

Abstract
In this article, we present a new technique for the rapid and precise docking of peptides to MHC class I and class II receptors. Our docking procedure consists of three steps: (1) peptide residues near the ends of the binding groove are docked by using an efficient pseudo-Brownian rigid body docking procedure followed by (2) loop closure of the intervening backbone structure by satisfaction of spatial constraints, and subsequently, (3) the refinement of the entire backbone and ligand interacting side chains and receptor side chains experiencing atomic clash at the MHC receptor–peptide interface. The method was tested by remodeling of 40 nonredundant complexes of at least 3.00 Å resolution for which three-dimensional structural information is available and independently for docking peptides derived from 15 nonredundant complexes into a single template structure. In the first test, 33 out of 40 MHC class I and class II peptides and in the second test, 11 out of 15 MHC–peptide complexes were modeled with a Cα RMSD < 1.00 Å.
Keywords: major histocompatibility complex, epitope prediction, flexible docking, immunology, Monte Carlo
Major histocompatibility complex (MHC) molecules are highly polymorphic cell surface molecules that present antigenic peptides to cells of the T-cell compartment of the immune system. Two classes of MHC molecules are distinguished, class I and class II.
MHC class I molecules are heterodimers, consisting of a heavy α-chain of about 45 kDa, and a light chain, 32-microglobulin (32M) of about 12 kDa (Klein 1986). Class I ligands are derived from endogenously expressed proteins that are degraded by cytosolic proteinases with typical length of 8–12 amino acids. The proteolytic fragments are transported into the endoplasmic reticulum in an ATP-dependent fashion by the transporter associated with antigen processing (TAP), where they bind to vacant receptor sites on newly synthesized MHC class I molecules. The MHC–peptide complex is subsequently transported to the cell surface and presented for recognition by the T-cell receptors (TCRs) of CD8+ cytotoxic T-cells (CTL).
Class II peptides are heterodimeric glycoproteins consisting of an α-chain (34 kDa) and β-chain (29 kDa) with very similar overall quaternary structure to that of class I molecules (Brown et al. 1993; Stern et al. 1994; Stern and Wiley 1994). Class II ligands are of variable length, usually of 9–25 amino acids and are derived mainly from exogenous or transmembrane proteins, as well as from cytosolic proteins that are degraded by various proteinases originating from the lysosomal compartment. After displacing the endogenous MHC class II ligand known as the CLIP peptide in the late endosomal/lysosomal compartment, this MHC-peptide complex is also transported to the cell surface and presented for recognition by the TCRs of CD4+ T cells.
In the design of molecular vaccines for the treatment of diseases, identification of T-cell epitopes from immunologically relevant antigens is an important prerequisite. The first step in T-cell-mediated immune response is the binding of antigenic peptides to MHC receptors, which serves as a necessary although not sufficient condition for epitope recognition (Flower 2003). The second step is the recognition and binding of T-cell receptors, which then initiate the immune response cascade. The experimental identification of T-cell epitopes is a time-consuming and expensive process due to the large number and diverse nature of MHC alleles and candidate peptides. Current computational techniques focus on the identification of potential MHC-binding candidate peptides, and can be broadly classified into two categories: (1) sequence-based approaches such as sequence motifs (Falk et al. 1991), matrix models (Parker et al. 1994; Davenport et al. 1995; Gulukota et al. 1997; Godkin et al. 1998; Rammensee et al. 1999), Artificial Neural Network (Brusic et al. 1998), Hidden Markov Model (Lim et al. 1996; Mamitsuka 1998; Brusic et al. 2002), and Support Vector Machine (Dönnes and Elofsson 2002; Bhasin and Raghava 2004) for large-scale screening of potential T-cell epitopes from protein sequence databanks; and (2) structure-based approaches such as homology modeling (Lim et al. 1996; Michielin et al. 2000), protein threading (Altuvia et al. 1995), and docking techniques (Caflisch et al. 1992; Rosenfeld et al. 1993, 1995; Sezerman et al. 1996; Rognan et al. 1999; Desmet et al. 2000; Michielin and Karplus 2002), which utilize three-dimensional data for the detailed structural analysis of interactions between the MHC and the bound short antigenic peptides. The former are more suitable for large-scale screening of potential T-cell epitopes, while the latter are better suited for detailed analysis of short immunogenic regions of antigens. Although sequence-based techniques are well established, a severe limitation of such approaches is the heavy reliance on the availability of large comprehensive training sets of peptides. This approach is not suitable for accurate prediction of situations where insufficient experimental data are available. As such, the coverage of sequence-based techniques is limited to subsets of binding peptides that belong to the most numerous groups and cannot generate reliable data for peptides that are least represented in the dataset. To date, developments of structure-based techniques are poorly developed and lagging far behind sequence-based procedures due to the relatively higher complexity in their development and excessive computational costs. Despite their slow progress, structure-based techniques are highly promising (Altuvia et al. 1995) and play a significant role as a predictive tool in detailed selection of peptides for binding studies, planning of experiments, and better understanding of biological processes involved in the stimulation of T-cell-mediated immune response. A preliminary analysis of the structural descriptors defining the MHC–peptide interactions has been carried out by our group (Govindarajan et al. 2003). The ability of structure-based approaches to reliably predict MHC binding peptides and thereby potential T-cell epitopes clearly has major implications for clinical immunology, particularly in the area of vaccine design. However, prior to predicting binding peptides based on structural approaches, it is crucial to improve the speed and accuracy of docking a known antigenic peptide to its cognate receptor. Once this problem has been successfully addressed, the methodology will be applied to predict antigenic regions from a protein sequence and understand which peptides are promiscuous and which ones are allele-specific.
In this article, we present a novel computational protocol for rapid and precise modeling of the bound conformations of flexible peptides to MHC receptors and apply it primarily to the remodeling of 40 (29 class I and 11 class II) nonredundant MHC–peptide complexes for which crystal structures are available in the Protein Data Bank (PDB). Our docking protocol consists of three steps: (1) peptide residues near the ends of the binding groove are docked by using an efficient pseudo-Brownian rigid body docking procedure followed by (2) loop closure of the remaining backbone structure by satisfaction of spatial constraints, and subsequently, (3) refinement of the entire backbone and the interacting side chains of the ligand and the receptor experiencing atomic clash at the MHC–peptide interface. In our preliminary experiments, the proposed docking procedure generated as many as 33 out of 40 peptides with Cα RMSD of less than 1.00 Å from the crystal structures. To the best of our knowledge, these results represent up to fivefold increase in accuracy compared to available flexible docking techniques in the remodeling of MHC–peptide complexes, establishing the efficacy of out procedure to model highly accurate MHC–peptide complex structures and permitting the conformational sampling of the peptide in the binding groove. As a second test, we have attempted docking new peptides onto a single template structure, to verify whether our method can accurately model the bound conformations of novel peptides bound to a specific MHC allele. With 15 peptides derived from Class I and Class II complexes and three structural templates, the Cα conformation of the docked peptides was within an RMSD of 1.00 Å in 11 structures.
Results
At the outset, a systematic structural analysis of 41 Class I and 42 Class II MHC–peptide complexes (listed in Table 1) was carried out to determine which regions of the bound peptide are conserved in the groove. The Cα RMSD values of the N and C termini reveal their relatively fixed locations within the groove across both classes of MHC–peptide complexes. Leveraging on this observation, a three-step docking procedure (detailed in Materials and Methods) as shown in Figure 1 ▶ has been developed as a generalized method for obtaining the conformation of peptides in the MHC groove, in a rapid yet accurate manner. Evaluation of our modeling procedure is performed systematically in the following two tests: (1) Redocking 40 test case complexes; and (2) docking of 15 solved peptides into templates of appropriate alleles.
Table 1.
Structural comparison of the bound peptides of MHC Class I and Class II complex crystal structures respectively
| Cα RMSD | |||||||
| Class | MHC allele | PDB | Peptide sequence | Length | Resolution (Å) | Head | Tail |
| I | HLA-A*0201 | 1AO7 | LLFGYPVYV | 9 | 2.60 | 0.03 | 0.10 |
| I | HLA-A*0201 | 1AKJ | ILKEPVHGV | 9 | 2.65 | 0.05 | 0.03 |
| I | HLA-A*0201 | 1BD2 | LLFGYPVYV | 9 | 2.50 | 0.02 | 0.13 |
| I | HLA-A*0201 | 1B0G | ALWGFFPVL | 9 | 2.50 | 0.05 | 0.09 |
| I | HLA-A*0201 | 1DUY | LFGYPVYV | 8 | 2.15 | 0.07 | 0.16 |
| I | HLA-A*0201 | 1DUZ | LLFGYPVYV | 9 | 1.80 | 0.05 | 0.13 |
| I | HLA-A*0201 | 1HHG | TLTSCNTSV | 9 | 2.60 | 0.02 | 0.07 |
| I | HLA-A*0201 | 1HHH | FLPSDFFPSV | 10 | 3.00 | 0.04 | 0.16 |
| I | HLA-A*0201 | 1HHI | GILGFVFTL | 9 | 2.50 | 0.14 | 0.07 |
| I | HLA-A*0201 | 1HHJ | ILKEPVHGV | 9 | 2.50 | 0.06 | 0.08 |
| I | HLA-A*0201 | 1HHK | LLFGYPVYV | 9 | 2.50 | 0.06 | 0.13 |
| I | HLA-A*0201 | 1I1F | FLKEPVHGV | 9 | 2.80 | 0.05 | 0.07 |
| I | HLA-A*0201 | 1I1Y | YLKEPVHGV | 9 | 2.20 | 0.06 | 0.03 |
| I | HLA-A*0201 | 1I4F | GVYDGREHTV | 10 | 1.40 | 0.00 | 0.00 |
| I | HLA-A*0201 | 1I7R | FAPGFFPYL | 9 | 2.20 | 0.08 | 0.07 |
| I | HLA-A*0201 | 1I7T | ALWGVFPVL | 9 | 2.80 | 0.11 | 0.07 |
| I | HLA-A*0201 | 1I7U | ALWGFVPVL | 9 | 1.80 | 0.03 | 0.03 |
| I | HLA-A*0201 | 1IM3 | LLFGYPVYV | 9 | 2.20 | 0.04 | 0.14 |
| I | HLA-A*0201 | 1JF1 | ELAGIGILTV | 10 | 1.85 | 0.08 | 0.00 |
| I | HLA-A*0201 | 1JHT | ALGIGILTV | 9 | 2.15 | 0.06 | 0.10 |
| I | HLA-A*0201 | 1OGA | GILGFVFTL | 9 | 1.40 | 0.06 | 0.10 |
| I | HLA-A*0201 | 1QRN | LLFGYAVYV | 9 | 2.80 | 0.05 | 0.12 |
| I | HLA-A*0201 | 1QSE | LLFGYPRYV | 9 | 2.80 | 0.04 | 0.18 |
| I | HLA-A*0201 | 1QSF | LLFGYPVAV | 9 | 2.80 | 0.07 | 0.11 |
| I | HLA-A*0201 | 2CLR | MLLSVPLLLG | 10 | 2.00 | 0.05 | 0.08 |
| I | HLA-A*6801 | 1TMC | EVAPPEYHRK | 10 | 2.30 | 0.07 | 0.09 |
| I | HLA-B*0801 | 1AGB | GGRKKYKL | 8 | 2.20 | 0.15 | 0.15 |
| I | HLA-B*0801 | 1AGC | GGKKKYQL | 8 | 2.10 | 0.03 | 0.19 |
| I | HLA-B*0801 | 1AGD | GGKKKYKL | 8 | 2.05 | 0.03 | 0.20 |
| I | HLA-B*0801 | 1AGE | GGKKKYRL | 8 | 2.30 | 0.02 | 0.18 |
| I | HLA-B*0801 | 1AGF | GGKKRYKL | 8 | 2.20 | 0.08 | 0.16 |
| I | HLA-B*2705 | 1HSA | ARAAAAAAA | 9 | 2.10 | 0.02 | 0.12 |
| I | HLA-B*3501 | 1A1N | VPLRPMTY | 8 | 2.00 | 0.03 | 0.18 |
| I | HLA-B*3501 | 1A9B | LPPLDITPY | 9 | 3.20 | 0.13 | 0.09 |
| I | HLA-B*3501 | 1A9E | LPPLDITPY | 9 | 2.50 | 0.08 | 0.09 |
| I | HLA-B*5101 | 1E27 | LPPVVAKEI | 9 | 2.20 | 0.13 | 0.04 |
| I | HLA-B*5101 | 1E28 | TAFTIPSI | 8 | 3.00 | 0.13 | 0.10 |
| I | HLA-B*5301 | 1A1M | TPYDINQML | 9 | 2.30 | 0.07 | 0.19 |
| I | HLA-B*5301 | 1A1O | KPIVQYDNF | 9 | 2.30 | 0.07 | 0.09 |
| I | HLA-Cw3 | 1EFX | GAVDPLLAL | 9 | 3.00 | 0.08 | 0.06 |
| I | HLA-Cw4 | 1IM9 | QYDDAVYKL | 9 | 2.80 | 0.20 | 0.29 |
| I | H2-Db | 1BZ9 | FAPGVFPYM | 9 | 2.80 | 0.09 | 0.10 |
| 1 | H2-Db | 1CE6 | FAPGNYPAL | 9 | 2.90 | 0.04 | 0.10 |
| 1 | H2-Db | 1FG2 | KAVYNFATC | 9 | 2.75 | 0.04 | 0.12 |
| I | H2-Db | 1JPF | SGVENPGGYCL | 11 | 2.18 | 0.04 | 0.12 |
| I | H2-Db | 1JPG | FQPQNGQFI | 9 | 2.20 | 0.32 | 0.07 |
| I | H2-Db | 1QLF | FAPSNYPAL | 9 | 2.65 | 0.04 | 0.13 |
| I | H2-Db | 1N5A | KAVYNFATM | 9 | 2.85 | 0.06 | 0.15 |
| I | H2-Kb | 1FO0 | INFDFNTI | 8 | 2.50 | 0.03 | 0.13 |
| I | H2-Kb | 1NAM | RGYVYQGL | 8 | 2.70 | 0.03 | 0.08 |
| I | H2-Kb | 1FZJ | RGYVQGL | 8 | 1.90 | 0.09 | 0.16 |
| I | H2-Kb | 1FZK | FAPGNYPAL | 9 | 1.70 | 0.04 | 0.08 |
| I | H2-Kb | 1FZM | RGYVYQGL | 8 | 1.80 | 0.16 | 0.13 |
| I | H2-Kb | 1FZO | FAPGNYPAL | 9 | 1.80 | 0.03 | 0.08 |
| I | H2-Kb | 1G6R | SIYRYYGL | 8 | 2.80 | 0.07 | 0.03 |
| I | H2-Kb | 1KBG | RGYVYXGL | 8 | 2.20 | 0.05 | 0.14 |
| I | H2-Kb | 1OSZ | RGYLYQGL | 8 | 2.10 | 0.05 | 0.14 |
| I | H2-Kb | 1VAC | SIINFEKL | 8 | 2.50 | 0.10 | 0.15 |
| I | H2-Kb | 1VAD | SRDHSRTPM | 9 | 2.50 | 0.09 | 0.08 |
| I | H2-Kb | 1KJ3 | KVITFIDL | 8 | 2.30 | 0.09 | 0.16 |
| I | H2-Kb | 1G7P | SRDHSRTPM | 9 | 1.50 | 0.03 | 0.05 |
| I | H2-Kb | 1G7Q | SAPDTRPA | 8 | 1.60 | 0.12 | 0.25 |
| I | H2-Kb | 2VAA | RGYVYQGL | 8 | 2.30 | 0.04 | 0.11 |
| I | H2-Kb | 2VAB | FAPGNYPAL | 9 | 2.50 | 0.05 | 0.09 |
| I | H2-Kb | 2CKB | EQYKFYSV | 8 | 3.20 | 0.23 | 0.36 |
| I | H2-Ld | 1LD9 | YPNVNIHNF | 9 | 2.40 | 0.11 | 0.14 |
| I | H2-Ld | 1LDP | APAAAAAAM | 9 | 3.10 | 0.29 | 0.16 |
| I | H2-Ld | 1LDP | QLSPFPFDL | 9 | 3.10 | 0.22 | 0.16 |
| I | RT1-Aa | 1ED3 | ILFPSSERLISNR | 13 | 2.55 | 0.14 | 0.11 |
| II | HLA-DR1 | 1AQD | DWRFLRGYHQ | 10 | 2.45 | 0.22 | 0.13 |
| II | HLA-DR1 | 1AQD | DWRFLRGYHQ | 10 | 2.45 | 0.13 | 0.08 |
| II | HLA-DR2 | 1BX2 | VVHFFKNIVT | 10 | 2.60 | 0.05 | 0.18 |
| II | HLA-DR2 | 1BX2 | VVHFFKNIVT | 10 | 2.60 | 0.07 | 0.10 |
| II | HLA-DR2 | 1FV1 | HFFKNIVTPR | 10 | 1.90 | 0.01 | 0.04 |
| II | HLA-DR1 | 1FV1 | HFFKNIVTPR | 10 | 1.90 | 0.07 | 0.20 |
| II | HLA-DR3 | 1A6A | KMRMATPLLM | 15 | 2.75 | 0.14 | 0.02 |
| II | HLA-DR4 | 1J8H | KYVKQNTLKL | 13 | 2.40 | 0.09 | 0.08 |
| II | HLA I-Ak | 1IAK | TDYGILQINS | 13 | 1.90 | 0.16 | 0.02 |
| II | HLA I-Ak | 1F3J | KRHGLDNYRG | 14 | 3.10 | 0.12 | 0.11 |
| II | HLA I-Ak | 1D9K | SHRGAIEWEG | 16 | 3.20 | 0.22 | 0.18 |
| II | HLA-DQ8 | 1JK8 | VEALYLVCGE | 14 | 2.40 | 0.19 | 0.20 |
| II | HLA-DR51 | 1H15 | VYHFVKKHVH | 14 | 3.10 | 0.10 | 0.27 |
| II | I-Ak | 1JL4 | SHRGAIEWEG | 16 | 4.30 | 0.22 | 0.18 |
Three residues at the N (“head”) and C (“tail”) termini of the peptide within the MHC groove are compared.
Figure 1.

Flow chart of the three-step docking procedure used in this work.
Redocking bound peptides to MHC molecules
To validate our docking procedures, we first applied our technique to the rebuilding of 40 nonredundant MHC–peptide complexes (refer to the Selection of the MHC–peptide complexes section) by docking peptides extracted from MHC–peptide complexes back into their respective binding grooves. This initial experiment is an important first step for testing the capability of our technique to model peptides into their cognate MHC receptors. Peptides were separated from experimental structures and remodeled back into their own bound states. A correct docking result is defined as a complex with not more than 2.50 Å CRMSD from the known experimental structure. Our procedure generated 33 α out of 40 nonredundant complexes (extracted from a larger dataset listed in Table 1) within a Cα RMSD of 1.00 Å. The RMSD for the near-native solution ranges from 0.09 Å (complex 1G7Q) to 1.53 Å (complex 1JF1). Table 2 and Figure 2 ▶ detail the results obtained from this validation test.
Table 2.
Comparison of the position the bound peptide in the original crystal structure and after docking back into the MHC groove
| Class | Allele | PDB | Res (Å) | Length | RMSD (Å) | Sequence |
| I | HLA-A*0201 | 1DUZ | 1.80 | 9 | 0.33 | LLFGYPVYV |
| I | HLA-A*0201 | 1HHG | 2.60 | 9 | 0.46 | TLTSCNTSV |
| I | HLA-A*0201 | 1HHJ | 2.50 | 9 | 0.87 | ILKEPVHGV |
| I | HLA-A*0201 | 1HHH | 3.00 | 10 | 1.10 | FLPSDFFPSV |
| I | HLA-A*0201 | 1I1Y | 2.20 | 9 | 0.70 | YLKEPVHGV |
| I | HLA-A*0201 | 1I4F | 1.40 | 10 | 0.49 | GVYDGREHTV |
| I | HLA-A*0201 | 1I7R | 2.20 | 9 | 0.59 | FAPGFFPYL |
| I | HLA-A*0201 | 1I7U | 1.80 | 9 | 0.32 | ALWGFVPVL |
| I | HLA-A*0201 | 1JF1 | 1.85 | 10 | 1.53 | ELAGIGILTV |
| I | HLA-A*0201 | 1JHT | 2.15 | 9 | 0.54 | ALGIGILTV |
| I | HLA-A*0201 | 1OGA | 1.40 | 9 | 0.32 | GILGFVFTL |
| I | HLA-A*0201 | 1QRN | 2.80 | 9 | 0.46 | LLFGYAVYV |
| I | HLA-A*0201 | 1QSE | 2.80 | 9 | 0.26 | LLFGYPRYV |
| I | HLA-A*0201 | 1QSF | 2.80 | 9 | 0.54 | LLFGYPVAV |
| I | HLA-A*6801 | 1TMC | 2.30 | 10 | 0.52 | EVAPPEYHRK |
| I | HLA-B*0801 | 1AGD | 2.05 | 8 | 0.28 | GGKKKYKL |
| I | HLA-B*0801 | 1AGF | 2.20 | 8 | 0.66 | GGKKRYKL |
| I | HLA-B*3501 | 1A1N | 2.00 | 8 | 0.10 | VPLRPMTY |
| I | HLA-B*3501 | 1A9E | 2.50 | 9 | 1.09 | LPPLDITPY |
| I | HLA-B*5101 | 1E27 | 2.20 | 9 | 1.27 | LPPVVAKEI |
| I | HLA-B*5301 | 1A1M | 2.30 | 9 | 0.59 | TPYDINQML |
| I | HLA-B*5301 | 1A1O | 2.30 | 9 | 0.78 | KPIVQYDNF |
| I | H2-Db | 1JPF | 2.18 | 11 | 1.14 | SGVENPGGYCL |
| I | H2-Db | 1JPG | 2.20 | 9 | 0.33 | FQPQNGQFI |
| I | H2-Dd | 1BII | 2.40 | 10 | 1.49 | RGPGRAFVTI |
| I | H2-Kb | 1FZM | 1.80 | 8 | 0.32 | RGYVYQGL |
| I | H2-Kb | 1FZO | 1.80 | 9 | 0.40 | FAPGNYPAL |
| I | H2-Kb | 1G7P | 1.50 | 9 | 0.97 | SRDHSRTPM |
| I | H2-Kb | 1G7Q | 1.60 | 8 | 0.09 | SAPDTRPA |
| II | HLA-DR1 | 1AQD | 2.45 | 10 | 0.63 | DWRFLRGYHQ |
| II | HLA-DR1 | 1AQD | 2.45 | 10 | 1.08 | DWRFLRGYHQ |
| II | HLA-DR2 | 1BX2 | 2.60 | 10 | 0.60 | VVHFFKNIVT |
| II | HLA-DR2 | 1BX2 | 2.60 | 10 | 0.81 | VVHFFKNIVT |
| II | HLA-DR2 | 1FV1 | 1.90 | 10 | 0.47 | HFFKNIVTPR |
| II | HLA-DR2 | 1FV1 | 1.90 | 10 | 0.58 | HFFKNIVTPR |
| II | HLA-DR3 | 1A6A | 2.75 | 10 | 0.38 | KMRMATPLLM |
| II | HLA-DR4 | 1J8H | 2.40 | 10 | 0.59 | KYVKQNTLKL |
| II | HLA-DR4 | 2SEB | 2.50 | 10 | 0.43 | YMRADAAAGG |
| II | HLA I-Ak | 1IAK | 1.90 | 10 | 0.42 | TDYGILQINS |
| II | HLA-DQ8 | 1JK8 | 2.40 | 10 | 0.21 | VEALYLVCGE |
RMSD values are calculated for the ligand interface Cα atoms of the lowest energy solution, superimposed onto the experimental structure.
Figure 2.

Representations of selected lowest energy solutions in the binding grooves obtained after redocking the peptides into the respective MHC grooves in the first benchmarking test (Table 2). Experimental peptide structures are represented as bold dark lines and remodeled structures as thin gray lines, showing all heavy atoms for MHC Class I (A–H) and class II (I–R) complexes. The relative orientations of the peptide side chains with respect to the floor of the binding groove are indicated by arrows pointing either up (away from the groove) or down (towards the groove).
This preliminary experiment establishes the validity of our approach, using the three-step proposed procedure. Encouraged by these results, we next apply our procedure to a more practical problem in allele-specific vaccine design, that is, the prediction of MHC–peptide complexes resulting from multiple peptides binding to a single MHC allele template.
Docking MHC-binding peptides onto a single template
We next applied our technique to the modeling of 15 nonredundant peptides (13 class I and 2 class II) for which crystal structures are available into a single template. This stage of the testing is critical to determine the capability of our procedure to model unknown peptides onto available templates. Due to the deficiency of available class II crystal structures, only two class II peptides are tested in this stage. Our procedure constantly found a solution with RMSD below 1.48 Å. Table 3 shows the results obtained from this experiment.
Table 3.
Comparison between modeled peptides and relevant crystal structures after docking onto a single template
| Class | Allele | PDB | Length | Template | RMSD (Å) | Sequence |
| I | HLA-A*0201 | 1DUZ | 9 | 1I4F | 0.69 | LLFGYPVYV |
| I | HLA-A*0201 | 1HHG | 9 | 1I4F | 0.58 | TLTSCNTSV |
| I | HLA-A*0201 | 1HHJ | 9 | 1I4F | 0.73 | ILKEPVHGV |
| I | HLA-A*0201 | 1HHH | 10 | 1I4F | 1.48 | FLPSDFFPSV |
| I | HLA-A*0201 | 1I1Y | 9 | 1I4F | 0.77 | YLKEPVHGV |
| I | HLA-A*0201 | 1I7R | 9 | 1I4F | 0.60 | FAPGFFPYL |
| I | HLA-A*0201 | 1I7U | 9 | 1I4F | 0.70 | ALWGFVPVL |
| I | HLA-A*0201 | 1JF1 | 10 | 1I4F | 1.20 | ELAGIGILTV |
| I | HLA-A*0201 | 1JHT | 9 | 1I4F | 1.09 | ALGIGILTV |
| I | HLA-A*0201 | 1OGA | 9 | 1I4F | 0.38 | GILGFVFTL |
| I | HLA-A*0201 | 1QRN | 9 | 1I4F | 0.81 | LLFGYAVYV |
| I | HLA-A*0201 | 1QSE | 9 | 1I4F | 0.52 | LLFGYPRYV |
| I | HLA-A*0201 | 1QSF | 9 | 1I4F | 0.57 | LLFGYPVAV |
| II | HLA-DR2 | 1BX2 | 10 | 1FV1 | 1.22 | VVHFFKNIVT |
| II | HLA-DR4 | 2SEB | 10 | 1J8H | 0.42 | KYVKQNTLKL |
RMSD values calculated for the ligand interface Cα atoms of the lowest energy solution superimposed onto the experimental PDB structure are listed.
Benchmarking
To determine the validity and accuracy of our procedure, we benchmark our technique with four previously published studies involving MHC class I peptide modeling as detailed in Table 4. As there was no previously reported accuracy for MHC class II peptide modeling, no benchmarking could be performed on the modeled MHC class II peptides. It is notable that validation process by Rognan (Rognan et al. 1999), Desmet (Desmet et al. 2000), and Sezerman (Sezerman et al. 1996) involved remodeling peptides back into their original crystal structure. Using this criterion, our procedure is either comparable or outperforms the three earlier studies (Sezerman et al. 1996; Rognan et al. 1999; Desmet et al. 2000) in terms of the Cα RMSD of the modeled peptides.
Table 4.
Benchmarking of our MHC-peptide procedure with previously published studies in MHC class I peptide modeling
| Peptide sequence | Technique | Literature reference | Ref RMSD(Å) | Current RMSD (Å)a |
| TLTSCNTSV | Simulated annealing | Rognan et al. 1999 | 1.04 | 0.46, 0.58 |
| FLPSDFFPSV | Simulated annealing | Rognan et al. 1999 | 1.59 | 1.10, 1.48 |
| GILGFVFTL | Simulated annealing | Rognan et al. 1999 | 0.46 | 0.32, 0.38 |
| ILKEPVHGV | Simulated annealing | Rognan et al. 1999 | 0.87 | 0.87, 0.73 |
| LLFGYPVYV | Simulated annealing | Rognan et al. 1999 | 0.78 | 0.33, 0.69 |
| RGYVYQGL | Combinatorial buildup algorithm | Desmet et al. 2000 | 0.56 | 0.32, 0.66 |
| FAPGNYPAL | Multiple copy algorithm | Rosenfeld et al. 1993, 1995 | 2.70 | 0.40, 0.90 |
| GILGFVFTL | Multiple copy algorithm | Rosenfeld et al. 1993, 1995 | 1.40 | 0.32, 0.38 |
| LLFGYPVYV | Combinatorial buildup algorithm | Sezerman et al. 1996 | 1.40 | 0.33, 0.69 |
| ILKGPVHGV | Combinatorial buildup algorithm | Sezerman et al. 1996 | 1.30 | 0.87, 0.73 |
| GILGFVFTL | Combinatorial buildup algorithm | Sezerman et al. 1996 | 1.60 | 0.32, 0.38 |
| TLTSCNTSV | Combinatorial buildup algorithm | Sezerman et al. 1996 | 2.20 | 0.46, 0.58 |
aRMSD of peptide Cα atoms obtained in our work from redocking bound complexes and docking into single template, respectively.
Discussion
Modeling the bound conformation of MHC-binding peptides is a complex problem in the field of immunology. In this work, we present a generic protocol for the modeling of both MHC class I and class II complexes. The proposed procedure forms a basis for the prediction of peptides that will bind to specific MHC alleles and hence vaccine design, based on computational immunological methods. To the best of our knowledge, the current study presents one of the most accurate MHC–peptide flexible docking techniques to date. Our procedure has been assessed against a large dataset of nonredundant MHC–peptide complexes in which 3D information is available. Out of 40 peptides considered in this study (Table 2), we have consistently obtained a Cα RMSD below 1.00 Å for 33 peptides by remodeling peptide-bound MHC structures. The worst structure was generated from the remodeling of the bound peptide ELAGIG ILTV from complex 1JF1 with Cα RMSD of 1.53 Å. The loop formed around residues 5 to 7 was erroneously predicted, and this disorientation is a direct consequence of missing water molecules positioned around the loop in the template, which resulted in incorrect positioning of interacting residues. In the absence of explicit water molecules, the predicted conformation of our peptide is energetically more favorable than the crystal conformation. Nonetheless, our procedure can correctly predict the conformation of residues that extends into the binding cleft and identify essential contacts with the MHC receptor as shown in Figure 3 ▶. Although water molecules and other common biological ions such as phosphate and chloride may mediate MHC–peptide interactions, they were left out in our preliminary experiments to determine the generic prediction capability of our docking protocol using a single template for each allele because the significance and contributions of these molecules varies between different peptides and the respective alleles. It is possible that for some MHC–peptide complexes, appropriate addition of mediating molecules or considerations of solvent effects may lead to an improvement in prediction accuracy.
Figure 3.

Comparison of the predicted and experimental structures of the ELAGIGILTV peptide in the 1JF1 complex (Table 2). The crystal structure (in black) and modeled structure (in gray) are shown in Cα trace representation (A), and stick representation (B) of all heavy atoms.
The performance of our method, in terms of computational time, is highly efficient, and requires approximately 11 min for the complete modeling of one peptide (with the first rigid-body docking step of ~3.5 min, loop closure of ~12 sec and the final refinement step of ~7 min) on a 4-CPU SGI Origin 3200 workstation. Rapid modeling of the bound peptide conformation is possible by restraining the conformational spaces to be sampled in the early phase of our modeling protocol (please refer to the Rigid docking of residues at the ends of binding groove section for details). Large-scale modeling and scanning of potential MHC sequences is possible through automation for all steps. Our docking procedure also proved to be capable of accurate modeling of MHC–peptide complexes in the absence of essential anchor residues by exploiting the highly conserved backbone conformation of bound MHC class I and class II peptide termini. Furthermore, our procedure has the added advantage over other techniques such as Artificial Neural Networks, Support Vector Machines, and Hidden Markov Models in that only a suitable template for a particular allele is required and training of experimental data is unnecessary.
Materials and methods
Selection of the MHC–peptide complexes
We have tested the docking procedure on a nonredundant data set of 40 (29 class I and 11 class II) MHC–peptide complexes at and below 3.00 Å for which 3D structures are available. When more than one complex with the same bound peptide is found, the highest quality structure (with the highest resolution) is selected. When more than one bound peptide is available in the selected crystal structure, all bound peptides in that crystal structure are analyzed. Table 2 details the complexes used in this study. When more than one allele is available as template for docking of peptides into a single template, the highest resolution allele was selected.
The peptide-docking procedure
Our peptide-docking procedure exploits the highly conserved backbone conformation of peptide termini, originally noted for HLA-Aw68 (Guo et al. 1992), and confirmed by the analysis of all available high-quality crystal structures in the Protein Data Bank (PDB) for both MHC class I and class II complexes (Table 1). Structural comparison of all available MHC class I crystal structures to date reveals highly conserved backbone of peptide termini residues with Cα RMSD of 0.02–0.29 Å and 0.00–0.25 Å for the peptide N- and C-terminal ends, respectively. A similar highly conserved backbone conformation is observed at the ends of the core peptide fragments in the binding cleft of MHC class II alleles with the Cα RMSD in the range 0.01–0.22 Å and 0.02–0.27 Å for the two peptide termini, respectively. The structure comparison results are detailed in Table 1.
Our docking procedure (Fig. 1 ▶) for MHC class I peptides and the core residues of MHC class II peptides is performed in three steps: (1) rigid docking of residues at the ends of the binding groove; (2) loop closure of central residues by satisfaction of spatial constraints; (3) followed by iterative ab initio refinements of backbone and ligand interacting side-chain dihedral angles to eliminate or minimize atomic clash regions at the MHC receptor–peptide interface, using a Monte Carlo procedure. The first two steps were used to generate an initial model, which is further refined in the last step to produce the final product.
Rigid docking of residues at the ends of binding groove
The backbone of peptide termini as shown in Table 1 is highly conserved over peptides in different MHC alleles (class I and class II), but due to the allelic variability in the binding grooves, there are subtle differences in the position of these peptide termini. This rigid docking step is adopted to ensure a best fit of the peptide termini before the loop closure procedure in the next step. A fast soft interaction energy function (Fernández-Recio et al. 2002) is utilized to sample different positions and orientations of peptide fragments at the ends of the binding grooves using an Internal Coordinate Mechanics (ICM) global optimization algorithm, with flexible ligand interface side chains and a grid map representation of the receptor energy localized to small cubic regions of side 1.00 Å from the backbone of the ligand.
The ligand side-chain torsions within the grid map were changed in each random step using a Biased Monte Carlo procedure, which begins by pseudorandomly selecting a set of torsion angles in the ligand and subsequently finding the local energy minimum about those angles. New conformations are adopted upon satisfaction of the Metropolis criteria with probability min(1,exp[−ΔG/RT]), where R is the universal gas constant and T is the absolute temperature of the simulation. For the current study, T was set to 300 K. Loose restraints were imposed on the positional variables of the ligand molecule to keep it close to the starting conformation. The optimal energy function adopted for our stimulations consisted of the internal energy of the ligand and the intermolecular energy of the optimized potential maps:
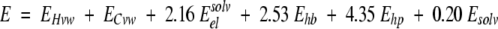 |
The internal energy function incorporates internal van der Waals interactions, hydrogen bonding, and torsion energy (calculated using an extension of the Empirical Conformational Energy Program for Peptides 3 (ECEPP/3; Nemethy et al. 1992; force field parameters), as well as a electrostatic energy with a distance-dependent dielectric constant (e = 4r; Fernández-Recio et al. 2002). The final energy includes the configurational entropy of side chains and the surface-based solvation energy to select the best-refined solutions.
Loop closure of center residues
An initial conformation of the central loop is generated using the program MODELLER (Sali and Blundell 1993) by satisfaction of spatial constraints based on the allowed subspace for backbone dihedrals in accordance with the conformations of peptides docked into the ends of the binding groove. A comprehensive coverage of modeling by satisfaction of spatial restraints is given in the literature (Sali and Blundell 1993). In brief, this is performed in three steps: (1) distance and dihedral angle restraints on the entire peptide sequence are derived from its alignment with the sequences of probes docked into the binding groove; (2) the restraints on spatial features of the unknown center residues are derived by extrapolation from the known 3D structures of probes in the alignment, expressed as probability density functions; and (3) spatial restraints on the unknown center residues are satisfied by optimization of the molecular probability density function using a variable target function technique that applies the conjugate gradients algorithm to positions of all nonhydrogen atoms.
Model refinements
To improve the accuracy of the initial model, partial refinement was performed for the ligand backbone and side chains as well as detected atomic clash regions at the receptor-ligand interface, using the ICM Biased Monte Carlo procedure (Abagyan and Totrov 1999). The initial stages of the refinement attempt to overcome the penalty derived from the preliminary rigid docking of terminal residues by introducing partial flexibility to the ligand backbone. Restraints are imposed upon the positional variables of the Cα atoms of probes to keep it close to the starting conformation. The energy function adopted for this refinement step is:
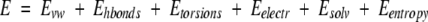 |
Refinements of ligand and receptor side-chain torsions in the vicinity of 4.00 Å from the receptor were performed upon the final backbone structure.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04631204.
References
- Abagyan, R. and Totrov, M. 1999. Ab initio folding of peptides by the optimalbias Monte Carlo minimization procedure. J. Comput. Phys. 151 402–421. [Google Scholar]
- Altuvia, Y., Schueler, O., and Margalit, H. 1995. Ranking potential binding peptides to MHC molecules by a computational threading approach. J. Mol. Biol. 249 244–250. [DOI] [PubMed] [Google Scholar]
- Bhasin, M. and Raghava, G.P.S. 2004. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 13 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.H., Jardetzky, T.S., Gorga, J.C., Stern, L.J., Urban, R.G., Strominger, J.L., and Wiley, D.C. 1993. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364 33–39. [DOI] [PubMed] [Google Scholar]
- Brusic, V., Rudy, G., Honeyman, M., Hammer, J., and Harrison, L. 1998. Prediction of MHC class II-binding peptides using an evolutionary algorithm and artificial neural network. 1998. Bioinformatics 14 121–130. [DOI] [PubMed] [Google Scholar]
- Brusic, V., Petrovsky, N., Zhang, G., and Bajic, V.B. 2002. Prediction of promiscuous peptides that bind HLA class I molecules. Immunol. Cell Biol. 80 280–285. [DOI] [PubMed] [Google Scholar]
- Caflisch, A., Niederer, P., and Anliker, M. 1992. Monte Carlo docking of oligopeptides to proteins. Proteins 13 223–230. [DOI] [PubMed] [Google Scholar]
- Davenport, M.P., Ho Shon, I., and Hill, A.V. 1995. An empirical method for the prediction of T-cell epitopes. Immunogenetics 42 392–397. [DOI] [PubMed] [Google Scholar]
- Desmet, J., Maeyer, M.D., Spriet, J., and Lasters, I. 2000. Flexible docking of peptide ligands to proteins. Methods Mol. Biol. 143 359–376. [DOI] [PubMed] [Google Scholar]
- Dönnes, P. and Elofsson, A. 2002. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics 3 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk, K., Rötzschke, O., Stevanovic, S., Jung, G., and Rammensee, H.G. 1991. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351 290–296. [DOI] [PubMed] [Google Scholar]
- Fernández-Recio, J., Totrov, M., and Abagyan, R. 2002. Soft protein–protein docking in internal coordinates. Protein Sci. 11 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower, D.R. 2003. Databases and data mining for computational vaccinology. Curr. Opin. Drug Discov. Devel. 6 396–400. [PubMed] [Google Scholar]
- Godkin, A.J., Davenport, M.P., Willis, A., Jewell, D.P., and Hill, A.V. 1998. Use of complete eluted peptide sequence data from HLA-DR and -DQ molecules to predict T cell epitopes and the influence of the nonbinding terminal regions of ligands in epitope selection. J. Immunol. 161 850–858. [PubMed] [Google Scholar]
- Govindarajan, K.R., Kangueane, P., Tan, T.W., and Ranganathan, S. 2003. MPID: MHC-peptide interaction database for sequence-structure-function information on peptides binding to MHC molecules. Bioinformatics 19 309–310. [DOI] [PubMed] [Google Scholar]
- Gulukota, K., Sidney, J., Sette, A., and DeLisi, C. 1997. Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J. Mol. Biol. 267 1258–1267. [DOI] [PubMed] [Google Scholar]
- Guo, H.C., Jardetzky, T.S., Garrett, T.P.J., Lane, W.S., Strominger, J.L., and Wiley, D.C. 1992. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature 360 364–366. [DOI] [PubMed] [Google Scholar]
- Klein, J. 1986. Natural history of the major histocompatibility complex. J. Wiley & Sons, New York.
- Lim, S.K., Kim, S., Lee, H.G., Lee, K.Y., Kwon, T.J., and Kim, K. 1996. Selection of peptides that bind to the HLA-A2.1 molecule by molecular modeling. Mol. Immunol. 33 221–230. [DOI] [PubMed] [Google Scholar]
- Mamitsuka, H. 1998. Predicting peptides that bind to MHC molecules using supervised learning of Hidden Markov Models. Proteins 33 460–474. [DOI] [PubMed] [Google Scholar]
- Michielin, O. and Karplus, M. 2002. Binding free energy differences in a TCR–peptide–MHC complex induced by a peptide mutation: A stimulation analysis. J. Mol. Biol. 324 547–569. [DOI] [PubMed] [Google Scholar]
- Michielin, O., Luescher, I., and Karplus, M. 2000. Modeling the TCR–MHC–peptide complex. J. Mol. Biol. 300 1205–1235. [DOI] [PubMed] [Google Scholar]
- Nemethy, G., Gibson, K.D., Palmer, K.A., Yoon, C.N., Paterlini, G., Zagari, A., Rumsey, S., and Scheraga, H.A. 1992. Energy parameters in polypeptides, 10: Improved geometric parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to praline-containing peptides. J. Phys. Chem. 96 6472–6484. [Google Scholar]
- Parker, K.C., Bednarek, M.A., and Coligan, J.E. 1994. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 152 163–175. [PubMed] [Google Scholar]
- Rammensee, H., Bachmann, J., Emmerich, N.P., Bachor, O.A., and Stevanović, S. 1999. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 50 213–219. [DOI] [PubMed] [Google Scholar]
- Rognan, D., Laumoeller, S.L., Holm, A., Buus, S., and Tschinke, V. 1999. Predicting binding affinities of protein ligands from three-dimensional models: Application to peptide binding to class I major histocompatibility proteins. J. Med. Chem. 42 4650–4658. [DOI] [PubMed] [Google Scholar]
- Rosenfeld, R., Zheng, Q., Vajda, S., and DeLisi, C. 1993. Computing the structure of bound peptides: Application to antigen recognition by class I major histocompatibility complex receptors. J. Mol. Biol. 234 515–521. [DOI] [PubMed] [Google Scholar]
- ———. 1995. Flexible docking of peptides to class I major-histocompatibility-complex receptors. Genet. Anal. 12 1–21. [DOI] [PubMed] [Google Scholar]
- Sali, A. and Blundell, T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Sezerman, U., Vajda, S., and DeLisi, C. 1996. Free energy mapping of class I MHC molecules and structural determination of bound peptides. Protein Sci. 5 1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, L.J. and Wiley, D.C. 1994. Antigenic peptide binding by class I and class II histocompatibility proteins. Structure 2 245–251. [DOI] [PubMed] [Google Scholar]
- Stern, L.J., Brown, J.H., Jardetzky, T.S., Gorga, J.C., Urban, R.G., Strominger, J.L., and Wiley, D.C. 1994. Crystal structure of the human class II MHC protein HLA–DR1 complexed with an influenza virus peptide. Nature 368 215–221. [DOI] [PubMed] [Google Scholar]