

Abstract
Colicin E3 is a cytotoxic ribonuclease that specifically cleaves 16S rRNA at the ribosomal A-site to abolish protein synthesis in sensitive Escherichia coli cells. We have performed extensive mutagenesis of the 96-residue colicin E3 cytotoxic domain (E3 rRNase), assayed mutant colicins for in vivo cytotoxicity, and tested the corresponding E3 rRNase domains for their ability to inactivate ribosome function in vitro. From 21 alanine mutants, we identified five positions where mutation resulted in a colicin with no measurable cytotoxicity (Y52, D55, H58, E62, and Y64) and four positions (R40, R42, E60, and R90) where mutation caused a significant reduction in cytotoxicity. Mutations that were found to have large in vivo and in vitro effects were tested for structural integrity through circular dichroism and fluorescence spectroscopy using purified rRNase domains. Our data indicate that H58 and E62 likely act as the acid–base pair during catalysis with other residues likely involved in transition state stabilization. Both the Y52 and Y64 mutants were found to be highly destabilized and this is the likely origin of the loss of their cytotoxicity. The identification of important active site residues and sequence alignments of known rRNase homologs has allowed us to identify other proteins containing the putative rRNase active site motif. Proteins that contained this active site motif included three hemagglutinin-type adhesins and we speculate that these have evolved to deliver a cytotoxic rRNase into eukaryotic cells during pathogenesis.
Keywords: colicin E3, ribonuclease, ribosome, active site, mutagenesis
Colicins are plasmid-encoded multidomain protein antibiotics that are produced by and are active against Escherichia coli and closely related bacteria. Production of the toxin is induced under conditions that cause damage to the bacterial chromosome and forms part of the bacterial SOS response (Walker 1996). Accordingly, colicin production is believed to aid in competition for limited nutrient resources, but has also been implicated in the maintenance of biodiversity (Kerr et al. 2002) and in bacterial adaptation (Walker et al. 2004). Most colicins characterized to date possess either a poreforming activity that causes depolarization of the cytoplasmic membrane or a lethal nuclease activity that specifically targets rRNA, tRNA, or chromosomal DNA. The DNase-type colicins (E2, E7, E8, and E9) kill cells through nonspecific, magnesium-dependent cleavage of chromosomal DNA (Pommer et al. 2001; Walker et al. 2002). These enzymes belong to the HNH family of endonucleases that includes the caspase-activated DNases, responsible for the degradation of chromatin in eukaryotic apoptosis (Walker et al. 2002; Scholz et al. 2003). The RNase-type colicins D and E5 cleave specific, albeit different, tRNA species (Ogawa et al. 1999; Tomita et al. 2000), and colicin E3 (and by homology colicins E4 and E6 and cloacin DF13) specifically cleaves at the A-site of the 16S rRNA, between bases A1493 and G1494. This inactivates the ribosome and arrests protein synthesis, inducing a cold shock-like transcriptional response prior to cell death (Bowman et al. 1971; Walker et al. 2004).
Cleavage of 16S rRNA by colicin E3 has been reported to occur only in the context of the intact 70S ribosome (Boon 1972), although other studies indicate that at high concentrations of the toxin, both isolated 30S subunits (Ohno-Iwashita and Imahori 1977) and even naked 16S rRNA (Ohno and Imahori 1978) are cleaved, although in the latter case with a lack of specificity. Colicin E3 is cytotoxic only against E. coli and closely related bacteria because more distantly related bacteria lack a suitable receptor and/or translocation system for uptake of the colicin. However, in vitro colicin E3 is capable of inactivating ribosomes from distantly related bacterial species (Sidikaro and Nomura 1973). Further to this, colicin E3 has been shown to inactivate chloroplast ribosomes from Euglena gracilis (Steege et al. 1982) and has a cytostatic activity against leukemic cells (Smarda et al. 2001). Like other cytotoxic microbial ribonucleases, colicin E3 is of potential interest as a novel chemotherapeutic agent (for review, see Makarov and Ilinskya 2003).
The nuclease-type colicins are coexpressed and released in complex with a specific immunity protein that binds with high affinity to the cytotoxic domain and abolishes its enzymatic activity. The equilibrium dissociation constant (Kd) for both the colicin E3-Im3 and colicin E9-Im9 complexes has been measured as 10−14 M in 200 mM salt (Wallis et al. 1995; Walker et al. 2003). However, the affinity of the complex formed between the isolated rRNase domain and Im3 is two orders of magnitude weaker (10−12 M) and this reflects additional weak interactions between Im3 and the translocation domain of the toxin as revealed by the structure of the colicin E3-Im3 complex (Soelaiman et al. 2001). Structures for the E3 rRNase domain Im3 complex and the full-length colicin E3-Im3 complex have been solved at 2.4 Å and 3.0 Å, respectively (Carr et al. 2000; Soelaiman et al. 2001). The location of the active site of colicin E3 was originally predicted through the similar structural arrangement of H58 and E62 of E3 rRNase with the catalytic residues of the nonspecific ribonuclease barnase (Carr et al. 2000). Subsequent mutation of these residues to alanine in the full-length toxin showed that these mutations did indeed abolish colicin cytotoxicity in a cell extract containing the protein (Soelaiman et al. 2001). This study did not, however, rule out the possibility that these mutants are either structurally compromised or indeed that mutation abolishes the ability of the cytotoxic domain to reach its cellular target in the bacterial cytoplasm. Because little is known about the mechanism of inner-membrane translocation by E3 rRNase, the latter is of particular concern.
Here we extend this work to assign unambiguously active site residues through extensive site-directed mutagenesis and in vivo cytotoxicity assays by using purified mutant colicins. Mutant rRNase domains were tested in in vitro ribosome inactivation assays and their structural integrity and stability assessed through spectroscopic methods. Our findings support the idea that the rRNase activity of colicin E3 is mediated in a similar way to that of barnase and other members of the T1 ribonuclease family, with H58 and E62 of E3 rRNase acting as the acid–base pair. We identify other residues, the mutations of which abolish or significantly reduce cytotoxicity without significant perturbation of the rRNase structure, giving a fuller picture of the arrangement of the E3 rRNase active site. In addition, we identified a further class of mutations that led to a loss of cytotoxicity through destabilization of the rRNase structure. Defining the E3 rRNase active site has allowed us to identify a minimal catalytic motif used to identify other E3-like rRNase domains involved in biological processes such as plant–microbe interactions.
Results
Identification of Col− mutants
Mutant colicin E3 proteins were purified in complex with the immunity protein Im3 and tested for cytotoxicity against colicin-sensitive E. coli cells in an agar plate spot test assay (Fig. 1 ▶). In this assay, serial fivefold dilutions of the toxin are spotted onto a growing lawn of E. coli cells and cytotoxicity indicated by a clear or hazy zone in the cell lawn. For this in vivo activity assay, the intact colicin E3-Im3 complex was used because it displays a substantially greater cytotoxic activity than the uncomplexed colicin (Walker et al. 2003). We initially targeted residues identified by Carr et al. (2000) as likely involved in catalysis or substrate binding through a structural similarity of the putative E3 rRNase active site with the active site of barnase. In total, 21 mutant colicin complexes were purified and tested for cytotoxicity by this method.
Figure 1.
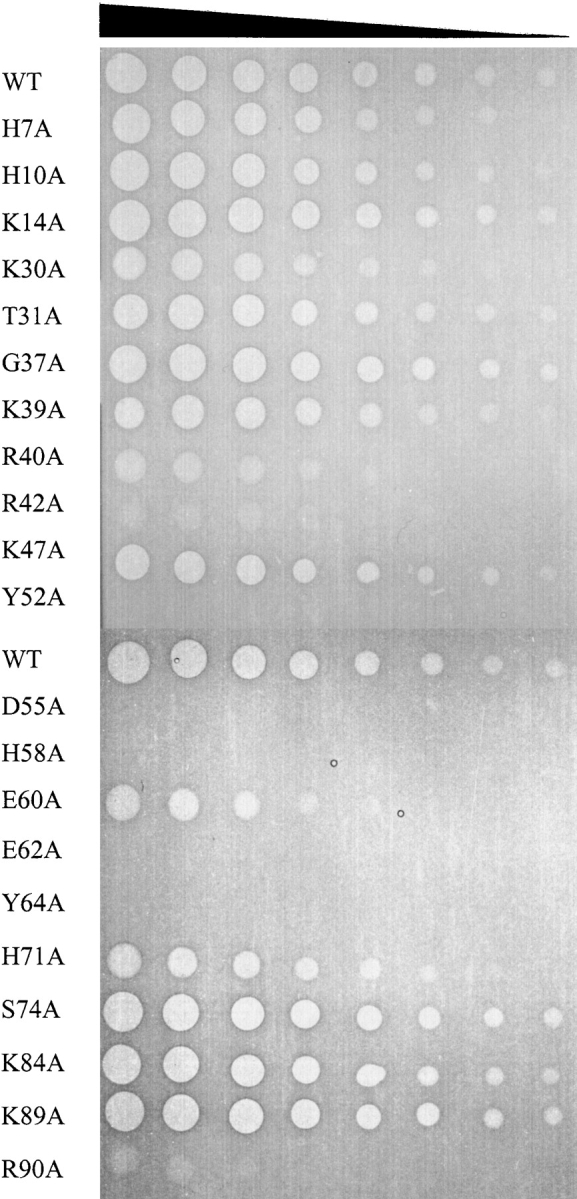
In vivo cytotoxicity assays of mutant colicin E3-Im3 complexes. Cytotoxicity of the purified colicin E3 alanine mutants was assessed by spotting a fivefold serial dilution of purified colicin E3-Im3 complex from a starting concentration of 1 mg mL−1 onto a growing lawn of E. coli JM83 cells (see Materials and Methods). Clear zones in the cell lawn indicate cell killing by colicin E3, and hazy zones indicate partial toxicity.
Spot tests showed that the mutant colicins Y52A, D55A, H58A, E62A, and Y64A are devoid of cytotoxic activity and the activity of the R40A, R42A, E60A, and R90A mutants are diminished to <1% of the wild-type protein (Fig. 1 ▶). This is consistent with predictions based on structural alignments and previous mutational studies that implicated H58 and E62 in the catalytic center of the protein (Carr et al. 2000; Soelaiman et al. 2001). This analysis predicted that H58 and E62 were analogous to the catalytic residues of barnase H102 and E73 that act as the general acid and general base in the transesterification step of RNA hydrolysis (Carr et al. 2000). The use of histidine and glutamate side chains as the catalytic residues for the acid–base hydrolysis of RNA is thought to be universal for members of the T1 ribonuclease family (Irie 1997) and is in contrast to the mechanism used by members of the RNase A family, where these roles are fulfilled by two histidine residues (Cuchillo et al. 1997). Of the four histidine residues in E3 rRNase, only mutation of H58 was found to be important for cytotoxicity, with mutation of H7, H10, and H71 having no significant effect on colicin activity (Fig. 1 ▶).
Purification of mutant rRNase domains
The significant reduction or, in some cases, abolition of colicin activity of nine of the mutations is in agreement with previous data and predictions based on structural studies (Carr et al. 2000; Soelaiman et al. 2001), but does not rule out the possibility that the identified mutations have an effect on colicin import rather than directly on substrate binding and catalysis. Destabilization of the rRNase domain could also be a significant factor in reducing or even abolishing the catalytic activity of the toxin. Destabilizing mutational effects on the toxin may well be masked by the presence of the immunity protein Im3, which binds strongly to the rRNase domain, and has been shown to considerably reduce its susceptibility to proteolysis (Ohno et al. 1977; Walker et al. 2003). Little is known about how the E3 rRNase domain is able to cross the E. coli inner membrane. In the case of the colicin E9 DNase domain, it has been shown that this enzyme is capable of forming channels in planar lipid bilayers and this is thought to be directly related to the ability of the enzyme to cross the inner membrane of susceptible cells (Mosbahi et al. 2002). This may not be the case for the E3 rRNase, but in the absence of a clear mechanism of inner-membrane translocation, it cannot be ruled out that a point mutation may impair this function.
Because we were interested in the location of the E3 rRNase active site, it was necessary to exclude both the possibility that the mutant proteins were impaired in their ability to enter the cytoplasm or that they were significantly destabilized in the absence of the immunity protein. In order to address these points, we assessed the in vitro RNase activity of mutant rRNase domains and used fluorescence and CD to probe their structure and stability.
We have reported previously that the purification of the colicin E3 rRNase domain is readily accomplished using a tandem expression system in which the cytotoxic domain is coexpressed with its inhibitor Im3 followed by denaturation/renaturation of the isolated domain (Walker et al. 2003). Difficulty purifying the rRNase R42A and H71A mutants by this method excluded them from further study and suggested that these mutations cause structural changes to the enzyme. In addition, we could only obtain the inactive E62A mutant in low yield. The other mutant rRNase domains, H7A, H10A, K14A, T31A, G37A, R40A, K47A, Y52A, D55A, H58A, E60A, Y64A, K84A, K89A, and R90A, could all be purified in similar or greater quantity than the wild-type protein. Molecular weights for the mutant rRNase domains obtained by electrospray ionization mass spectrometry were within 2 Da of those expected, showing that the mutant rRNase domains were not degraded in the producing cell or during the purification process (data not shown).
Correlation between in vitro enzymatic activity and in vivo cytotoxicity
We tested the ability of purified mutant rRNase domains to inactivate E. coli ribosomes in vitro through a coupled transcription-translation assay. We were primarily interested in determining if the mutations that caused a significant or total loss of cytotoxicity in the full-length colicin had a corresponding effect on the in vitro activity of the rRNase domain. In this assay, wild-type E3 rRNase domain was found to be active down to around nanomolar concentrations (data not shown). We assayed the mutant rRNase domains at a single concentration of 10 nM. Activity was assessed through the production of a reporter protein, firefly luciferase, which was quantitated by measuring its enzymatic activity through chemiluminescence (see Materials and Methods). Mutant rRNase domains were considered to be enzymatically active if the production of firefly luciferase was <1% of the buffer control. We found an excellent correlation between cytotoxicity and in vitro enzymatic activity, with the rRNase domain mutants R40A, Y52A, D55A, H58A E60A, E62A, Y64A, and R90A all showing lowered or absent in vitro enzymatic activity at 10 nM (Fig. 2 ▶). In the case of the D55A, H58A, and E62A mutants, we observed an apparent increase in translational activity. The reason for this is not known, although we note that the standard deviations in these cases are relatively large. In addition, we tested the mutant rRNase domains H7A, H10A, K14A, T31A, G37A, K47A, K84A, and K89A. These mutations, which had little effect on colicin cytotoxicity, had no measurable effect on in vitro rRNase activity under the conditions of the assay. Because mutations that had a significant effect on colicin activity had a corresponding effect on the in vitro enzymatic activity of the isolated rRNase domain, we can conclude that these mutations directly affect the ability of the colicin to cleave 16S rRNA. Thus, it can be ruled out that the absent or reduced cytotoxicity of the inactive and partially active mutant proteins is due to a loss of the ability of the rRNase domain to translocate into the cytoplasm.
Figure 2.

In vitro ribosome inactivation by E3 rRNase mutants. Mutant rRNase domains were assessed for the ability to inactivate ribosomes in vitro through a coupled transcription-translation assay at an rRNase concentration of 10 nM. Ribosome function was monitored through the production of a firefly luciferase reporter protein. Data are presented as the translational activity of the in vitro system after colicin E3 treatment normalized to the no E3 rRNase control. Error bars represent the standard deviation from two independent experiments (see Materials and Methods).
Structure and stability of mutant E3 rRNase domains
In order to determine if the structure and stability of mutant rRNase domains had been significantly perturbed by mutation, we used fluorescence and far-UV CD measurements to look for changes in secondary and tertiary structure. It would be expected that any effects would be most readily observed in the context of the isolated E3 rRNase domain, because even highly destabilizing mutations in the >500 residue full-length colicin may lead to only minor changes in fluorescence or CD spectra.
E3 rRNase has a fluorescence emission spectrum characteristic of a folded protein with tryptophan side chains buried in the protein core, displaying a λmax of 330 nm (Walker et al. 2003). Spectra for all mutant rRNase domains were found to be similar to those of the wild-type protein in all but two cases. These mutants, Y52A and Y64A, both show λmax values of 354 nm and are enzymatically inactive (Fig. 3A ▶). This λmax value is similar to that of free tryptophan in solution or thermally denatured E3 rRNase and is indicative of solvent exposure of the E3 rRNase tryptophan residues, indicating that the Y52A and Y64A mutants are highly destabilized. For the R40, D55A, H58A, E60, E62A, and R90A mutants, all of which are partially or completely inactive, the intrinsic tryptophan fluorescence was not altered significantly from that of the wild type, indicating a similarity in tertiary structure.
Figure 3.

Structural properties of purified mutant E3 rRNase domains probed by intrinsic tryptophan fluorescence and CD. (A) Tryptophan fluorescence emission spectra of wild-type and mutant rRNase domains. Fluorescence spectra (λex = 295 nm) were recorded at a protein concentration of 1 μM in 10 mM KPi (pH 7.0) at 25°C. All mutant rRNase domains except Y52A and Y64A displayed emission maxima of 330 (±1) nm, essentially identical to that of the wild-type protein. Y52A and Y64A rRNase show emission maxima of 354 nm, which is indicative of solvent-exposed tryptophan residues, indicating that these proteins are destabilized and do not possess a wild-type-like structure under these conditions. (B) Far-UV CD spectra of wild-type and mutant rRNase domains. (C) Thermal denaturation profiles of wild-type and mutant E3 rRNase domains. Comparison of thermal denaturation profiles for wild-type E3 rRNase, H58A, and Y52A monitored by the change in the far-UV CD signal at 227 nm.
The far-UV CD spectrum of wild-type E3 rRNase domain displays maxima and minima at 226 and 195 nm, respectively (Fig. 3B ▶). Deconvolution of the spectrum by using the program CDNN (Bohm et al. 1992) predicts a secondary structure content (helix 7%, β-sheet 26%, random coil 45%) similar to that observed for E3 rRNase in the crystal structure of the E3 rRNase-Im3 complex (helix 5%, β-sheet 25%, and random coil 31%). For the mutant rRNase domains D55A, H58A, and E62A, far-UV CD spectra, indistinguishable from the wild-type protein, were obtained, whereas for those Y52A and Y64A differ significantly from the wild-type spectrum with the maxima at 226 nm absent and the minima shifted from 195 nm to 198 nm (Fig. 3B ▶). Calculated secondary structure predictions for Y52A and Y64A E3 rRNase domains were consistent with a large reduction in β-sheet structure (data not shown). Thus, consistent with the fluorescence data, CD measurements indicate that the structures of Y52A and Y64A E3 rRNase domains are significantly perturbed. It is probable that the loss of enzymatic activity in these mutants is primarily due to a loss of the wild-type protein structure, although this does not rule out the possibility that these residues may also interact with the substrate.
Thermal denaturation profiles were used to determine if loss of E3 rRNase function could correlate with decreased stability of protein structure. Thermal denaturation of wild-type E3 rRNase, monitored by the change in CD signal at 227 nm, gave a sigmoidal curve characteristic of a single cooperative transition from the folded to unfolded state from which a melting temperature (Tm) of 48°C was obtained (Fig. 3C ▶). A similar value (50°C) was obtained from monitoring the change in λmax of the intrinsic tryptophan fluorescence with increasing temperature, indicating that fluorescence and CD are monitoring the same cooperative transition (data not shown). The thermal denaturation profile of the inactive mutant protein H58A was found to be very similar to the wild-type protein with a Tm of 48°C, indicating that the protein structure has been affected little by this mutation (Fig. 3C ▶). In contrast, Y52A and Y64A do not unfold in a cooperative manner, as indicated by their linear denaturation profiles. Interestingly, and despite the fact that both the fluorescence and CD indicate that Y52A and Y64A are highly destabilized, they retain their ability to bind the immunity protein Im3 (L. Lancaster and C. Kleanthous, unpubl.).
Functional organization of the E3 rRNase domain
The data presented here strongly support the hypothesis that the H58 and E62 constitute the catalytic pair of the enzyme. These amino acids lie close to the side chains of R40, D55, E60, and R90, residues that were also found to be important to the enzymatic function of E3 rRNase but had little effect on its overall structure (Fig. 4A ▶). From our observations, it appears that the active center of the protein is located between residues 40 and 90 of the E3 rRNase domain. Clearly, the active center lies away from the immunity protein binding site in the tertiary structure of E3 rRNase (Fig. 4A ▶). In the primary sequence, the immunity protein binding site constitutes two sets of almost contiguous sequence from residues 2–15 and 25–41 of E3 rRNase (Carr et al. 2000). With the exception of R40, mutation of residues within the N-terminal 41 amino acids (H7, H10, K14, K30, T31, G37, K39) did not cause a significant decrease in E3 rRNase function (Fig. 4A ▶). Because all of these mutants were isolated bound to Im3, through the tandem expression strategy, this further implies that binding to Im3 has not been grossly destabilized. From our observations, it appears that the E3 rRNase domain can be nominally split into two functional regions, with residues that contact the immunity protein located in an N-terminal portion of the protein and an active site composed mainly or completely of C-terminal residues.
Figure 4.
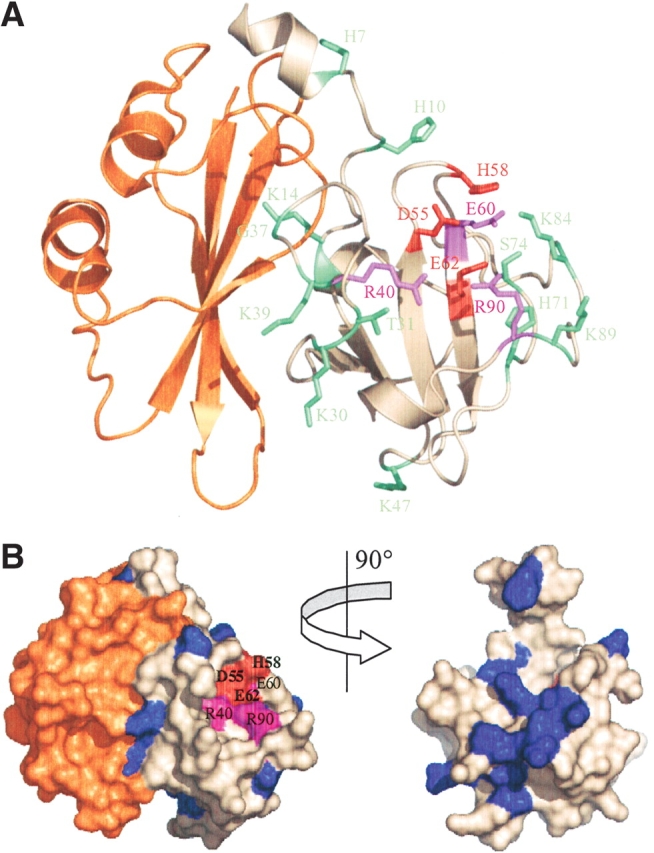
Structure of the E3 rRNase-Im3 complex highlighting the effect of alanine substitution on colicin E3 cytotoxicity and sequence conservation among rRNase-like bacteriocins. (A) Residues colored according to the effect of mutation on colicin E3 cytotoxicity; red indicates no detectable cytotoxicity, purple indicates severe impairment of cytotoxic activity, and green indicates little change relative to wild-type colicin E3 (see Fig. 1 ▶). The immunity protein (Im3) is colored orange. (B) Surface representation with E3 rRNase variable residues colored blue and conserved residues shown in white. The active site residues D55, H58, and E62 (all conserved) are colored red, and R40, E60, and R90 are colored purple. The right figure shows the E3 rRNase domain alone rotated ~90° to show sequence variability at the E3 rRNase-Im3 interface. This figure was prepared using the program Pymol v2.1 (PDB code 1E44; Carr et al. 2000).
Discussion
In this work we have identified residues of the colicin E3 rRNase domain likely to be involved directly in its highly specific rRNase activity. The arrangement of these residues (R40, D55, H58, E60, E62, and R90) in the active center of the toxin is shown in Figure 4 ▶. Our data support the prediction that H58 and E62 act as the acid–base pair during a putative transphosphorylation reaction (Carr et al. 2000). Mutation of either to alanine leads to a complete loss of colicin cytotoxicity but does not perturb the E3 rRNase structure. Mutation of D55 to alanine also leads to a complete loss of function, indicating this residue also plays a key role in catalysis. D55 lies within hydrogen bonding distance of H58 (2.9 Å) and may serve to stabilize its positive charge during its role as the proton donor in the transphosphorylation reaction. A similar arrangement of residues occurs in RNase A, where D121 lies in close proximity to the general acid H119 (Cuchillo et al. 1997). In this case, it has been proposed that the major effect of D121 is to orient the proper tautomer of H119 for catalysis (Schultz et al. 1998). It was also predicted by analogy with the barnase active site that R40, R90, and K84 of E3 rRNase would play a role in E3 rRNase activity (Carr et al. 2000) and, although the R40A and R90A mutants were found to have a considerably reduced activity, mutation of K84 to alanine was found to have no effect on colicin E3 activity (Fig. 1 ▶). The location of R90 and R40 and their substantial effect on colicin activity suggest that these residues do indeed play a role in substrate binding/catalysis, perhaps in transition state stabilization. The proposed structural effect of mutating Y52 or Y64 to alanine, resulting in an inactive colicin, was confirmed by mutation of these residues to phenylalanine. Both the Y52F and Y64F colicin E3 mutants were found to be active toxins (J. Leigh, L. Lancaster, and C. Kleanthous, unpubl.). Mapping the degree of sequence conservation among colicin rRNase domains along with proposed active site residues onto the surface of the rRNase domain-Im3 complex confirms that the putative active site cleft is highly conserved (Fig. 4B ▶). As was predicted previously, the immunity protein-binding site of E3 rRNase lies away from the active site and there is no obvious overlap between the two. An almost identical relationship between the immunity protein-binding site and catalytic site occurs in the colicin E9 DNase-Im9 complex (Kleanthous et al. 1999). Kleanthous and Walker (2001) have suggested that this arrangement, which appears common to structurally unrelated complexes, engenders a selective advantage for the producing organism through the ability to generate sequence variation at the interface of the colicin cytotoxic domain and immunity protein. For colicin E3, we mutated some of the residues that contact the immunity protein (H7, H10, K14, K30, G37, and K39) and showed that these have little or no effect on colicin cytotoxicity (Fig. 1 ▶). When the variation in sequence within the colicin E3 rRNase family is mapped onto the structure of the domain, it is clear that the immunity protein-binding site is indeed the most sequence-variable region and so, like the E9 DNase, a ‘hypervariable exosite’ governs inhibitor binding and specificity (Fig. 4B ▶). Such an exosite mechanism allows for the evolution of protein inhibitors with distinct specificities for enzymes carrying identical active sites (Kleanthous and Walker 2001).
Despite the probable mechanistic similarities between members of the T1 ribonuclease family and E3 rRNase, they share no sequence similarity. Indeed, the rRNase domains of colicin E3 and related bacteriocins do not, on the basis of their primary sequence, readily fit into any wider RNase family thus far described. Sequence alignment of E3 rRNase with homologous bacteriocin rRNase domains from colicins E3, E4, and E6; cloacin DF13; Lum1A; and Pflu4757 shows that, as expected, the region of the primary sequence around the active site is the most highly conserved region of the enzyme (Fig. 5 ▶). Of the residues of E3 rRNase that were found to be important for cytotoxicity, R40, R42, D55, E62, and R90 are completely conserved. However, in one sequence (Pfu2957, which is not included in Fig. 5 ▶) the proposed catalytic histidine (H58) is substituted for threonine (Parret and De Mott 2002). We speculate that this may represent an inactive pseudogene, an idea strengthened by the observation that, unlike all other identified E3 rRNase-like genes, Pflu2957 has no recognizable rRNase immunity protein gene associated with it (Parret and De Mott 2002).
Figure 5.

Sequence alignment of E3 rRNase with known and putative rRNase domains. Fully conserved positions are highlighted in dark gray and semiconserved in light gray. Asterisks denote residues identified as important for cytotoxicity. Sequences were obtained from the NCBI: E3 (gi_147 19502), E4 (gi_1071871), E6 (gi_116063), Ccl (gi_6176538), Lum1A (gi_16416909), Pflu4757 (gi_23062707), CV2871 (gi_34498326), Haso120001 (gi_32029964), HecA (gi_23573417).
Blast searches identified a further three proteins: CV2871 from Chromobacterium violaceum, Haso120001 from Haemophilus somnus, and HecA from Erwinia chrysanthemi, all classified as hemagglutinin-like adhesins that also carry putative rRNase domains with an apparent full complement of catalytic residues (Fig. 5 ▶). In the case of HecA, sequence similarity is limited to the C-terminal region of E3 rRNase, although, as discussed earlier, the N-terminal portion of the protein does not appear to be directly involved in catalysis. For CV2871 and Haso120001, sequence similarity to E3 rRNase is extensive. The best characterized of these hem-agglutinins is HecA, which contributes to virulence phenotypes of the plant pathogen E. chrysanthemi (Rojas et al. 2002). Indeed, hemagglutinin-type adhesins are generally associated with pathogenicity, although to our knowledge have not previously been identified as possessing cytotoxic catalytic activities. It is interesting to note that in all three of the hemagglutinins, the E3 rRNase-like sequence is located at the extreme C terminus of the protein, as is the case in the colicins themselves.
If the hemagglutinins identified here do in fact carry an active E3-like rRNase domain, what then is the role of this enzymatic domain in animal or plant pathogenicity? Interestingly, colicin E3 has previously been shown to be active against chloroplast ribosomes (Steege et al. 1982), which more closely resemble the prokaryotic ribosome than cytosolic eukaryotic ribosomes, and we speculate that chloroplast or mitochondrial ribosomes may be the cellular targets of these novel E3 rRNase-like domains. Further to this, the supposition that the rRNase-like domains of the hemagglutinins are not active against host ribosomes is supported by the absence of endogenous immunity-protein-like genes.
Materials and methods
Plasmid construction and protein purification
Site-directed mutagenesis, ligation, and transformation of competent E. coli cells were performed using standard molecular biology procedures. Wild-type and mutant E3 rRNase domains were tandemly expressed with Im3 from the plasmid pDW1 and the complex isolated by nickel affinity chromatography through an engineered six-histidine tag located at the C terminus of Im3, as described previously (Walker et al. 2003). E3 rRNase was eluted from the column by denaturation of the complex in 6 M guanidine hydrochloride, the isolated rRNase refolded by extensive dialysis into water (5 L), followed by 50 mM KPi (pH 7.0; 5 L). A final gel filtration step (Superdex-75) in 50 mM KPi (pH 7.0) was used to remove any remaining contaminants (Walker et al. 2003). Vectors to express colicin E3-Im3 complex (referred to as wild-type colicin E3-Im3 complex in this paper) were constructed by excision of the NcoI-XhoI fragment from pDW1 and ligation into the complementary sites of the pCS4 vector that carries the nuclease E colicin translocation and receptor binding domains (Garinot-Schneider et al. 1996). Wild-type and mutant colicin E3-Im3 complexes were purified by nickel affinity chromatography in a similar way to the E3 rRNase domain, except the complex was eluted from the column with a 5-mM to 500-mM imidazole gradient (100 mL on a 5-mL Ni affinity column). A final gel filtration step (Superdex-75) in 50 mM KPi (pH 7.0) was used to remove any remaining contaminants. Im3 was purified as described previously (Walker et al. 2003).
Cell-killing assays
Cell-killing assays were performed by spotting a fivefold serial dilution of purified colicin E3-Im3 complex, from a starting concentration of 1 mg mL−1, onto a growing lawn of E. coli JM83 cells containing the plasmid pTrc99a that confers ampicillin resistance. Plates were prepared by inoculation of E. coli JM83 cells, 1 : 100, into 0.7% molten agar at 42°C, which was overlaid onto LB-agar plates containing ampicillin (100 μg mL−1). Colicin E3-Im3 complex (2 μL) was then spotted onto the plates, which were incubated overnight at 37°C.
In vitro activity assays
The activity of the purified E3 rRNase domains directed against E. coli ribosomes was assessed through a coupled in vitro transcription-translation assay by using the E. coli S30 extract system for circular DNA (Promega). The activity of the mutant E3 rRNase domains was determined by the presence or absence of a reporter protein (firefly luciferase) encoded by pBESTluc vector (Pro-mega). Components of the transcription-translation system were set up according to the manufacturer’s instructions, and E3 rRNase to a concentration of 10−8 M was added and the mixture incubated for 5 min at 37°C prior to the addition of 3 μg of the template DNA and then incubated for a further hour at the same temperature. The quantity of firefly luciferase produced was determined by chemiluminescence using the Bright-Glo luciferase assay system (Promega) and measuring the light produced on a TopCount luminometer. The rRNase domains were considered active at this concentration if the measured light was <1% of that determined in buffer control experiments.
Spectroscopy
Fluorescence emission spectra were recorded on a Spex-Fluoro-Max-3 spectrofluorimeter. Spectra were recorded in 10 mM KPi (pH 7.0) at a protein concentration of 1 μM by using an excitation wavelength of 295 nm with excitation and emission slit widths set at 3 nm. CD spectra of E3 rRNase domains were recorded on a Jasco J-810 spectropolarimeter equipped with a Jasco Peltier temperature controller (PFD-4255). Far-UV CD spectra (190–260 nm) were recorded in 10 mM KPi (pH 7.0) at protein concentrations of 8.5 μM in a 1-mm path length quartz cuvette at 25°C. Spectra shown are the averaged data from five runs with the spectrum of the buffer subtracted. For thermal denaturation experiments, spectra were run at a protein concentration of 1.7 μM in a 10-mm path length quartz cuvette with magnetic stirring.
Acknowledgments
This work was supported by the Wellcome Trust and the BBSRC. The authors thank Andrew Leech (York) for performing electrospray mass spectrometry measurements and members of the CK lab for helpful discussion.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
E3 rRNase, the ribonuclease domain of colicin E3
Im3, immunity protein of colicin E3
E9 DNase, the endonuclease domain of colicin E9
Im9, immunity protein of colicin E9
Col−, a noncytotoxic colicin phenotype
KPi, potassium phosphate
CD, circular dichroism
λmax, wavelength of maximum fluorescence intensity
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04658504.
References
- Bohm, G., Muhr, R., and Jaenicke, R. 1992. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5 191–195. [DOI] [PubMed] [Google Scholar]
- Boon, T. 1972. Inactivation of ribosomes in vitro by colicin E3 and its mechanism of action. Proc. Natl. Acad. Sci. 69 549–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman, C.M., Dahlberg, J.E., Ikemura, T., Konisky, J., and Nomura, M. 1971. Specific inactivation of 16S ribosomal RNA induced by colicin E3 in vivo. Proc. Natl. Acad. Sci. 68 964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, S., Walker, D., James, R., Kleanthous, C., and Hemmings, A.M. 2000. Inhibition of a ribosome-inactivating ribonuclease: The crystal structure of the cytotoxic domain of colicin E3 in complex with its immunity protein. Structure Fold. Des. 8 949–960. [DOI] [PubMed] [Google Scholar]
- Cuchillo, C.M., Vilanova, M., and Nogues, M.V. 1997. Pancreatic ribonucleases. In Ribonucleases: Structures and functions (eds. G. D’Alessio and J.F. Riordan), pp. 272–297. Academic Press, New York.
- Garinot-Schneider, C., Pommer, A.J., Moore, G.R., Kleanthous, C., and James, R. 1996. Identification of putative active-site residues in the DNase domain of colicin E9 by random mutagenesis. J. Mol. Biol. 260 731–742. [DOI] [PubMed] [Google Scholar]
- Irie, M. 1997. RNase T1/RNase T2 family RNases. In Ribonucleases: Structures and functions (eds. G. D’Alessio and J.F. Riordan), pp. 101–124. Academic Press, New York.
- Kerr, B., Riley, M.A., Feldman, M.W., and Bohannan, B.J.M. 2002. Local dispersal promotes biodiversity in a real-life game of rock–paper–scissors. Nature 418 171–174. [DOI] [PubMed] [Google Scholar]
- Kleanthous, C. and Walker, D. 2001. Immunity proteins: Enzyme inhibitors that avoid the active site. Trends Biochem. Sci. 26 624–631. [DOI] [PubMed] [Google Scholar]
- Kleanthous, C., Kuhlmann, U.C., Pommer, A.J., Ferguson, N., Radford, S.E., Moore G.R., James, R., and Hemmings, A.M. 1999. Structural and mechanistic basis of immunity toward endonuclease colicins. Nat. Struct. Biol. 6 243–252. [DOI] [PubMed] [Google Scholar]
- Makarov, A.A. and Ilinskaya, O.N. 2003. Cytotoxic ribonucleases: Molecular weapons and their targets. FEBS Lett. 540 15–20. [DOI] [PubMed] [Google Scholar]
- Mosbahi, K., Lemaitre, C., Keeble, A.H., Mobasheri, H., Morel, B., James, R., Moore, G.R., Lea, E.J., and Kleanthous, C. 2002. The cytotoxic domain of colicin E9 is a channel-forming endonuclease. Nat. Struct. Biol. 9 476–484. [DOI] [PubMed] [Google Scholar]
- Ogawa, T., Tomita, K., Ueda, T., Watanabe, K., Uozumi, T., and Masaki, H. 1999. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science 283 2097–2100. [DOI] [PubMed] [Google Scholar]
- Ohno, S. and Imahori, K. 1978. Colicin E3 is an endonuclease. J. Biochem. 84 1637–1640. [DOI] [PubMed] [Google Scholar]
- Ohno, S., Ohno-Iwashita, Y., Suzuki, K., and Imahori, K. 1977. Purification and characterization of active component and active fragment of colicin E3. J. Biochem. 82 1045–1053. [DOI] [PubMed] [Google Scholar]
- Ohno-Iwashita, Y. and Imahori, K. 1977. Colicin E3 induced cleavage of 16S rRNA of isolated 30S ribosomal subunits. J. Biochem. 82 919–922. [DOI] [PubMed] [Google Scholar]
- Parret, A.H.A. and De Mott, R. 2002. Bacteria killing their own kind: Novel bacteriocins of Pseudomonas and other γ-proteobacteria. Trends Microbiol. 10 107–112. [DOI] [PubMed] [Google Scholar]
- Pommer, A.J., Cal, S., Keeble, A.H., Walker, D., Evans, S.J., Kuhlmann, U.C., Cooper, A., Connolly, B.A., Hemmings, A.M., Moore, G.R. et al. 2001. Mechanism and cleavage specificity of the H-N-H endonuclease colicin E9. J. Mol. Biol. 314 735–749. [DOI] [PubMed] [Google Scholar]
- Rojas, C.M., Ham, J.H., Deng, W.L., Doyle, J.J., and Collmer, A. 2002. HecA, a member of a class of adhesins produced by diverse pathogenic bacteria, contributes to the attachment, aggregation, epidermal cell killing, and virulence phenotypes of Erwinia chrysanthemi EC16 on Nicotiana clevelandii seedlings. Proc. Natl. Acad. Sci. 99 13142–13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz, S.R., Korn, C., Bujnicki, J.M., Gimadutdinow, O., Pingoud, A., and Meiss, G. 2003. Experimental evidence for a ββα-Me-finger nuclease motif to represent the active site of the caspase-activated DNase. Biochemistry 42 9288–9294. [DOI] [PubMed] [Google Scholar]
- Schultz, L.W., Quirk, D.J., and Raines, R.T. 1998. His. . .Asp catalytic dyad of ribonuclease A: Structure and function of the wild-type, D121N, and D121A enzymes. Biochemistry 37 8886–8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidikaro, J. and Nomura, M. 1973. Colicin E3-induced in vitro inactivation of ribosomes from colicin-insensitive bacterial species. FEBS Lett. 29 15–19. [DOI] [PubMed] [Google Scholar]
- Smarda, J., Fialova, M., and Smarda Jr., J. 2001. Cytotoxic effects of colicins E1 and E3 on v-myb-transformed chicken monoblasts. Folia Biol. 47 11–13. [PubMed] [Google Scholar]
- Soelaiman, S., Jakes, K., Wu, N., Li, C.M., and Shoham, M. 2001. Crystal structure of colicin E3: Implications for cell entry and ribosome inactivation. Mol. Cell 8 1053–1062. [DOI] [PubMed] [Google Scholar]
- Steege, D.A., Graves, M.C., and Spremulli, L.L. 1982. Euglena gracilis chloroplast small subunit rRNA. Sequence and base pairing potential of the 3′ terminus, cleavage by colicin E3. J. Biol. Chem. 257 10430–10439. [PubMed] [Google Scholar]
- Tomita, K., Ogawa, T., Uozumi, T., Watanabe, K., and Masaki, H. 2000. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc. Natl. Acad. Sci. 97 8278–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, G.C. 1996. The SOS response of Escherichia coli. In Escherichia coli and Salmonella: Cellular and molecular biology (ed. F.C. Neidhardt), pp. 1400–1416. American Society for Microbiology Press, Washington, DC.
- Walker, D.C., Georgiou, T., Pommer, A.J., Walker, D., Moore, G.R., Kleanthous, C., and James, R. 2002. Mutagenic scan of the H-N-H motif of colicin E9: Implications for the mechanistic enzymology of colicins, homing enzymes and apoptotic endonucleases. Nucleic Acids Res. 30 3225–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, D., Moore, G.R., James, R., and Kleanthous, C. 2003. Thermodynamic consequences of bipartite immunity protein binding to the ribosomal ribonuclease colicin E3. Biochemistry 42 4161–4171. [DOI] [PubMed] [Google Scholar]
- Walker, D., Rolfe, M., Thompson, A., Moore, G.R., James, R., Hinton, J.C.D., and Kleanthous, C. 2004. Transcriptional profiling of colicin-induced cell death of Escherichia coli MG1655 identifies potential mechanisms by which bacteriocins promote bacterial diversity. J. Bacteriol. 186 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis, R., Moore, G.R., James, R., and Kleanthous, C. 1995. Protein–protein interactions in colicin E9 DNase-immunity protein complexes. 1. Diffusion-controlled association and femtomolar binding for the cognate complex. Biochemistry 34 13743–13750. [DOI] [PubMed] [Google Scholar]