

Abstract
Pseudomonas aeruginosa alcohol dehydrogenase (PaADH; ADH, EC 1.1.1.1) catalyzes the reversible oxidation of primary and secondary alcohols to the corresponding aldehydes and ketones, using NAD as coenzyme. We crystallized the ternary complex of PaADH with its coenzyme and a substrate molecule and determined its structure at a resolution of 2.3 Å, using the molecular replacement method. The PaADH tetramer comprises four identical chains of 342 amino acid residues each and obeys ~222-point symmetry. The PaADH monomer is structurally similar to alcohol dehydrogenase monomers from vertebrates, archaea, and bacteria. The stabilization of the ternary complex of PaADH, the coenzyme, and the poor substrate ethylene glycol (kcat = 4.5 sec−1; Km > 200 mM) was due to the blocked exit of the coenzyme in the crystalline state, combined with a high (2.5 M) concentration of the substrate. The structure of the ternary complex presents the precise geometry of the Zn coordination complex, the proton-shuttling system, and the hydride transfer path. The ternary complex structure also suggests that the low efficiency of ethylene glycol as a substrate results from the presence of a second hydroxyl group in this molecule.
Keywords: alcohol dehydrogenase, Pseudomonas aeruginosa, ternary complex, NADH, ethylene glycol, ion-pair network, proton shuttling, 3D structure
ADHs are oxidoreductases that catalyze the reversible oxidation of a wide range of primary and secondary alcohols into the corresponding carbonyl compounds, using NAD+ or NADP+ as coenzymes. These ubiquitous enzymes are present virtually in all life forms, from bacteria to vertebrates, and can be classified by size into three major classes (Jornvall 1994): (I) nonmetallic, short-chain ADHs having about 250 amino acid residues per polypeptide chain, for example, Drosophila ADH (Villarroya et al. 1989; Benach et al. 1998); (II) medium-chain, mostly zinc-containing ADHs having 350 to 375 residues per chain, for example, bacterial ADH (Bogin et al. 1998; Karlsson et al. 2003), archaeon ADH (Ammendola et al. 1992), yeast ADH (Jornvall 1977), and mammalian ADH (Jornvall 1970); and (III) long-chain ADHs having more than 700 residues per polypeptide chain, for example, Acetobacter acetii ADH (Inoue et al. 1989) and Escherichia coli ADH (Goodlove et al. 1989). Several representative enzymes of the medium-chain zinc-containing ADH family have been characterized structurally, for example, bacteria (Korkhin et al. 1998), archaea (Esposito et al. 2002; Guy et al. 2003), cod (Ramaswamy et al. 1996), and human (Hurly et al. 1994; Xie et al. 1997; Niederhut et al. 2001), with horse liver ADH (Eklund et al. 1977) serving as the archetype of this class. Medium-chain ADHs are either homotetramers, as in bacteria, archaea, and yeast, or homodimers, as in plants and vertebrates.
Here we describe a novel structure of the medium-chain, tetrameric NAD-dependent PaADH (EC 1.1.1.1), obtained from an opportunistic bacterial pathogen. The enzyme catalyzes the reversible oxidation of primary and secondary alcohols to the corresponding aldehydes and ketones, using NAD as coenzyme, and shares about 30% sequence identity with other structurally characterized ADHs. The PaADH structure shows two interesting, unique features: a five-member ion-pair network at the intersection of three monomers and a breakdown of the twofold symmetry initiated by hydrogen bond donor–acceptor recognition at one of the dimer interfaces.
The mechanism of action of horse liver ADH (HLADH) has been studied in detail with a variety of benzyl- and halogen-substituted alcohols (for reviews, see, e.g., Eklund et al. 1994; Plapp 1994; Fersht 1999). The catalytic reaction proceeds predominantly by the ordered bi-bi mechanism: E →E–NAD+ →E–NAD+–RCH2OH →E–NADH–RCHO →E–NADH →E, but at high alcohol concentration, the reaction is inhibited by formation of an abortive E–NADH–RCH2OH complex (Shearer et al. 1993). The alcohols directly coordinate the catalytic Zn as alcoholates, first suggested by the observation of a linear dependence of the ionization pKs of the alcohols in binary complexes on the pKs of the free alcohols (Kvassman and Pettersson 1980; Andersson et al. 1984) and later observed structurally for the ADH–NAD+–p-bromobenzyl alcohol complex (Eklund et al. 1982). The structure of the ternary complex shows that the arrangement of the catalytic Zn atom ligands (Cys 46, His 67, and Cys 174) adopts an approximate tetrahedral symmetry. Certain reports suggested, on the basis of the structure of the ternary complex and proved by chemical modifications (Hennecke and Plapp 1983) and site-directed mutagenesis (Ehrig et al. 1991), that the proton abstracted from the alcohol substrate during catalysis is transferred to the free solvent via a proton-relay system that includes Ser 48, the 2′OH of the nicotinamide-ribose of the bound NAD+, and His 51. The structure of the ternary complex of HLADH with NAD+ and with the substrate analog trifluoroethanol (Bahnson et al. 1997) demonstrated the mechanism of pro-R hydride-ion transfer from the substrate to the C4 atom of the coenzyme nicotinamide ring. The structure also proved the critical dependence of the catalytic efficacy on the nature of the protein residue adjacent to the pro-S face of the nicotinamide ring.
Although the mechanism of ADH catalysis is well characterized, structural information has not yet emerged regarding the productive complexes between the enzyme and ethanol, the major ADH substrate in human alcohol metabolism and the major product in yeast anaerobic fermentation. The observation of such complexes is prevented by the rapid course of the ethanol oxidation reaction; for example, the turnover number for ethanol of HLADH is 140 sec−1 at physiological conditions. Here we present the 3D structure of a ternary complex between a bacterial ADH, a coenzyme (NADH), and a substrate (EG, a very close analog of ethanol differing by a single oxygen atom). The study of this complex became possible because of blocking of the coenzyme exit by crystalline packing, combined with high EG concentration in the crystal, which led to formation of an abortive enzyme–coenzyme–alcohol complex. The proton-relay system in PaADH comprises hydrogen bonds between Thr 46, the 2′ hydroxyl of the nicotinamide ribose, and the imidazole of His 49. The ternary complex structure also revealed a hydride-ion transfer path from the C1 atom of the substrate to the C4 atom of the nicotinamide ring, with a C1–C4 distance similar to that found in other ADHs. Structural aspects of inefficiency of the EG substrate are also discussed.
Results and Discussion
The structure of the PaADH monomer
The monomer of PaADH (Fig. 1A–C ▶) consists of 342 amino acid residues that fold into a catalytic domain and a coenzyme-binding domain. Residues 1–157 and 292–342 form the catalytic domain, and the coenzyme-binding domain is formed by residues 158–291, exhibiting a Rossmann fold motif characteristic of nicotinamide-adenine coenzyme-binding proteins (Rossmann et al. 1974). The two domains are connected by the α4-helix (residues 155–166; Fig. 1B,C ▶) and by a stretch of seven amino acid residues, 289–295, positioned at a 5.5 Å-distance from the helix, thus forming an interdomain “hinge”. Rotation around this hinge is essential for intersubunit rearrangements, which occur on coenzyme binding (Eklund et al. 1982; Korkhin et al. 1998; Esposito et al. 2003). The NAD+ coenzyme and the substrate bind to the protein in a deep cleft located between the two domains. Inside the cleft, the catalytic Zn2+ ion is coordinated by the triad Cys 44, His 67, and Cys 154. The catalytic domain is formed by eight-stranded β-sheets, three α-helices, and a long “protruding lobe” (residues 96–116), which harbors the structural Zn atom, ligated by cysteines 98, 101, 104, and 112 (Fig. 1C ▶).
Figure 1.

(A) The ribbon model of PaADH monomer. The coenzyme is shown in balls and sticks. Catalytic and structural Zn2+ are shown as gray balls. (B) The amino acid sequence and the secondary structure of PaADH. Secondary structure elements are shown in white for the catalytic domain and in gray for the coenzyme-binding domain. Residues participating in the intersubunit interactions are labeled by circles for a five-member ion-pair network between subunits A and D, by triangles for a single ion pair between subunits A and B, and by a rhomb for Thr 275 participating in the A–B interaction. (C) Stereo view of the PaADH Cα trace. Every tenth residue is labeled by a full sphere.
Although the PaADH protein has low sequence identity with other ADHs of known structure, the structural homology among all ADH proteins is relatively high. The PaADH monomer can be superimposed over the closed form of human type γ2 ADH (Niederhut et al. 2001) with an r.m.s. deviation of 1.62 Å for the 301 matching Cα atoms as exhibited by the DEJAVUE program (Kleywegt and Jones 1997). The major structural difference occurs in the catalytic domain, where PaADH lacks two additional β-sheets (residues 122–138; human ADH nomenclature) that are present in the human enzyme. Although the protruding lobes of these two enzymes are also different (r.m.s. deviation of 2.1 Å), the geometry of the ligands binding the structural Zn is conserved within 0.21 Å (SPASM, Kleywegt 1999). The catalytic triads of these two proteins are also structurally conserved with an r.m.s. deviation of 0.27 Å.
The superposition with the NADP-dependent ADH from TbADH, used as a model for solving the present structure by molecular replacement, results in an r.m.s. deviation of 1.85 Å for the 288 matching Cα atoms. PaADH lacks the loop in the protruding lobe existing in TbADH (residues 105–109, TbADH nomenclature), and the α9-helix in PaADH is two residues shorter. In TbADH these two regions come together to make a van der Waals contact between subunits A and B, whereas in PaADH such a contact is missing.
In each of the eight monomers in the asymmetric unit, clear electron density of the dinucleotide NAD molecule was observed (Fig. 2 ▶). The coenzyme electron density, showing a slight puckering of the nicotinamide ring and the nonplanarity of its N1 atom, suggests the presence of NADH (reduced) rather than NAD+ (oxidized) as the coenzyme in the structure. A reduced form of the coenzyme was also observed in the crystal structure of holo-HLADH (Meijers et al. 2001).
Figure 2.

Stereo view of the |Fo-Fc| omit electron-density map, used for modeling the NADH coenzyme and EG. The map, calculated without noncrystallographic symmetry restraints to avoid the averaging of individual conformations, was contoured at 3σ. For clarity, the cover-radius option (as implemented in Bobscript [Esnouf 1997]) was used to exclude regions of the map placed farther than 2 Å from the coenzyme or the EG. Catalytic Zn2+ is shown in gray.
The coenzyme is fixed in the cleft between the catalytic and the coenzyme-binding domains through the interactions of the pyrophosphates with Arg 337 and with His 45 and the interaction of the adenosine ribose with the conserved Asp 201 (Fig. 3 ▶). The coenzyme also interacts with PaADH through a number of interactions with the protein main chain. The carboxamide group forms three hydrogen bonds with the main-chain atoms of Val 266, Ser 290, and Val 292 (Fig. 3 ▶) homologous to Val 292, Ala 317, and Phe 319 of HLADH. Colby and colleagues (Colby et al. 1998) suggested that the three hydrogen bonds are required to maintain the closed conformation of the HLADH enzyme. Val 292 is in the hinge region between the two domains of the ADH, and this hydrogen bond leads to the slightly tensed φ/ψ angles of this valine, placing it in a disallowed region of the Ramachandran plot (data not shown).
Figure 3.

Hydrogen bonding interactions between PaADH and the coenzyme. Interactions within 3.3 Å are shown. The proton-shuttling pathway is labeled by arrows.
In PaADH, the characteristic ADHs proton-relay system includes hydrogen bonds between Thr 46, the 2′ hydroxyl of the nicotinamide ribose, and the imidazole of His 49 (see arrows in Fig. 3 ▶).
The quaternary organization and stability of PaADH
The overall quaternary organization of PaADH is similar to that of other tetrameric ADHs (for NADP-dependent bacterial ADHs, see Korkhin et al. 1998; for NAD-dependent archaeon ADH, see Esposito et al. 2002; Fig. 4a ▶). This homotetramer obeys ~222-point symmetry. The four coenzyme-binding domains form the core of the protein tetramer, whereas the four catalytic domains point outside, leaving a certain degree of flexibility in the hinge regions and keeping the interdomain clefts accessible to the substrates and to the coenzyme. The tetramer can be represented as a dimer of two identical dimers: AB and CD (see Fig. 4a ▶ for subunit nomenclature), each dimer being similar to the classical HLADH dimer. In each dimer, the monomer–monomer contact occurs along the antiparallel β-strands (β14 residues 285–289 in each monomer), and the subunits associate into a 12-stranded β-sheet (6 β-strands of the Rossmann fold motifs in each monomer). The surface of the extended β-sheet structure is nonpolar and forms a hydrophobic patch, together with the adjacent α-helix (α8, residues 247–255) and β-strand (β13, residues 272–277). This patch is shared by both A and B monomers, thus stabilizing the region of interaction between them. The hydrophobic patch is “clipped” on both sides by the two symmetric intersubunit salt bridges: Arg 258A–Glu 102B and Arg 258B–Glu 102A, as well as Arg 258C–Glu 102D and Arg 258D–Glu 102C (Fig. 4b ▶). These ion pairs bind the protruding structural lobes to the neighboring subunits.
Figure 4.

(a) The tetramer of PaADH. Subunits A, B, C, and D are colored purple, green, orange, and blue, respectively. The catalytic and structural Zn2+ ions are shown in yellow. (b) The ion-pair network at the interface of three subunits of PaADH. The long ion-pair network consists of Lys 161A-Asp 297A-Lys 164A-Glu 300A and Arg 169D (superscripts denote the different subunits). The short ion pair includes Arg 258D and Glu 102C. The ion pairs are shown as thick dotted green lines. Subunit assignment and coloring are as in the legend to Figure 4a, above. (c) The interface of subunits A and B. At the dimer interface, a distortion of the noncrystallographic twofold symmetry is due to the asymmetrical, donor–acceptor nature of the intersubunit Thr 275A–Thr 275B hydrogen bond. The hydrogen bonds are shown in green dashed lines; the 4.6 Å distance between the Thr 275A hydroxyl and the Phe 273A carbonyl is shown in red (dashed line).
In other ADHs (e.g., TbADH), this type of binding is provided only by van der Waals interactions, which are most probably sufficient for somewhat larger protruding lobes. The ion-pair binding in PaADH is probably needed, because the protruding lobe in PaADH is narrower than in other ADHs and therefore cannot be fixed in its place by van der Waals interaction with subunit B, as in TbADH (where it interacts with helix α9).
The exceptions to this hydrophobicity are two polar Thr 275 residues, which point into the hydrophobic patch. Unfavorable interactions caused by introducing the two-hydroxyl groups into a hydrophobic environment are apparently partially compensated by an intersubunit hydrogen bond between the adjacent Thr 275A and Thr 275B (Fig. 4c ▶). Thr 275A and Thr 275B violate the noncrystallographic twofold symmetry. Precise twofold symmetry in this case is not possible because the donor and acceptor properties of the hydrogen bond are resolved, ordered, and coupled with the specific residues (Thr 275A donates its proton to Thr 275B). The violation of the twofold symmetry near the twofold axis is manifested by the asymmetry in the neighboring side-chain orientation: Thr 275B donates its proton to the neighboring main-chain carbonyl of Phe 273B (3 Å apart) and accepts the proton of Thr 275A. Therefore, a symmetric hydrogen bond between Thr 275A and the carbonyl of Phe 273A cannot be formed, and the carbonyl is turned away 4.6 Å, causing the whole residue to rotate. This rotation causes the asymmetry between the Phe 250A and the Phe 250B side chains (Fig. 4c ▶). This is a very unusual and interesting case in which a hydrogen atom causes a local energetic instability, which is relaxed only 10 Å apart.
Other intersubunit interactions are less extensive. Subunits A and C of PaADH associate solely through van der Waals interactions within a limited area, lacking any inter-subunit ion pairs or hydrogen bonds. This association is contrary to those found in the thermophilic TbADH and the mesophilic CbADH, in which the A and C subunits of the tetramer are connected by the ion pairs Arg 91A–Asp 128C (Korkhin et al. 1998).
The interaction between subunits A and D of PaADH is very similar to that of TbADH. In PaADH, a five-member intersubunit ion-pair network connects subunits A and D: a “long” intersubunit ion-pair network in subunit A comprising four members (Lys 161-Asp 297-Lys 164-Glu 300) is extended by one additional intrasubunit ion pair between Glu 300A and Arg 169D (Fig. 4b ▶). This network resembles the four-member ion-pair network on the interface of subunits A and D in TbADH (Korkhin et al. 1998). The five-member ion-pair network of PaADH lies in close proximity to the intersubunit salt bridge Arg 258D–Glu 102C, which serves as an “ion-pair clip” at one end of the hydrophobic patch discussed earlier. In this way, the ion-pair network is located in the strategic point where three subunits come together, leading to stabilization of the protein.
The active site of PaADH
The composition and conformation of the elements of the active site, interpreted as a complex of PaADH and NADH with EG, are clearly visible in the electron-density maps. In the catalytic site of PaADH, the Zn ion exhibits approximately tetrahedral symmetry (tetrahedral angles of 97°, 110°, 103°, 118°) and is coordinated by the sulfur atoms of Cys 44 and Cys 154 and by the imidazole nitrogen of His 67, the ion-ligand distances being 2.3 Å, 2.5 Å, and 2.2 Å, respectively (Fig. 5A ▶). The fourth ligand was clearly discernible and identified as the EG molecule.
Figure 5.
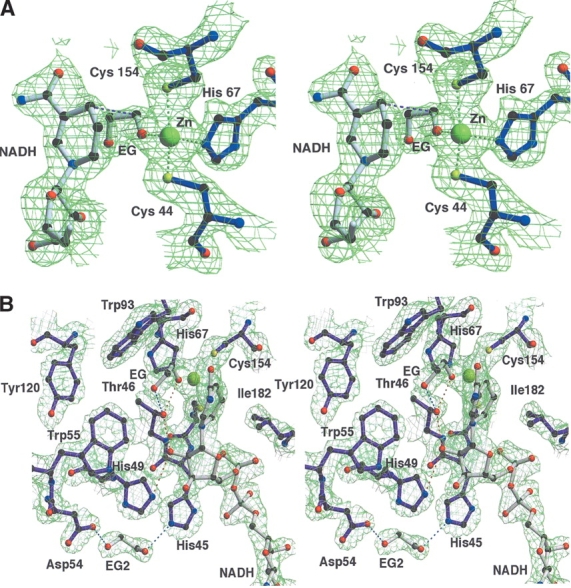
(A) Stereo view of the |2Fo-Fc| electron-density map of the catalytic site region of PaADH contoured at 1.5σ. The Zn2+ ion (in yellow) is coordinated by the sulfur atoms of Cys 44 and Cys 154, by the nitrogen of His 67, and by the oxygen of EG; the ion-ligand distances are 2.3 Å, 2.5 Å, 2.2 Å, and 2.2 Å, respectively. The distance between the C1 atom of the substrate and the C4 of NADH, assumed for the hydride ion transfer, is shown in pink (3.7 Å). For clarity, the cover-radius option was used to exclude from the map any regions placed farther than 1.8 Å from the selected atoms. (B) Stereo view of the proton-shuttling pathway of PaADH. A proton shuttles from the substrate’s EG hydroxyl group, ligated by the Zn2+ ion via Thr 46, 2′OH of the nicotinamide ribose, and His 49. The substrate is stabilized by an additional H-bond with Thr 46 (shown in blue). A second EG molecule at the proposed “proton exit” near Asp 54 is stabilized by an H-bond with His 45 and Asp 54.
Before the X-ray data collection, EG (used for cryoprotection) was added to the crystallization solution at a concentration of 2.5 M. Being a primary alcohol, EG serves as a weak substrate for PaADH, and its kinetic parameters have been estimated. PaADH binds EG with Km > 200mM, which is at least 120× weaker than the Km of ethanol (1.65 mM), and its turnover number, kcatEG = 4.5 sec−1, is about 4.5× slower than ethanol.
EG binds PaADH in a cis-conformation, with a torsion angle of 60°, forming the coordination bond to the catalytic Zn (the distance Zn–O is 2.2 Å) and fitting perfectly into the electron-density map. In two protein subunits of each of the crystallographic-independent PaADH molecules, the exit of the coenzyme is blocked by crystalline packing: The neighboring protein molecule forms several hydrogen bonds, salt bridges, and van der Waals contacts with the exit of the NADH-binding cleft. In particular, N6 of the adenine forms a water-mediated contact with Glu 52 of a neighboring protein molecule. In these subunits, the coenzyme and the substrate are well ordered, and the average temperature factors are 17.9 Å2 for NADH and 19.2 Å2 for EG. The observation that the enzyme–coenzyme–substrate complex is clearly present in the PaADH crystal supports the findings of Shearer et al. (1993) of an abortive complex pathway in the ADHs. These authors demonstrated that the coenzyme dissociated from the HLADH–NADH–alcohol complex at the slow rate of 0.3 sec−1, followed by a faster (6.3 sec−1) dissociation of the alcohol. In these particular protein subunits of PaADH, the crystal packing hampers the dissociation of the coenzyme; consequently, the EG substrate remains bound to the protein complex.
In the other two protein subunits of each of the crystallographic-independent PaADH molecules, the exit of the NADH coenzyme is not blocked. In these subunits, the electron density for the coenzyme and the substrate is also clearly observed. The density of the substrate in these two subunits is slightly shorter than that in the subunits with the blocked coenzyme exit. Nevertheless, both the substrate and the coenzyme are clearly visible, with respective average temperature factors of 39.9 Å2 and 26.5 Å2. The higher temperature factors suggest that the abortive complex is also observed in these subunits, but with lower occupancy.
The 3D structure of the ternary complex offers an explanation for the inefficiency of the enzymatic conversion of EG by PaADH. The second hydroxyl group of EG is pointing away from the Zn atom; thus, the mode of binding of the diol is unexpectedly different from that observed for glycerol in the glycerol dehydrogenase structure of Bacillus stearothermophilus (Ruzheinikov et al. 2001). This second hydroxyl group interferes with Thr 46, a member of the proton-shuttling system that serves to abstract a proton from the substrate (Fig. 5B ▶). This interference probably causes the slow release of EG from the ternary complex, resulting in a low turnover number. On the basis of the structure, we propose that the poor affinity (high Km) of the EG substrate could be due to the difficulty in inserting two polar hydroxyl groups of EG into the hydrophobic pocket of the active site of PaADH.
In the ternary complex, EG is positioned such that it is predisposed for a hydride transfer to the Re (pro-R hydrogen) face of the nicotinamide ring. The C1 atom of EG is at a distance of 3.7 Å from the C4 atom of the nicotinamide ring (Fig. 5A ▶), similar to 3.9 Å as found for the hydride transfer in murine ADH (Svensson et al. 2000), and to 4.0 Å observed for the mutant V203A-HLADH (Bahnson et al. 1997). In the latter study, the donor–acceptor distance, as well as the catalytic efficacy of the enzyme, was found to be critically dependent on the presence of van der Waals contacts between the nicotinamide ring and a hydrophobic residue adjacent to the opposite (S) face of the nicotinamide ring. When this contact is present (as in F93W mutant of HLADH), then the ring tilts toward the substrate, and the donor–acceptor distance can become as short as 3.2 Å, whereas in its absence (as in V203A mutant of HLADH) the distance is 4.0 Å. The results of the present study support this proposal. In PaADH, no van der Waals contacts were detected between the protein residues and the pro-S face of the nicotinamide ring, and the distance between the C1 atom of the substrate and the C4 atom of the nicotinamide ring is as long as 3.7 Å.
Conclusion
Here we have determined the 3D structure of ADH from a pathogenic bacterial source, Pseudomonas aeruginosa. The structure is a ternary complex of this homotetrameric NAD-linked ADH with the coenzyme and a weak substrate, EG. The complex was best observed in the protein subunits, in which crystal packing prevented the possible exit of the coenzyme. Along with the classic studies on ADH ternary complexes with brombenzyl alcohol and the recent studies of ternary complexes of ADHs with smaller alcohols, such as the substrate analog trifluorethanol (Bahnson et al. 1997; Colby et al. 1998) and the substrate 2-ethoxyethanol (Esposito et al. 2003), the present study of an ADH ternary complex with NAD and EG maps the precise geometry of the active site of ADHs.
Materials and methods
Protein expression and purification
The BSp80PaADH vector harboring the PaADH gene was transformed into E. coli cells, TG-1 strain. A single colony was inoculated into 2YT medium supplemented with 100 μg/mL ampicillin and cultured at 37°C. After 12 h, an additional amount of ampicillin (100 μg/mL) was added and cells were cultured for an additional 4 h. Cells were then harvested by centrifugation at 7000g for 10 min and stored at −80°C. For protein purification, the cells were resuspended in buffer A (25 mM Tris-HCl, 0.1 mM DTT, 0.1 mM EDTA, 0.1 mM benzamidine, 0.02% sodium azide, 10% glycerol at pH 7.3) and supplemented with phenylmethylsulfonyl fluoride and trypsin inhibitor. After disruption by pulsed sonication, cell debris was removed by ultracentrifugation (100,000g for 1 h). The clear cell extract was loaded onto a DEAE-52 cellulose column (Whatman Chemicals), which was extensively washed with 80 mM NaCl in buffer A. The retained protein was eluted with two column volumes of 150 mM NaCl in buffer A, and the enzyme was further purified on Phenyl Sepharose (Pharmacia) by using a linear gradient (0.3 to 0 M) of ammonium sulfate in buffer A. Enzymatically active fractions were pulled, concentrated to a protein concentration of 2 mg/mL, and loaded onto a High Load Superdex 200 FPLC column (Pharmacia). The purified protein eluted from this column in a single peak in 150 mM NaCl, 50 mM Tris-HCl, 0.1 mM DTT, 5 μM ZnCl2 (pH 8.4). The dialyzed (50 mM Tris-HCl, 0.1 mM DTT, 5 μM ZnCl2 at pH 8.4) protein was concentrated on a Vivaspin concentrator (Vivascience) to a final protein concentration of 3.8 mg/mL. All purification, concentration, and dialysis steps were performed at 4°C. The specific activity of the purified protein was estimated as 40 U/mg.
Enzyme assay and kinetic experiments
The catalytic activity of the ADH was measured at 40°C by following the reduction of NAD+ (and monitoring the formation of NADH) at 340 nm (ɛ340 = 6.2 mM−1cm−1). The standard assay mixture contained 150 mM 2-propanol, 0.5 mM NAD+, and 100 mM Tris-HCl (pH 8.8) in a total volume of 1 mL. One unit of the ADH (U) is defined as the amount of enzyme that catalyzes the oxidation of 1 μmole of 2-propanol/min under initial velocity at the earlier-mentioned conditions. Kinetic parameters were measured by using a Beckman DU-7500 spectrophotometer, equipped with a thermostated circulating water bath. The Km values for ethanol, 2-propanol, and EG were determined by using different concentrations of the alcohols (0.05 to 40 mM for ethanol and 2-propanol and 36 mM to 1.7 M for EG), the enzyme (5 to 120 nM), and 0.5 mM NAD+ (which is above saturation concentration of the coenzyme) in 100 mM Tris-HCl (pH 8.8). The reported values represent the average of three experiments; individual measurements were within 10% of the quoted mean. The Michaelis-Menten parameters (Vmax and Km) were derived by nonlinear (hyperbolic) regression. Protein content was determined by using Bradford and Lowry methods, with bovine serum albumin as standard.
Crystallization and data collection
Crystals of PaADH were grown at 19°C by using the hanging-drop method. The reservoir solution contained 14% polyethylene glycol monomethyl ether 2000, 12 mM sodium potassium tartarate, 100 mM Tris, 0.1 mM DTT, 0.007% sodium azide (pH 8.6). Drops containing 4 μL of protein at 3.8 mg/mL in the presence of 2 mM NAD+ were mixed with an equal amount of precipitation solution. Initial crystals had a needle shape and diffracted to very low resolution. The crystal morphology changed to rectangular plates after the addition of 16 mM spermine (Sigma) to the well solution. The final crystals of triclinic symmetry, up to five per drop with a typical dimension of 0.4 × 0.2 × 0.04 mm, were obtained in pre-stabilized, streak-seeded drops (Stura and Wilson 1991) from smaller crystalline plates that were grown in less successful crystallization trials. The washed and dissolved crystals exhibited enzymatic activity. For data collection at cryogenic conditions (T = 120K), the crystals were gradually transferred to a modified mother liquor solution containing 15% EG in addition to the initial constitution and flash cooled mounted on a cryogenic loop (Teng 1990) by using an Oxford Cryostream (Cosier and Glaser 1986) low-temperature device. A complete data set was collected from a single crystal on a Rigaku R-AXIS IV++ imaging plate area detector mounted on a Rigaku RU-H3R rotating anode equipped with multilayer focusing mirrors from OSMIC, using Cu Xray radiation (λ = 1.5418 Å). The diffraction data were integrated, scaled, and reduced using the HKL (Otwinowski and Minor 1997) program package (relevant data are shown in Table 1).
Table 1.
Data collection and refinement statistics
| Data collection | |
| Space group | P1 |
| Unit cell | 74.2Å × 86.3Å × 125.7Å; 79.3°, 78.7°, 71.6° |
| Resolutiona (Å) | 20.0–2.3 (2.34–2.3) |
| No. of unique reflections | 123,136 |
| Completeness | 96.4 (94.2) |
| 〈I〉/〈σ(I)〉 | 12.1 (2.8) |
| Rmerge (%) | 10.8 (46.3) |
| Redundancy | 3.4 (3.3) |
| Refinement | |
| No. of reflections in the test set | 12,264 |
| Atoms used in refinement | 5,133 |
| Water molecules | 262 |
| R & Rfree factor (%) | 22.9; 24.6 |
| Mean B-factor | 31.3 |
| Deviation from ideal geometry | |
| RMSD bond distance (Å) | 0.007 |
| RMSD bond angles (deg) | 1.5 |
| RMSD dihedral angles (deg) | 24.4 |
| RMSD improper angles (deg) | 1.18 |
| Ramachandran plot | |
| Most favored | 91% |
| Additional allowed | 7.9% |
| Generously allowed | 0.9% |
| Disallowed | 0.2% |
aNumber in parentheses is for uppermost shell.
Structure determination and refinement
Molecular replacement
The structure of PaADH was solved by using a combined molecular replacement method and noncrystallographic (NCS) density averaging. Assuming a calculated molecular weight of 40.6 kD and two tetramers in the asymmetric unit, the Vm value was estimated to be 2.2 Å3/Da, which is in the normal range for globular proteins (Matthews 1968). The calculation of self-rotation function, Patterson function, and molecular replacement search were then performed, using 32,740 reflections in the 20–3.6 Å-resolution shell.
A yield of three, rather than the expected six, strong peaks of the self-rotation function at the 18σ level (next highest peak 6.5σ), suggests that the two tetramers are identically oriented. A prominent nonorigin Patterson peak of 21σ (9% of the origin peak) at the end of the 0.64, 0.75, 0.48 vector provided the relative position of the tetramers.
For molecular replacement, the tetramer of ADH from TbADH (Korkhin et al. 1998), which shares 26% sequence homology with PaADH, was used as a model after the truncation of its side chains to alanine. The cross-rotation function, calculated with AMoRe (Navaza 2001), yielded four relatively strong peaks (about 8σ, next highest peak 4σ), related by noncrystallographic 222 symmetry with the three axes oriented as previously found by the self-rotation function. Two tetramers were oriented according to the cross-rotation function solution. The first tetramer was fixed at the origin, using an arbitrary origin choice in the triclinic cell, and the phased translation function of AMoRe was applied to verify the position of the second tetramer, previously found from the Patter-son function. The resulting best translation function solution (peak of 17σ, next highest peak 5σ) was identical to that found from Patterson function analysis.
Rigid body refinement was performed with CNS (Brunger et al. 1998), using 110,753 reflections in the 20–2.3 Å-resolution shell, leaving 10% of reflections for Rfree calculation. Refinement of individual monomers and of individual monomer domains yielded R-factor = 55% (Rfree = 55%), good packing contacts but an uninterpretable map. Further improvement of the molecular replacement solution was then performed, using the algorithm suggested in Hamiaux et al. (1999). The position of the first tetramer was fixed, and the phased translation function was used to position separately the four monomers of the second tetramer, exploiting the rotations of the TbADH-monomer model. After the second monomer was built and its position fixed, the positions of the monomers in the first tetramer were found in the same way. This approach allowed a correction for mutual orientations of individual monomers, and, after a rigid body refinement, a drop of the R-factor value to 52% (Rfree = 52%) yielded a map with continuous density at most secondary structure elements and at certain loops.
NCS averaging and the final model
An extensive revision of the monomer structure was then performed through the following iterative procedure, implementing NCS averaging over eight independent monomers: (1) envelope construction, using a model of the monomer (CNS) followed by the removal of overlaps between NCS-related envelopes, using MAMA (Kleywegt 1999); (2) NCS averaging of the electron-density map and the solvent flattening, using CNS (Brunger et al. 1998) and SOLOMON (Abrahams and Leslie 1996) as implemented in CNS; (3) model rebuilding in accordance with the averaged map, using O (Jones et al. 1991); (4) generating full asymmetric unit content, rigid body and conjugate gradient minimization refinement, and recalculating the NCS operators.
After several iterations, the R-factor dropped to 45%, the full trace of the main chain could be outlined, and the two Zn atoms were unambiguously placed. Then residues 2–342 were built into the model of the monomer. At this stage, the NADH coenzyme and the EG substrate moieties were clearly revealed by combined omit electron-density map. It seemed unlikely that the density assigned to EG belonged to PEG monomethyl ether 2000, which was used as a crystallizing agent at a concentration of 14%. (PEG)4 and longer PEG chains have been observed previously in complexes with various enzymes (Greenblatt et al. 1999). The observation that in all eight independent monomers, no electron density was evident beyond the four atoms in this moiety supports the finding that this density belongs to EG rather than to PEG.
To check the presence of these ligands in each individual monomer, and to avoid averaging of possible different conformations, we first placed the ligands into the electron-density map, calculated without NCS symmetry (Fig. 2 ▶). NADH and EG then were added to the refinement, yielding R = 33%, Rfree = 34%.
At this stage, the omit electron-density map (calculated over the region of dimerization between the coenzyme binding domains–Rossmann fold motif [Rossmann et al. 1974], with the release of NCS constraints), clearly displayed differences in the structures of the individual monomers. The main differences occurred in the position of the side chain of Phe 273, placed almost exactly on the noncrystallographic twofold axes, and the side chains of the adjacent Phe 250 and Gln 247. The eight monomers could be divided into two groups, according to the side-chain and the coenzyme conformation similarity of the individual monomers.
Further refinement was achieved by using the coordinates of the AB dimer (nomenclature as in Fig. 4a ▶), replicated to both tetramers by four NCS operators. Refinement of the structure was then performed by using strict NCS, including simulated annealing, group, and individual B-factor refinement and implementing bulk solvent correction on all stages. The further gradual reduction of NCS symmetry was probed, substituting strict NCS constraints by NCS restraints with a restraint weight of 250 kcal/mole/Å2. Because this approach led to poor electron density for residues 5–25 and 316–342 in the monomers generated from monomer B, this substitution was abandoned and strict NCS symmetry constraints were kept. For ligand refinement, geometry and energy parameters from Uppsala Hetero-compound Information Centre (Kleywegt and Jones 1998) were used. The parameters set for EG have been derived from Addlagatta et al. (2001) and for NADH from Meijers et al. (2001). To verify the exact oxidation states of the EG and the coenzyme, we modeled this ligand both in oxidized (aldehyde) and reduced (alcohol) states, using Refmac5 (Murshudov et al. 1997). Both forms fitted the density equally well, and release of restraints on the C1-O bond length could not confirm either of the oxidation states. The release of the restraints on the ethylene torsion angle led to steady convergence of this angle to 60°. Release of the restraints on the nicotinamide ring torsion angles refined to a slightly nonplanar ring geometry. Both coenzyme and EG molecules were thus modeled in reduced state, suitable for the abortive complex pathway. Water molecules were added based on the basis of the peaks higher than 2.7σ of the |Fo-Fc| map and reasonably positioned relative to the protein and other water structures. The final model consists of residues A2–A342 and B2–B342, two NADH coenzyme molecules, three EG moieties, and 262 water molecules, yielding a crystallographic R-factor of 22.9% and Rfree of 24.6%. In both independent monomers, no density was observed for Met 1. Model verification was performed with WHATIF (Vriend 1990) and PROCHECK (Bailey 1994). The figures were composed with Bobscript (Esnouf 1997) and rendered with Raster3D (Merritt and Murphy 1994). The AB dimer, which was used in all crystallographic calculations, was expanded to the two enzymatically active tetramer molecules, ABCD and EFGH, and then deposited in the Brookhaven Protein Data Bank under entry code 1LLU.
Acknowledgments
This study was supported in part by grant no. 296–00 (to Y.B.) from the Israel Science Foundation. Y.B. is the Maynard Wishner professor of bio-organic chemistry and malignant diseases research. We thank Prof. M. Safro and Dr. I. Agmon for their valuable suggestions, and Dr. Virginia Buchner for helpful remarks during the preparation of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ADH, alcohol dehydrogenase
CbADH, Clostridium beijerinckii ADH
EG, ethylene glycol
HLADH, horse liver ADH
NCS, noncrystallographic symmetry
PaADH, Pseudomonas aeruginosa ADH
PEG, polyethylene glycol
TbADH, Thermoanaerobacter brockii ADH
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03531404.
References
- Abrahams, J. and Leslie, A. 1996. Methods used in the structure determination of bovine mitochondrial F-1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52 30–42. [DOI] [PubMed] [Google Scholar]
- Addlagatta, A., Krzywda, S., Czapinska, H., Otlewski, J., and Jaskolski, M. 2001. Ultrahigh-resolution structure of a BPTI mutant. Acta Crystallogr. D Biol. Crystallogr. 57 649–663. [DOI] [PubMed] [Google Scholar]
- Ammendola, S., Raia, C.A., Caruso, C., Camardella, L., Dauria, S., Derosa, M., and Rossi, M. 1992. Thermostable Nad+-dependent alcohol dehydrogenase from Sulfolobus solfataricus. Gene and protein sequence determination and relationship to other alcohol dehydrogenases. Biochemistry 31 12514–12523. [DOI] [PubMed] [Google Scholar]
- Andersson, P., Kvassman, J., Olden, B., and Pettersson, G. 1984. Synergism between coenzyme and alcohol binding to liver alcohol dehydrogenase. Eur. J. Biochem. 144 317–324. [DOI] [PubMed] [Google Scholar]
- Bahnson, B.J., Colby, T.D., Chin, J.K., Goldstein, B.M., and Klinman, J.P. 1997. A link between protein structure and enzyme catalyzed hydrogen tunneling. Proc. Natl. Acad. Sci. 94 12797–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, S. 1994. The CCP4 Suite—Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Benach, J., Atrian, S., Gonzalez-Duarte, R., and Ladenstein, R. 1998. The refined crystal structure of Drosophila lebanonensis alcohol dehydrogenase at 1.9 Å resolution. J. Mol. Biol. 282 383–399. [DOI] [PubMed] [Google Scholar]
- Bogin, O., Peretz, M., Hacham, Y., Korkhin, Y., Frolow, F., Kalb, A.J., and Burstein, Y. 1998. Enhanced thermal stability of Clostridium beijerinckii alcohol dehydrogenase after strategic substitution of amino acid residues with prolines from the homologous thermophilic Thermoanaerobacter brockii alcohol dehydrogenase. Protein Sci. 7 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macro-molecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Colby, T.D., Bahnson, B.J., Chin, J.K., Klinman, J.P., and Goldstein, B.M. 1998. Active site modifications in a double mutant of liver alcohol dehydrogenase: Structural studies of two enzyme–ligand complexes. Biochemistry 37 9295–9304. [DOI] [PubMed] [Google Scholar]
- Cosier, J. and Glaser, A.M. 1986. A nitrogen-gas-stream cryostat for general x-ray diffraction studies. J. Appl. Crystallogr. 19 105–117. [Google Scholar]
- Ehrig, T., Hurley, T.D., Edenberg, H.J., and Bosron, W.F. 1991. General base catalysis in a glutamine for histidine mutant at position-51 of human liver alcohol-dehydrogenase. Biochemistry 30 1062–1068. [DOI] [PubMed] [Google Scholar]
- Eklund, H., Nordstrom, B., Zeppezauer, E., Soderlund, G., Ohlsson, I., Boiwe, T., Soderberg, B., Tapia, O., Branden, C., and Akeson, A. 1977. Three-dimensional structure of horse liver alcohol dehydrogenase at 2.4 Å resolution. J. Mol. Biol. 102 27–59. [DOI] [PubMed] [Google Scholar]
- Eklund, H., Plapp, B., Samama, J., and Branden, C. 1982. Binding of substrate in a ternary complex of horse liver alcohol dehydrogenase. J. Biol. Chem. 257 14349–14358. [PubMed] [Google Scholar]
- Eklund, H., Plapp, B.V., and Jornvall, H. 1994. Crystallographic investigations of alcohol dehydrogenases. In Towards a molecular basis of alcohol use and abuse (eds. B. Jannson et al.), pp. 269–277. Birkhause Verlag, Basel, Switzerland. [DOI] [PubMed]
- Esnouf, R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15 132–134. [DOI] [PubMed] [Google Scholar]
- Esposito, L., Sica, F., Raia, C.A., Giordano, A., Rossi, M., Mazzarella, L., and Zagari, A. 2002. Crystal structure of the alcohol dehydrogenase from the hyperthermophilic archaeon Sulfolobus solfataricus at 1.85 Å resolution. J. Mol. Biol. 318 463–477. [DOI] [PubMed] [Google Scholar]
- Esposito, L., Bruno, F., Sica, F., Raia, C.A., Giordano, A., Rossi, M., Mazzarella, L., and Zagari, A. 2003. Crystal structure of a ternary complex of the alcohol dehydrogenase from Sulfolobus solfataricus. Biochemistry 42 14397–14407. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R., ed. 1999. The alcohol dehydrogenases. In Structure and mechanism in protein science, pp. 460–465. W.H. Freeman and Company, New York.
- Goodlove, P.E., Cunningham, P.R., Parker, J., and Clark, D.P. 1989. Cloning and sequence analysis of the fermentative alcohol dehydrogenase-encoding gene of Escherichia coli. Gene 85 209–214. [DOI] [PubMed] [Google Scholar]
- Greenblatt, H.M., Kryger, G., Lewis, T., Silman, I., and Sussman, J.L. 1999. Structure of acetylcholinesterase complexed with (-)- galanthamine at 2.3 Å resolution. FEBS Lett. 463 321–326. [DOI] [PubMed] [Google Scholar]
- Guy, J.E., Isupov, M.N., and Littlechild, J.A. 2003. Crystallization and preliminary X-ray diffraction studies of a novel alcohol dehydrogenase from the hyperthermophilic archaeon Aeropyrum pernix. Acta Crystallogr. D Biol. Crystallogr. 59 174–176. [DOI] [PubMed] [Google Scholar]
- Hamiaux, C., Prange, T., Ries-Kautt, M., Ducruix, A., Lafont, S., Astier, J.P., and Veesler, S. 1999. The decameric structure of bovine pancreatic trypsin inhibitor (BPTI) crystallized from thiocyanate at 2.7 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 55 103–113. [DOI] [PubMed] [Google Scholar]
- Hennecke, M. and Plapp, B. 1983. Involvement of histidine residues in the activity of horse liver alcohol dehydrogenase. Biochemistry 22 3721–3728. [DOI] [PubMed] [Google Scholar]
- Hurley, T.D., Bosron, W.F., Stone, C.L., and Amzel, L.M. 1994. Structures of three human β alcohol dehydrogenase variants. Correlations with their functional differences. J. Mol. Biol. 239 415–429. [DOI] [PubMed] [Google Scholar]
- Inoue, T., Sunagawa, M., Mori, A., Imai, C., Fukuda, M., Takagi, M., and Yano, K. 1989. Cloning and sequencing of the gene encoding the 72-kilodalton dehydrogenase subunit of alcohol dehydrogenase from Acetobacter acetii. J. Bacteriol. 171 3115–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Jornvall, H. 1970. Horse liver alcohol dehydrogenase—On primary structures of isoenzymes. Eur. J. Biochem. 16 41–49. [DOI] [PubMed] [Google Scholar]
- ———. 1977. The primary structure of yeast alcohol dehydrogenase. Eur. J. Biochem. 72 425–442. [DOI] [PubMed] [Google Scholar]
- ———. 1994. The alcohol dehydrogenase system. In Towards a molecular basis of alcohol use and abuse (eds. B. Jannson et al.), pp. 221–229. Birkhause Verlag, Basel, Switzerland.
- Karlsson, A., El-Ahmad, M., Johansson, K., Shafqat, J., Jornvall, H., Eklund, H., and Ramswamy, S. 2003. Tetrameric NAD-dependent alcohol dehydrogenase. Chem. Biol. Interact. 143–144 239–245. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. 1999. Recognition of spatial motifs in protein structures. J. Mol. Biol. 285 1887–1897. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. and Jones, T.A. 1997. Detecting folding motifs and similarities in protein structures. Methods Enzymol. 277 525–545. [DOI] [PubMed] [Google Scholar]
- ———. 1998. Databases in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 54 1119–1131. [DOI] [PubMed] [Google Scholar]
- Korkhin, Y., Kalb, A.J., Peretz, M., Bogin, O., Burstein, Y., and Frolow, F. 1998. NADP-dependent bacterial alcohol dehydrogenases: Crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J. Mol. Biol. 278 967–981. [DOI] [PubMed] [Google Scholar]
- Kvassman, J. and Pettersson, G. 1980. Unified mechanism for proton-transfer reactions affecting the catalytic activity of liver alcohol dehydrogenase. Eur. J. Biochem. 103 565–575. [DOI] [PubMed] [Google Scholar]
- Matthews, B. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Meijers, R., Morris, R.J., Adolph, H.W., Merli, A., Lamzin, V.S., and Cedergren-Zeppezauer, E.S. 2001. On the enzymatic activation of NADH. J. Biol. Chem. 276 9316–9321. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster3d Version-2.0—A program for photorealistic molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 50 869–873. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Niederhut, M.S., Gibbons, B.J., Perez-Miller, S., and Hurley, T.D. 2001. Three-dimensional structures of the three human class I alcohol dehydrogenases. Protein Sci. 10 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Plapp, B.V. 1994. Control of alcohol metabolism. In Towards a molecular basis of alcohol use and abuse (eds. B. Jannson et al.), pp. 311–322. Birkhause Verlag, Basel, Switzerland.
- Ramaswamy, S., ElAhmad, M., Danielsson, O., Jornvall, H., and Eklund, H. 1996. Crystal structure of cod liver class I alcohol dehydrogenase: Substrate pocket and structurally variable segments. Protein Sci. 5 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann, M.G., Moras, D., and Olsen, K.V. 1974. Chemical and biological evolution of nucleotide binding protein. Nature 250 194–199. [DOI] [PubMed] [Google Scholar]
- Ruzheinikov, S.N., Burke, J., Sedelnikova, S., Baker, P.J., Taylor, R., Bullough, P.A., Muir, N.M., Gore, M.G., and Rice, D.W. 2001. Glycerol dehydrogenase: Structure, specificity, and mechanism of a family III polyol dehydrogenase. Structure 9 789–802. [DOI] [PubMed] [Google Scholar]
- Shearer, G.L., Kim, K.Y., Lee, K.M., Wang, C.K., and Plapp, B.V. 1993. Alternative pathways and reactions of benzyl alcohol and benzaldehyde with horse liver alcohol dehydrogenase. Biochemistry 32 11186–11194. [DOI] [PubMed] [Google Scholar]
- Stura, E.A. and Wilson, I.A. 1991. Application of streak seeding technique in protein crystallization. J. Cryst. Growth 110 270–282. [Google Scholar]
- Svensson, S., Hoog, J.O., Schneider, G., and Sandalova, T. 2000. Crystal structures of mouse class II alcohol dehydrogenase reveal determinants of substrate specificity and catalytic efficiency. J. Mol. Biol. 302 441–453. [DOI] [PubMed] [Google Scholar]
- Teng, T.Y. 1990. Mounting of crystals for macromolecular crystallography in a freestanding thin-film. J. Appl. Crystallogr. 23 387–391. [Google Scholar]
- Villarroya, A., Juan, E., Egestad, B., and Jornvall, H. 1989. The primary structure of alcohol dehydrogenase from Drosophila lebanonensis extensive variation within insect short chain alcohol dehydrogenase lacking zinc. Eur. J. Biochem. 180 191–197. [DOI] [PubMed] [Google Scholar]
- Vriend, G. 1990. What If—A molecular modeling and drug design program. J. Mol. Graph. 8 52–56. [DOI] [PubMed] [Google Scholar]
- Xie, P., Parsons, S.H., Speckhard, D.C., Bosron, W.F., and Hurley, T.D. 1997. X-ray structure of human class IVσσ alcohol dehydrogenase. Structural basis for substrate specificity. J. Biol. Chem. 272 18558–18563. [DOI] [PubMed] [Google Scholar]