MluI and SalI, and used to replace the original fragment in the cloning vector (pT4-BLA [Osuna et al. 2002] containing the single-chain PGA gene). The PGA library was selected on penicillin G plates at antibiotic concentrations above the background levels (around 25 or 60 μg/ mL, depending on the strain used).
The pTHIOC library was made using the following strategy: Using the same DNA template previously described, we amplified the gene portion coding for the α-domain with the oligonucleotides ACLINK (see above) and ACTAPSPETAG (see below); and the gene portion coding for the β-domain was amplified with the oligonucleotides PSNDE (5′ GGGCCATATGAGTATTCAACAT TTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTT 3′ [Underlined nucleotides code for the start of the β-lactamase leader peptide and the nucleotides in italics indicate the NdeI restriction site.]) and 20B-C (see below). The two purified PCR products were combined to construct the complete single-chain gene by PCR using the external oligonucleotides PSNDE and ACTAPSPETAG. The final product was digested with NdeI and SpeI restriction enzymes and cloned into the PTHIOC vector prepared with the same restriction enzymes.
Expression studies with different single-chain PGA
XL1-Blue was transformed by electroporation with the different expression plasmids and grown with shaking at 30°C in 5 mL of LB medium containing the appropriate antibiotic. When the culture OD600 reached 0.6–0.8, the expression of the proteins was induced with 20 μM of IPTG, and growth was continued for an additional 3 h at 30°C. Before pelleting the cells, the cultures OD600 were measured and the cell pellets were resuspended in PBS at appropriate volumes (around 500 μL) to normalize all samples to the same OD600. The resuspended cell pellets were disrupted by sonication and total and soluble (collected by centrifugation) protein samples were taken.
Primer NDENEW (5′ GGAATTCCATATGAGTATTCAACAT TTCCG 3′) and primer ACTAPSPETAG (5′ AGCCGTGGATC GTGGACTAGTAGAAGCGTAGTCCGGAACGTCGTACGG GTATGCTGTTTGGGA 3′) were used to amplify the NEGM and DPAG single-chain variant genes. These primers are complementary (underlined parts in the previous primers) to the start of the β-lactamase gene coding for the leader peptide (NDENEW) and to the end of the circularly permuted penicillin acylase gene coding for the last amino acids of the α-domain (ACTAPSPETAG). Nucleotides in bold code for a decapeptide tag recognized by monoclonal antibody 12Ca5 (I.A. Wilson, The Scripps Research Institute). PCR products were digested with NdeI and SpeI restriction enzymes (recognition sites shown in italics in the previous primers) and cloned in the pTHIOC vector prepared with the same restriction enzymes.
To introduce the different leader peptides to the single-chain variants we designed the following oligonucleotides: SPA-ALA (5′ GTTACTGCTTCCCTGATGTATTATTGGAGCTTCCCTGC ACTGGCTAGCAATATGTGGGTG 3′) and SPA-NDE1 (5′ TC GTGGCATATGAAAAATCGCAATCGTATGATCGTGAACTG TGTTACTGCTTCCCTGATG 3′) were used in two sequential PCR reactions to introduce the wild-type leader peptide. (The underlined part in SPA-ALA, and in all the following oligonucleotides, is complementary to the start of the β-domain of the single-chain PGA gene. The NdeI restriction site in SPA-NDE1, and in all the following oligonucleotides, is shown in italics.) TORA1 (5′ GGCGGGCATGCTGGGTCCGAGCCTGCTGACCCCGCGCC GTGCGACCGCGAGCAATATGTGGGTG 3′), TORA2 (5′ TC TGGCGCAGCTGGGCGGTCTGACCGTGGCGGGCATGCTG GGT 3′), and TORA3 (5′ TCGTGGCATATGAACAACAATG ATCTGTTTCAGGCGAGCCGCCGTCGCTTTCTGGCGCAGC TG 3′) were used in three sequential PCR reactions to introduce the TorA leader peptide.
To finish the PCR fragments, all of the constructions described above were extended using the backward primer 20B-C (5′ GCAACACTTCCTGCGACTCC 3′) that is complementary to the part of the single-chain gene coding for the last amino acids of the β-domain. The final PCR products were digested with NdeI and BglII, and cloned into the respective vector (carrying the different single-chain variants) previously digested with the same restriction enzymes.
To construct the cytoplasm expressed single-chain variants, we carried out a PCR reaction with oligonucleotide NPSNDEI (5′ CGCGGCCATATGAGCAATATGTGGGTGATC 3′) and the above described 20B-C primer. The final PCR product was cloned as described above.
To construct the wild-type penicillin acylase gene in the pThioC vector, primer SPA-NDEI described above and primer PACTAG (5′ TCGTGGCTCGAGTCATTAAGAAGCGTAGTCCGGAA CGTCGTACGGGTATCTCTGAACGTG 3′) were used in a PCR reaction to amplify the wild-type gene. The underlined part in PACTAG is complementary to the end of the gene that codes for the last amino acids of the β-domain and the nucleotides in bold code for the decapeptide tag previously described. The final PCR product was digested with NdeI and XhoI, and cloned in the pTHIOC vector prepared with the same restriction enzymes. The final constructs were verified by nucleotide sequence.
Preparation of cell extracts and Western blotting
Cells from an overnight culture were used to inoculate 5 mL of LB medium containing the appropriate antibiotics. If needed, at around 0.8 OD of cell density, all cultures were induced with IPTG at a final concentration of 0.02 M. After 3 to 4 h of induction, the final OD of the culture was measured and the cells were collected by centrifugation. The cell pellet was resuspended with 300 to 500 μL of PBS buffer, depending on the OD reached by any individual culture. The cell suspension was sonicated (three 30-sec periods at 0°C) with a model 450 Branson sonifier. Total (T) protein samples were taken before subjecting the lysed cells to an extra centrifugation step aimed to remove any aggregated protein and the cell debris. After the centrifugation step, soluble (S) protein samples were taken to be used for the Western blotting or to measure the specific activity (see below).
Western blotting was performed according to general procedures (Towbin et al. 1979). BSA 3% in PBS was used to block any nonspecific sites in the nitrocellulose paper. For PGA protein detection, a mouse antibody specific for the decapeptide sequence N-YPYDVPDYAS-C (introduced at the end of the wild-type or single-chain PGAs, see above) was used as the first antibody. The PGA band was revealed using an antimouse secondary alkaline phosphatase-conjugated antibody and ready-to-use alkaline phosphatase substrates from ICN.
Purification of wild-type and single-chain PGA containing the linkers NEGM and DPAG
Single-chain proteins and wild-type PGA, modified to include a histidine tail at the C terminus, were expressed and purified following the manufacturer’s instructions (pET system; Novagene). Briefly, cells were grown at 30°C in M9 medium. When OD600 reached 0.4, 0.5 mM IPTG was added to the culture to induce gene expression. After 8 h of induction, cells were harvested by centrifugation. The pellet was resuspended in 20 mL of PBS by sonication and the cell debris removed by centrifugation at 4°C. The protein solutions were applied to nickel columns (Qiagen) previously equilibrated with PBS. The bound protein was exhaustively washed using the same buffer and then eluted with 250 mM imidazole. To improve the purity of the proteins, they were precipitated with 5% to 30% ammonium sulfate, depending on the protein. The precipitate containing PGA was resuspended in PBS. The protein solution was thoroughly dialyzed against HEPES 20 mM. The protein yields were around 1 mg of purified protein per liter of culture.
Active site titration and specific activity measurements
PGA active site molar concentration was determined by direct titration with phenylmethylsulphonyl fluoride (PMSF; Siewinsky et al. 1984). PGA was incubated at 25°C for 30 min with PMSF (from 0 to 1 μM) in 10 mM phosphate buffer pH 7.8. The residual activity measured for the active-site titration, and the specific activity assays using crude soluble-protein extracts were determined under substrate saturation (2% penicillin G final concentration, well above the reported Km values for the heterodimeric enzyme) by the PDAB method (Balasingham et al. 1972). Assays for PGA specific activity done with crude soluble-protein extracts coming from cells harboring the pThioC expression vector required a pre-incubation with clavulanic acid (10 mM final) to inhibit the β-lactamase encoded in the plasmid. Briefly, 80 μL of soluble protein was, when needed, incubated at 30°C for 10 min with 10 μL of a 100 mM solution of clavulanic acid. Later, 10 μL of a 20% solution of penicillin G was added and the incubation continued by 10 min. The reaction was stopped by the addition of 2.4 mL of absolute ethanol, and the 6-APA produced during the reaction was revealed by the addition of 1.25 mL of a 1% solution of paradimethyl amino-benzaldehyde. The reaction product between 6-APA and the PDAB was monitored in a DU-650 Beckman spectrophotometer at a wavelength of 415 nm. To quantify the amount of 6-APA produced in the different reactions a calibration curve made with known amounts of pure 6-APA was used to interpolate the produced 6-APA values. All measurements were made at least by duplicate.
Electronic supplemental material
Table 1 shows the different linker sequences joining the β- and α-subunits. Figure 1 ▶ describes the different gene construction strategies. Figure 2 ▶ shows the purification of two single-chain penicillin acylases.
Acknowledgments
We thank Paul Gaytan and Eugenio López for the oligonucleotides, Filiberto Sánchez for technical assistance, and René Hernandez and Maricela Olvera for DNA sequencing. Administrative help is highly appreciated to Nelly Mellado. G.F. was supported by a partial fellowship from DGAPA/UNAM. CONACyt grant NC230 and PAPIIT grant IN223199 are fully appreciated.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03436604.
Supplemental material: see www.proteinscience.org
References
- Alkema, W.B.L., Hensgens, C.M.H., Kroezinga, E.H., de Vries, E., Floris, R., van der Laan, J.-M., Dijkstra, B.W., and Janssen, D.B. 2000. Characterization of the β-lactam binding site of penicillin acylase of Escherichia coli by structural and site-directed mutagenesis studies. Protein Eng. 13 857–863. [DOI] [PubMed] [Google Scholar]
- Balasingham, K., Warburton, D., Dunnill, P., and Lilly, D. 1972. The isolation and kinetics of penicillin amidase from Escherichia coli. Biochem. Biophys. Acta 276 250–256. [DOI] [PubMed] [Google Scholar]
- Brannigan, J.A., Dodson, G., Duggleby, H.J., Moody, P.C.E., Smith, J.L., Tomchick, D.R., and Murzin, A.G. 1995. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 378 416–419. [DOI] [PubMed] [Google Scholar]
- Burtscher, H. and Schumacher, G. 1992. Reconstitution in vivo of penicillin G acylase activity from separately expressed subunits. Eur. J. Biochem. 205 77–83. [DOI] [PubMed] [Google Scholar]
- Chou, C.P., Yu, C-C., Lin, W-J., Kuo, B-Y., and Wang, W-C. 1999. Novel strategy for efficient screening and construction of host/vector systems to overproduce penicillin acylase in Escherichia coli. Biotechnol. Bioeng. 65 219–226. [PubMed] [Google Scholar]
- del Rio, G., Rodriguez, M.-E., Munguía, M.-E., López-Munguía, A., and Soberón, X. 1995. Mutant Escherichia coli penicillin acylase with enhanced stability at alkaline pH. Biotechnol. Bioeng. 48 141–148. [DOI] [PubMed] [Google Scholar]
- Done, S.H., Brannigan, J.A., Moody, P.C.E., and Hubbard, R.E. 1998. Ligand-induced conformational change in penicillin acylase. J. Mol. Biol. 284 463–475. [DOI] [PubMed] [Google Scholar]
- Duggleby, H.J., Tolley, S.P., Hill, C.P., Dodson, E.J., Dodson, G., and Moody, P.C.E. 1995. Penicillin acylase has a single-amino-acid catalytic centre. Nature 373 264–268. [DOI] [PubMed] [Google Scholar]
- Hewitt, L., Kasche, V., Lummer, K., Lewis, R.J., Murshudov, G.N., Verma, C.S., Dodson, G.G., and Wilson, K.S. 2000. Structure of a slow processing precursor penicillin acylase from Escherichia coli reveals the linker peptide blocking the active-site cleft. J. Mol. Biol. 302 887–898. [DOI] [PubMed] [Google Scholar]
- Ignatova, Z., Hörnle, C., Nurk, A., and Kasche, V. 2002. Unusual signal peptide directs penicillin acylase from Escherichia coli to the Tat translocation machinery. Biochem. Biophys. Res. Commun. 291 146–149. [DOI] [PubMed] [Google Scholar]
- Janssen, M.H.A., van Langen, L.M., Pereira, S.R.M., van Rantwijk, F., and Sheldon, R.A. 2002. Evaluation of the performance of immobilized penicillin G acylase using active-site titration. Biotechnol. Bioeng. 78 425–432. [DOI] [PubMed] [Google Scholar]
- Kasche, V., Lummer, K., Nurk, A., Piotraschke, E., Rieks, A., Stoeva, S., and Voelter, W. 1999. Intramolecular autoproteolysis initiates the maturation of penicillin amidase from Escherichia coli. Biochem. Biophys. Acta 1433 76–86. [DOI] [PubMed] [Google Scholar]
- Kebir, M.O. and Kendall, D.A. 2002. SecA specificity for different signal peptides. Biochemistry 41 5573–5580. [DOI] [PubMed] [Google Scholar]
- Lin, W.-J., Huang, S.-W., and Chou, C.P. 2001. DegP-coexpression minimizes inclusion-body formation upon overproduction of recombinant penicillin acylase in Escherichia coli. Biotechnol. Bioeng. 73 484–492. [DOI] [PubMed] [Google Scholar]
- Lindsay, C.D. and Pain, R.H. 1991. Refolding and assembly of penicillin acylase, an enzyme composed of two polypeptide chains that result from proteolytic activation. Biochemistry 30 9034–9040. [DOI] [PubMed] [Google Scholar]
- McVey, C.E., Walsh, M.A., Dodson, G.G., Wilson, K.S. and Brannigan, J.A. 2001. Crystal structures of penicillin acylase enzyme–substrate complexes: Structural insights into the catalytic mechanism. J. Mol. Biol. 313 139–150. [DOI] [PubMed] [Google Scholar]
- Osuna, J., Viadiu, H., Fink, A.L., and Soberón, X. 1995. Substitution of Asp for Asn at position 132 in the active site of TEM β-lactamase. J. Biol. Chem. 270 775–780. [PubMed] [Google Scholar]
- Osuna, J., Pérez-Blancas, A., and Soberón, X. 2002. Improving a circularly permuted TEM-1 β-lactamase by directed evolution. Protein Eng. 15 463–470. [DOI] [PubMed] [Google Scholar]
- Palmer, T. and Berks, B.C. 2003. Moving folded proteins across the bacterial cell membrane. Microbiology 149 547–556. [DOI] [PubMed] [Google Scholar]
- Palzkill, T., Le, Q-Q., Wong, A., and Botstein, D. 1994. Selection of functional signal peptide cleavage sites from a library of random sequences. J. Bacteriol. 176 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamolina, T.A. and Svedas, V.K. 2000. Reactivation of heterodimer and individual subunits of penicillin acylase from E. coli after inactivation in urea. Biochemistry (Moscow) 65 672–676. [PubMed] [Google Scholar]
- Siewinsky, M., Kuropatwa, M., and Szewczuk, A. 1984. Phenylalkylsulfonyl derivatives as covalent inhibitors of penicillin amidase. Hoppe Seyler’s Physiol. Chem. 635 829–837. [DOI] [PubMed] [Google Scholar]
- Stanley, N.R., Sargent, F., Buchanan, G., Shi, J., Stewart, V., Palmer, T., and Berks, B.C. 2002. Behaviour of topological marker proteins targeted to the Tat protein transport pathway. Mol. Microbiol. 43 1005–1021. [DOI] [PubMed] [Google Scholar]
- Towbin, H., Staehelin, T., and Gordon, J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. 76 4350–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Langen, L.M., Janssen, M.H.A., Oosthoek, N.H.P., Pereira, S.R.M., Svedas, V.K., van Rantwijk, F., and Sheldon, R.A. 2002. Active site titration as a tool for the evaluation of immobilization procedures of penicillin acylase. Biotechnol. Bioeng. 79 224–228. [DOI] [PubMed] [Google Scholar]
- Wild, J., Rossmeissl, P., Walter, W.A., and Gross, C.A. 1996. Involvement of the DnaK-DnaJ-GrpE chaperone team in protein secretion in Escherichia coli. J. Bacteriol. 178 3608–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn, R., Axmann, S.E., Rücknagel, K-P., Jaeger, E., Laminet, A.A., and Plückthun, A. 1994. GroEL recognizes the signal sequences of β-lactamase precursor. J. Mol. Biol. 242 150–164. [DOI] [PubMed] [Google Scholar]