

Abstract
Dystroglycan (DG) is an adhesion complex, expressed in a wide variety of tissues, formed by an extracellular and a transmembrane subunit, α-DG and β-DG, respectively, interacting noncovalently. Recently, we have shown that the recombinant ectodomain of β-DG, β-DG(654–750), behaves as a natively unfolded protein, as it is able to bind the C-terminal domain of α-DG, while not displaying a defined structural organization. We monitored the effect of a commonly used denaturing agent, the anionic detergent sodium dodecylsulphate (SDS), on β-DG(654–750) using a number of biophysical techniques. Very low concentrations of SDS (≤2 mM) affect both tryptophan fluorescence and circular dichroism of β-DG, and significantly perturb the interaction with the α-DG subunit as shown by solid-phase binding assays and fluorescence titrations in solution. This result confirms, as recently proposed for natively unfolded proteins, that β-DG(654–750) exists in a native state, which is crucial to fulfill its biological function. Two-dimensional NMR analysis shows that SDS does not induce any evident conformational rearrangement within the ectodomain of β-DG. Its first 70 amino acids, which show a lower degree of mobility, interact with the detergent, but this does not change the amount of secondary structure, whereas the highly flexible and mobile C-terminal region of β-DG(654–750) remains largely unaffected, even at a very high SDS concentration (up to 50 mM). Our data indicate that SDS can be used as a useful tool for investigating natively unfolded proteins, and confirm that the β-DG ectodomain is an interesting model system.
Keywords: dystroglycan, SDS, NMR spectroscopy, circular dichroism, fluorescence, natively unfolded protein
Dystroglycan (DG) is an adhesion complex composed of two subunits that interact tightly, but noncovalently. α-DG is extracellular and highly glycosylated, and binds with high affinity a number of basement membrane proteins such as laminins, agrin, perlecan, neurexin, and biglycan (Henry and Campbell 1999; Winder 2001), whereas the transmembrane β-DG interacts with dystrophin, utrophin, and other cytosolic proteins, such as rapsyn, caveolin-3, and Grb2 (Jung et al. 1995; Yang et al. 1995; Cartaud et al. 1998).
DG plays a central role within the dystrophin–glycoprotein complex (DGC), formed also by sarcoglycans, dystrobrevins, syntrophins, and sarcospan. The DGC represents, together with integrins, a major molecular bridge connecting the cytoskeleton to the surrounding extracellular matrix in skeletal muscle, as well as in a wide variety of tissues (Ervasti and Campbell 1993; Winder 2001), including the central and peripheral nervous system and epithelia (Durbeej et al. 1998). The integrity of the DG complex is highly compromised in those muscular dystrophies in which the absence of both DG subunits, or even of only α-DG, is frequently observed (Durbeej and Campbell 2002), whereas the DG gene disruption is lethal during mouse early embryogenesis (Williamson et al. 1997). DG acts also as a receptor for some infective agents (Cao et al. 1998), and it is seriously altered during tumorigenesis (Losasso et al. 2000).
Given the multifaceted biological and biomedical role of DG, the full elucidation of the molecular mechanism underlying the interaction between the two DG subunits should be considered of primary importance. We already demonstrated that the ectodomain of β-DG binds the C-terminal domain of α-DG independently from glycosylation, and that the binding epitope is located between amino acids 550 and 567 of α-DG (Bozzi et al. 2001a; Sciandra et al. 2001). Moreover, spectroscopic studies on the recombinant fragment spanning the extracellular domain of β-DG, β-DG(654–750), have previously shown that β-DG can be classified as a natively unfolded protein, as it is still able to bind both native and recombinant α-DG, despite the fact that its conformation seems to be essentially disordered (Boffi et al. 2001). NMR analysis has also revealed that the β-DG(654–750) polypeptide contains an extended N-terminal region with a restricted mobility and a C terminus of about 30 residues characterized by a high flexibility (Bozzi et al. 2003).
Paradigmatic experiments have shown that it is possible to energetically favor completely alternative secondary and tertiary structures within the same polymer (Cregut et al. 1999). Proteins generally show a large conformational versatility, and many different chemical (for example ionic compounds and alcohols) or physical agents are able to influence dramatically the stability and folding of polypeptides (Minor and Kim 1996; Fändrich et al. 2001; Torrent et al. 2003), especially those belonging to the natively unfolded family (Uversky et al. 2001). For this reason, we have decided to use sodium dodecylsulphate (SDS) to investigate, for the first time, the effects of an amphipatic molecule on the conformation of a natively disordered polypetide such as β-DG(654–750).
Results
Figure 1 ▶ shows the alignment obtained using ClustalW (Higgins et al. 1994) of the β-DG ectodomain sequences from different species as follows: Homo sapiens (NP_004384), Mus musculus (Q62165), Danio rerio (NP_775381), and Xenopus laevis (AAH46260). We have recombinantly expressed a peptide corresponding to the entire murine sequence (654–750), which also contains at its N terminus, two foreign residues (Gly–Ser) introduced for cloning purposes (Sciandra et al. 2001). The binding epitope for α-DG is comprised between amino acids 691 and 719 (Fig. 1 ▶, in bold; Bozzi et al. 2003). It should be noted that the N-terminal part of the alignment shows a higher degree of conservation (654–719, ≈71% of homology) than the C-terminal one (720–750, ≈55% of homology). This is relevant when considering the outcome of our NMR analysis (Bozzi et al. 2003; see below).
Figure 1.

Multiple alignment of the primary sequences corresponding to the ectodomain of β-DG from different species. Positions refer to the human DG sequence. The presence of an identical residue in all of the sequences analyzed is indicated by an asterisk (*); of conserved substitutions, by a colon (:); of semiconserved substitutions, by a period (.). The bold sequence corresponds to the binding epitope for α-DG (Bozzi et al. 2003).
CD experiments
Circular dichroism (CD) experiments were carried out in order to study the effect of the detergent SDS on the β-DG(654–750) conformation. Figure 2A ▶ shows that in the absence of detergent, the spectrum of β-DG(654–750) is typical of an unfolded polypeptide chain, with a minimum in the vicinity of 200 nm and the absence of any characteristic bands between 210 and 230 nm. The addition of SDS induces a shoulder at 222 nm and a decrease in the minimum of the ellipticity at 200 nm. This progressive change in signal takes place at increasing concentration of SDS between 0 and 1.5 mM. A further increase in SDS concentration above 1.5 mM has no effect on the CD spectrum of β-DG(654–750) (Fig. 2B ▶). The spectral changes induced by SDS may suggest that β-DG(654–750) undergoes a slight conformational rearrangement involving an eventual increase of canonical secondary structures, even if the random coil conformation is prominent, as shown by the more negative values of the ellipticity at 200 nm (Fig. 2B ▶).
Figure 2.
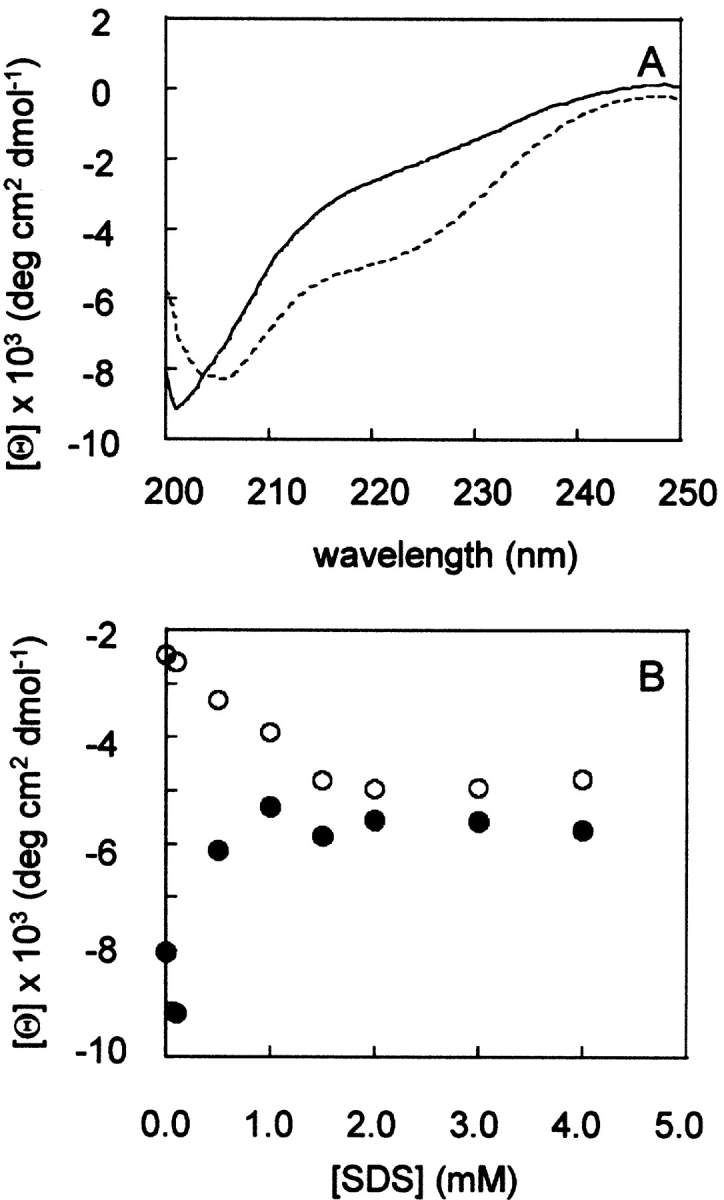
(A) CD spectra of 3 μM β-DG(654–750) in the absence (continuous line) and in the presence (dotted line) of 1.5 mM SDS. (B) Plot of the mean molar ellipticity measured at 222 nm (open circles) and at 200 nm (solid circles) vs. SDS concentration.
Fluorescence spectra
The titration of β-DG(654–750) with SDS has been recorded by fluorescence emission spectroscopy. In the absence of SDS, the emission spectrum of β-DG(654–750) has a maximum at 350 nm (Fig. 3A ▶), indicating that its single Trp 659 is solvent exposed, as expected for a disordered protein. Whereas SDS concentration changes within the low micromolar range (up to 50 μM) leave the fluorescence signal substantially unaffected, its increase above 100 μM causes a shift of the emission maximum toward lower wavelengths (336 nm, Fig. 3C ▶), indicating the presence of a more hydrophobic environment around Trp 659. The concomitant progressive fluorescence intensity decrease of about 45% (Fig. 3B ▶) cannot be easily explained; to do so, further structural data needed to be collected and analyzed (see below). A further increase of SDS concentration (up to 2 mM) has no additional effect on the β-DG(654–750) spectrum.
Figure 3.
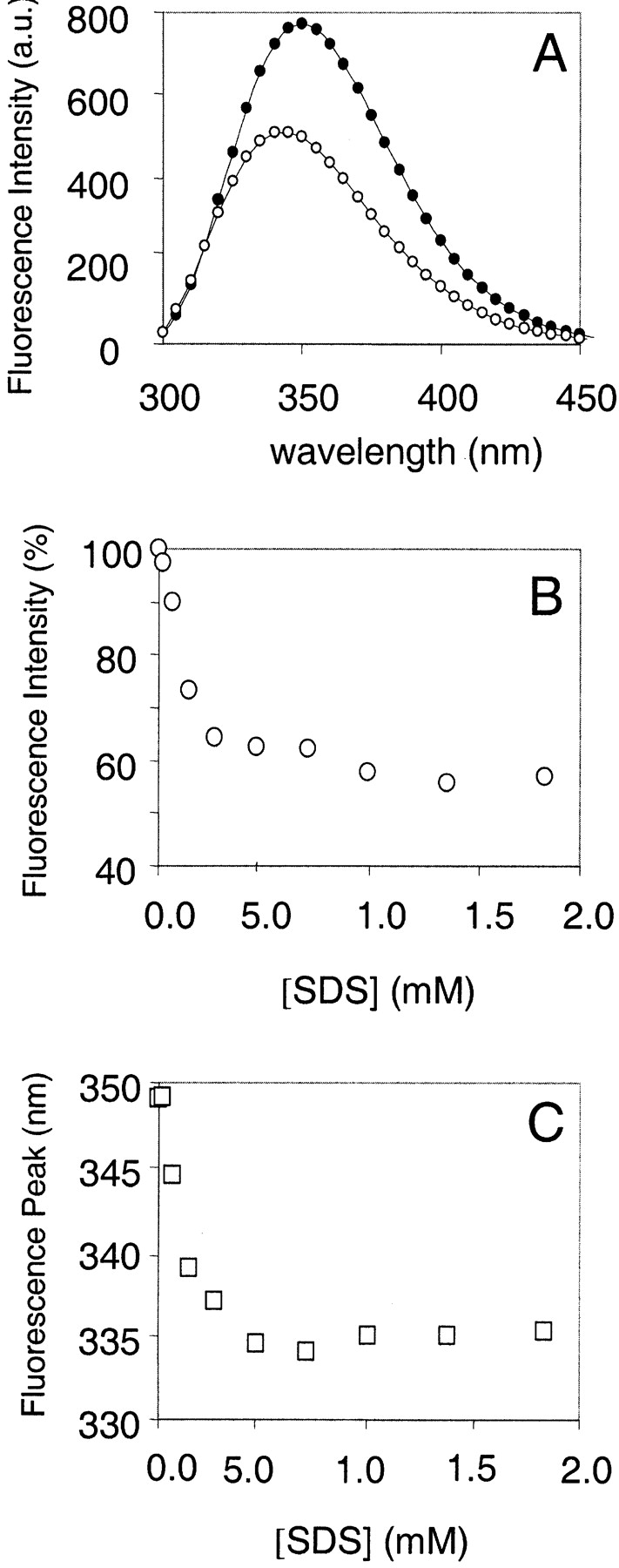
Titration of β-DG(654–750) with SDS, monitored by fluorescence spectroscopy. (A) Fluorescence emission spectra of β-DG(654–750) in the absence (solid circles) and in the presence of 1 mM SDS (open circles). (B) Intensity of the fluorescence signal of β-DG(654–750) as a function of SDS concentration. (C) Wavelength of the emission maximum peak of β-DG(654–750) as a function of SDS concentration.
NMR experiments
With the aim of obtaining a more detailed structural picture of the effects of SDS on the β-DG(654–750) conformation, a series of two-dimensional HSQC spectra of 15N β-DG(654–750), in the presence of increasing amounts of SDS, were recorded. Figure 4A ▶ reports the HSQC spectrum at 400 MHz of 15N β-DG(654–750). The complete assignment of the spectrum was obtained following the data deposited in the BioMagResBank (http://www.bmrb.wisc.edu) under accession number 5743 (Bozzi et al. 2003).
Figure 4.
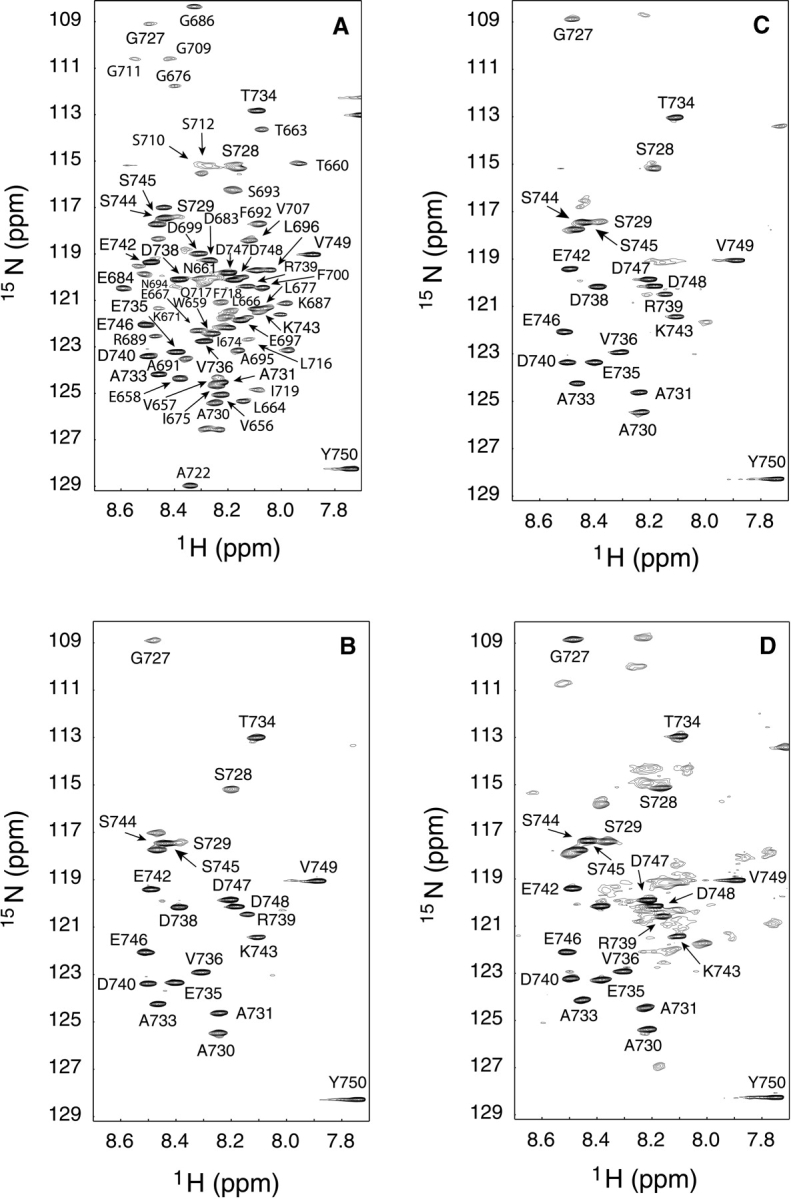
HSQC spectra (400 MHz) of 300 μM 15N β-DG(654–750) with increasing amounts of SDS: (A) 0 mM; (B) 1 mM; (C) 20 mM; (D) 50 mM.
Adding small amounts of SDS causes a general broadening of the cross-peaks related to the first 70 amino acids, until their complete disappearance at 1 mM SDS, whereas the residues belonging to the highly flexible C-terminal region of β-DG(654–750) maintain their narrow resonance cross-peaks (Fig. 4B ▶). When the SDS concentrations are within 5 and 15 mM, all cross-peaks disappear (data not shown), but by increasing the SDS concentration up to 20 mM, the presence of some new cross-peaks becomes apparent (Fig. 4C ▶). This process reaches a plateau at around 50 mM SDS, where most of the expected cross-peaks are observable, even if their line shape is very broad (Fig. 4D ▶), and the addition of SDS above 50 mM has no further effect on the 15N β-DG(654–750) spectrum (data not shown). Figure 4 ▶, B, C, and D show that SDS does not induce any spread of 15N β-DG(654–750) cross-peaks, suggesting that the protein maintains its disordered conformation. Although it is impossible to discriminate which kind of interactions the surfactant establishes with β-DG(654–750), it is noteworthy that SDS preferentially interacts with its first 70 residues. In fact, the cross-peaks corresponding to the C-terminal portion of β-DG(654–750), comprised between Gly 727 and Tyr 750 present the same chemical shift and line shape in the presence and in the absence of SDS, indicating that even high concentrations of detergent do not affect this region.
Solid-phase binding assays
To verify whether the interaction between β-DG(654–750) and SDS molecules affects the binding of β-DG(654–750) with α-DG, solid-phase binding experiments were carried out with increasing amounts of SDS. The biotinylated β-DG(654–750) was used as soluble ligand at increasing concentrations, whereas the recombinant fragment corresponding to the C-terminal region of α-DG, used as a fusion protein with thioredoxin, Trx-α-DG(485–600), was immobilized on microtiter plates. Trx alone was used as negative control, also in the absence of SDS. At a concentration as low as 50 μM, SDS already causes a substantial reduction of the affinity between β-DG(654–750) and Trx-α-DG(485–600) (Fig. 5 ▶; Table 1), as shown by an increase of the apparent equilibrium dissociation constant (Kd) from a value of 8.2 to 17.8 μM. A further increase in the SDS concentration (up to 100 μM) almost completely inhibits the interaction between Trx-α-DG(485–600) and β-DG(654–750) (Fig. 5 ▶; Table 1).
Figure 5.

Solid-phase biotinylated-ligand binding assay. Biotinylated β-DG(654–750) binding to Trx-α-DG(485–600) with increasing amounts of SDS: (open circles) 0 mM; (solid circles) 0.05 mM; (squares) 0.1 mM. Data refer to a representative experiment.
Table 1.
Equilibrium dissociation constants, Kd, measured at increasing SDS concentrations
| SDS (μM) | Kd (μM) | Kdapp* (μM) |
| 0 | 3.1 ± 0.3 | 8.2 |
| 50 | 6.3 ± 0.6 | 17.8 |
| 100 | 16.2 ± 1.6 | n.d. |
| 250 | n.d. | |
| 500 | n.d. | |
| 1000 | n.d. |
Equilibrium dissociation constants, Kd, obtained by fluorescence titrations of β-DG(654–750) with a synthetic peptide α-DG(549–567) (central column, standard errors are reported) and by solid-phase binding assays of biotinylated β-DG(654–750) with Trx-α-DG(485–600) (right column), carried out in the presence of increasing SDS concentrations. (*) Apparent equilibrium dissociation constants. Kd values represent an average of three independent experiments. (n.d.) Not determined.
Fluorescence titrations with the α-DG synthetic peptide
A further evidence of the reduced affinity of β-DG(654–750) for α-DG in the presence of SDS was obtained by a series of titrations of β-DG(654–750) with a synthetic peptide containing the binding epitope of α-DG, α-DG(549–567). Both β-DG(654–750) and α-DG(549–567) contain a single Trp residue, but the overall fluorescence intensity of β-DG(654–750) is about eight times higher than that of α-DG(549–567) at the same concentration. Therefore, the contribution of α-DG(549–567) to total fluorescence emission is very low, especially in the first part of the titration, where β-DG(654–750) is present in a stoichiometric excess with respect to α-DG(549–567). In fact, results obtained with and without correction for α-DG(549–567) are very similar. Moreover, it should be outlined that the emission spectrum of α-DG(549–567) alone is not influenced by the presence of SDS (data not shown). Each titration was carried out at a different SDS concentration and followed by monitoring the quenching of the intrinsic tryptophan fluorescence emission of β-DG(654–750) as a function of α-DG(549–567) concentration. Table 1 shows the increase of the equilibrium dissociation constant values with the increase of SDS concentrations. Above 250 μM SDS, no binding is detectable.
Discussion
In previous studies, we have classified β-DG(654–750) as a natively unfolded protein, as its polypeptide chain, although essentially disordered, is still able to bind its target protein, α-DG, in vitro (Di Stasio et al. 1999; Boffi et al. 2001; Sciandra et al. 2001). NMR analysis of the 15N-labeled domain shows that no major structural rearrangement occurs within the β-DG ectodomain induced by its interaction with α-DG, confirming that a natively unfolded macromolecule such as β-DG(654–750) needs a very high degree of conformational freedom, which is not compatible with a classical structural arrangement, to fulfill its biological role properly (Bozzi et al. 2003). However, using the aforementioned approach, we succeeded in narrowing down a continuous binding epitope for α-DG, located between β-DG positions 691–719 (Bozzi et al. 2003), which is very likely to undergo some local structural reorganization upon recognition and/or binding, and which need to be further investigated.
Within cells and tissues, the β-DG ectodomain is juxtaposed to the plasma membrane and, therefore, we decided to use an ionic detergent such as SDS, as it could somehow mimic the chemical properties of the plasma membrane environment in solution (Jones 1996). SDS has, in fact, been used successfully to obtain 1H-15N HSQC spectra with good peak dispersion and linewidth for two membrane proteins, namely, the subunit c from Bacillus pseudofirmus OF4 and the small multidrug resistance protein (Smr) from Staphylococcus aureus (Krueger-Koplin et al. 2004). Furthermore, it was interesting to analyze its potential effect on a natively unfolded polypeptide per se, considering how the biological role of this kind of protein is increasingly gaining importance (Wright and Dyson 1999; Dunker et al. 2002; Uversky 2002). The effect of SDS has been already analyzed on other proteins and enzymes (Bozzi et al. 2001b) and recently it was used, as other conventional denaturing agents, to also monitor the refolding pathway of globular proteins such as native cytochrome c (Chattopadhyay and Mazumdar 2003).
Both fluorescence and circular dichroism spectroscopy show that in a range of concentrations comprised between 0 and 2 mM, SDS is able to perturb the β-DG(654–750) conformation. The fluorescence emission spectrum of β-DG(654–750), obtained upon excitation at 280 nm, is mainly due to its unique Trp (in position 659; see Fig. 1 ▶). The emission maximum at 349 nm is typical of a solvent-exposed Trp, which, accordingly, does not change, even in the presence of increasing amounts of traditional denaturing agents such as Gdn/HCl (Di Stasio et al. 2004). The addition of SDS to β-DG(654–750) (although producing a quenching of its overall fluorescence intensity; see below) induces a shift of its emission maximum to lower wavelengths (Fig. 3 ▶), which suggests a progressive burying of the Trp indole ring. Interestingly, within the same SDS concentration range, the affinity of β-DG(654–750) for α-DG is strongly reduced, as shown both by solid-phase binding assays and by fluorescence titration experiments in solution (Fig. 5 ▶; Table 1). It is noteworthy that at ≈50 μM SDS concentration, we already observed a significant effect on the affinity constant. Therefore, SDS molecules at very low concentrations (submicellar) are already able to interact with β-DG(654–750) and significantly perturb its function. It is also remarkable that our fluorescence titrations experiments show a quenching of the overall fluorescence signal, which mainly originates from the β-DG ectodomain (see Results) upon binding of the α-DG(549–667) synthetic peptide, even in the absence of SDS. Together with the results of our previous NMR titration experiment (labeled β-DG vs. α-DG; see Bozzi et al. 2003), this would confirm the occurrence of some local structural rearrangements, influencing the Trp 659 fluorescence within the β-DG polypeptide upon α-DG binding. Interestingly, a reorganization process upon binding, eventually leading to an overall folded conformation, has been recently demonstrated for a natively unfolded domain belonging to bacterial colicin (Anderluh et al. 2004).
The changes of the β-DG(654–750) near-UV dichroic spectrum, induced by SDS in a range of concentrations comprised between 0 and 4 mM, indicate an increase of the residual secondary structure, as evidenced by the increase of the ellipticity at 222 nm and the parallel decrease of the signal at 200 nm. Together with the fluorescence behavior, this evidence at a first glance might be interpreted as a refolding process. Further details on the effects of SDS on the β-DG(654–750) chain are provided by NMR spectra carried out on 15N β-DG(654–750) as a function of increasing amounts of SDS. These experiments, together with the evidence of a decrease in the fluorescence intensity signal upon SDS titration, indicate that what we monitor cannot be considered as an ordered refolding of the β-DG ectodomain (see Fig. 3A, B ▶). On the contrary, an unspecific hydrophobic collapse leading to a disordered assembly of molecules and to the progressive burying of the Trp 659 moieties, with a reciprocal quenching of their fluorescence intensities, is very likely to take place.
A progressive broadening of the cross-peaks corresponding to the first 70 amino acids of β-DG(654–750), until their complete disappearance at 1 mM SDS can be observed. On the contrary, the cross-peaks related to the C-terminal region remain completely unaffected by the detergent (Fig. 4B ▶). It is worth mentioning that the 70 residues that are affected by SDS represent the same region that shows a restricted mobility with respect to the C terminus, which contains the putative-binding epitope (positions 691–719) for α-DG (Bozzi et al. 2003), and which is also more conserved in the various species analyzed (see Fig. 1 ▶). The different effect of SDS on the two β-DG(654–750) regions of different mobility sheds light on some structural characteristics concerning the β-DG ectodomain. The C-terminal segment, composed of about 30 residues, is unaffected by SDS, even at very high over-micellar concentrations (50 mM, see Fig. 4D ▶); therefore, it may be considered as a highly flexible Pro-rich-domain (see Fig. 1 ▶) displaying a random conformation. This flexibility would make the C terminus of β-DG(654–750) an ideal linker between the transmembrane sequence of β-DG and the N-terminal region of the ectodomain. On the contrary, the less mobile N-terminal domain (positions 659–722, Fig. 1 ▶), although lacking an overall ordered structure, is likely to exist in a native conformation, showing some residual secondary structures (Boffi et al. 2001) that can be perturbed by SDS. Accordingly, the disruption of such a native conformation makes the protein unable to fulfill its biological function (Fig. 5 ▶), that is, the interaction with the α-DG subunit or with other plasmalemmal receptor molecules such as sarcoglycans (Yoshida et al. 1998).
The sensitivity of β-DG(654–750) to SDS supports the hypothesis of other authors (Gunasekaran et al. 2003), who suggest that all polypeptides (disordered or stable) exist as an ensemble of conformers influenced by the environmental conditions of the solution (pH, ionic strength, temperature, dielectric constant). Their ligands bind only native conformers, stabilizing them and, therefore, shifting the equilibrium between native and non-native conformers (Anderluh et al. 2004). Every experimental condition, such as the presence of SDS in our case, driving the equilibrium toward non-native conformers, would then make the protein unable to interact with its biological partners (Gunasekaran et al. 2003; Baskakov et al. 2004). However, aside from the change induced by SDS in the solution properties, the effect that we have detected on the β-DG ectodomain can represent an interaction of the ionic detergent molecules with some protein loops directly and/or indirectly involved in the binding of the α-DG subunit.
The fact that SDS, either at low or at higher concentrations, does not apparently induce any ordered structural rearrangement of β-DG(654–750), should be particularly emphasized, as it is a further proof of its extreme conformational freedom. To our knowledge, this is the first study carried out on the effect of SDS on a natively unfolded protein domain. We believe that it will be interesting to also analyze other members of this protein family in order to test whether the behavior that we have found could be considered as a common feature. Another possible development can arise from the use of different detergents, which have been shown to provide an excellent model of the biological membrane environment (White et al. 2001).
Concluding remarks
Our results shed light on two important structural-functional aspects of DG. First, within the SDS concentration range in which the surfactant inhibits the binding between the DG subunits, the C terminus of the β-DG ectodomain (≈720–750) remains largely unaffected, reinforcing the notion that it is not involved in the interaction with α-DG (Bozzi et al. 2003).
Another important aspect deals with the structure of the β-DG ectodomain. Namely, the absence of a defined three-dimensional structure does not prevent the β-DG ectodomain to exist in a native ensemble of conformations, which, accordingly, can be perturbed by SDS. On the other hand, it is likely that its natively unfolded conformation would particularly favor modulation processes at the α/β interface, as also suggested by the relatively low affinity displayed for α-DG. Such a feature would make DG a good candidate to ferry signals within cells (Spence et al. 2004).
Materials and methods
Purification of DG recombinant fragments
All of the DNA manipulations required for the production of the recombinant proteins have been already described elsewhere (Sciandra et al. 2001). Murine DG recombinant fragments were expressed in Escherichia coli BL21(DE3) as thioredoxin (Trx) fusion proteins that also contain an N-terminal His6-tag and a thrombin cleavage site (Kammerer et al. 1998). Bacterial cells were cultured at 37°C in 1.2 L of normal LB medium until an OD of 0.8 at 600 nm; cells were induced with IPTG and harvested after 3 h. Cellular pellets were resuspended in a lysis buffer containing 5 mM imidazole, 0.5 M NaCl, 20 mM Tris-HCl and 1 mM PMSF (pH 7.9). Cell lysis was achieved by sonication. The fusion proteins were purified using nickel nitrilotriacetate affinity chromatography. Trx-α-DG(485–600) was used as it is, whereas β-DG(654–750), starting with foreign residues Gly–Ser, was obtained upon thrombin cleavage. The yield of the recombinant proteins was ~5 mg/L of bacterial culture.
The synthetic peptide α-DG(549–567) was purchased from Synt:em (France) and used without further purification.
Preparation of recombinant-labeled 15 Nβ-DG(654–750)
The labeled recombinant fragment 15N β-DG(654–750) was expressed in E. coli BL21(DE3) as thioredoxin fusion protein, which also contains an N-terminal His6-tag and a thrombin cleavage site. Bacterial cells were cultured at 37°C in 1.2 L of normal LB medium, until an OD of 0.5–0.6 at 600 nm. Cellular pellet was collected and resuspended in 400 mL of M9 1× (containing 48 mM Na2HPO4•7H2O, 22 mM KH2PO4, 8.5 mM NaCl, 18 mM 15NH4Cl) for two times. Cells were then resuspended in 300 mL of minimal medium containing M9 1×, 2 mM MgSO4, 0.1 mM CaCl2, 0.4% (w/v) D-glucose, and an appropriate amount of BME vitamins 100× solution purchased by ICN Biomedicals. After 1 h of growth at 37°C, cells were induced with IPTG and harvested after 3 h (Marley et al. 2001). Cell lysis and protein purification were carried out as already described. The yield of the recombinant labeled protein was ~1 mg/L of initial bacterial culture.
Circular dichroism experiments
Circular dichroism (CD) spectra were measured using a Jasco J-715 spectropolarimeter (Jasco). Spectra were recorded with the following setup: bandwidth, 2 nm; time constant, 2 sec; scan rate, 20 nm/min; temperature, 25°C. Samples were dyalized in 20 mM NaH2PO4, 150 mM NaCl (pH 6.5) and placed in a 0.1-cm quartz cell. The spectra were the average of multiple accumulations and were noise reduced using standard procedures. The CD signal was expressed as mean molar ellipticity according to the equation:
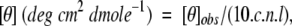 |
where [θ]obs is the measured ellipticity (mdeg), c is the protein concentration (M), n is the number of amino acid residues, and l is the path length of the cell (cm).
Fluorescence experiments
Fluorescence emission spectra were collected between 300 and 450 nm using an excitation wavelength of 260, 280, and 295 nm in a 1-cm quartz cell, using a Spex (Edison) FluoroMax spectrofluorimeter. Titrations of β-DG(654–750) with SDS were carried out in 20 mM NaH2PO4, 150 mM NaCl (pH 6.5) at 25°C and with increasing amounts of SDS ranging from 0 to 1 mM, as described elsewhere (Di Stasio et al. 2004). At the protein concentration used in our experiments (micromolar range), the best signal-to-noise ratio and spectra resolution were obtained using the 280-nm excitation wavelength. The fluorescence signal was corrected for buffer contribution and for protein dilution. To compensate for the decrease in fluorescence due to the progressive exposure of the sample to a high-intensity light beam, all measurements were corrected with a control experiment in which β-DG(654–750) was titrated with the buffer alone (Di Stasio et al. 2004).
The binding between β-DG(654–750) and the synthetic peptide, α-DG(549–567), in the presence of SDS, was studied monitoring the quenching of the intrinsic tryptophan fluorescence of β-DG(654–750) as a function of α-DG(549–567) concentration. The titrations were carried out using 1, 2, and 3 μM β-DG(654–750) with increasing amounts of α-DG(549–567) (from 0 to 8 μM) at different SDS concentrations ranging between 0 and 1 mM. Data were analyzed according to models proposed by others (Lohman and Bujalowski 1991; Eftink 1997) for single-site tight binding systems.
NMR experiments
15N β-DG(654–750) was dialyzed in a buffer solution containing 20 mM NaH2PO4 and 150 mM NaCl (pH 6.5), and concentrated by filtration to ≈300 μM; 5% D2O was added. We used 150 mM NaCl to increase the protein solubility, and it showed no influence on the chemical shifts of 15N β-DG(654–750) (data not shown).
The NMR data were recorded on a Bruker Avance 400 spectrometer equipped with pulsed-field gradient triple-resonance probes. The temperature was held constant at 298 K. Chemical shift for 1H is referred to DSS, whereas NH4Cl was used as standard for 15N calibration.
Two-dimensional HSQC spectra were recorded using a water flip-back version (Grzesiek and Bax 1993) as 100 (15N) × 1024 (1H) complex points data set, with eight scans for each fid and with acquisition times of 64 msec (15N) and 163 msec (1H). Data were processed using NMRPipe (Delaglio et al. 1995) and analyzed with the NMR View 5.0 software package (Johnson and Blevins 1994).
Solid-phase binding assays
Recombinant β-DG(654–750) was biotinylated in 5 mM sodium phosphate buffer (pH 7.4), with 0.5 mg/mL of sulfosuccidinimidobiotin (S-NHS-biotin) from Pierce. The reaction mixture was kept for 30 min in the dark in ice and dialysed overnight against 10 mM Tris/HCl buffer with 150 mM NaCl (pH 7.4; TBS). The optimal dilution for signal detection was determined by dot blot analysis. To assess the binding properties of recombinant β-DG(654–750) to a fusion protein formed by thioredoxin and the C-terminal region of α-DG, Trx-α-DG(485–600) with increasing amounts of SDS, solid-phase assays were performed as follows: 0.1–0.5 μg/well of Trx-α-DG(485–600) was immobilized on microtiter plates by overnight incubation at 4°C in coating buffer (50 mM NaHCO3 at pH 9.6). The plates were washed three times with TBS, 0.05% Tween-20 (T-TBS), containing 1.25 mM CaCl2 and 1 mM MgCl2 (Wash buffer), and then incubated for 1 h in T-TBS containing 3% milk powder for blocking unspecific binding sites. After extensive washing with Wash buffer, wells were incubated with biotinylated β-DG(654–750) in T-TBS, 1% BSA, 1% milk powder, 1.25 mM CaCl2, and 1 mM MgCl2 for 3 h at room temperature in the presence of SDS concentration ranging from 0 to 250 μM. The biotinylated β-DG(654–750) bound fraction was detected with the VECTASTAIN AB Complex from Vector Laboratories. A total of 5 mg of p-Nitrophenyl phosphate in 10 mL of 10 mM diethanolamine and 0.5 mM MgCl2 was used as substrate for the alkaline phosphatase, and quantitative measurements were carried out at 405 nm. The absorbance values were corrected for the signals obtained incubating biotinylated β-DG(654–750) only with BSA (≈0.5 μg/well). Data were fitted using a single class of binding sites, and the equation
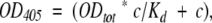 |
where OD405 represents absorbance, Kd is the binding dissociation constant, c is the concentration of biotinylated β-DG(654–750), and ODtot, absorbance at saturation.
Acknowledgments
We thank Fabio Bertocchi and Lorenzo Ferri for their valuable technical assistance and Maria Giulia Bigotti (Bristol) for helpful discussions and critical reading of the manuscript. The financial support of CNR, target project “Biotechnology” and Telethon-Italy (grant no. GGP030332) is gratefully acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
DG, dystroglycan
α-DG, α-dystroglycan
β-DG, β-dystroglycan
Trx, thioredoxin
NMR, nuclear magnetic resonance
HSQC, heteronuclear single-quantum coherence
CD, circular dichroism
SDS, sodium dodecylsulphate
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04762504.
References
- Anderluh, G., Gokce, I., and Lakey, J.H. 2004. A natively unfolded toxin domain uses its receptor as a folding template. J. Biol. Chem. 279 22002–22009. [DOI] [PubMed] [Google Scholar]
- Baskakov, I.V., Legname, B., Gryczynski, Z., and Prusiner, S.B. 2004. The peculiar nature of unfolding of the human prion protein. Protein Sci. 13 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffi, A., Bozzi, M., Sciandra, F., Woellner, C., Bigotti, M.G., Ilari, A., and Brancaccio, A. 2001. Plasticity of secondary structure in the N-terminal region of β-dystroglycan. Biochim. Biophys. Acta 1546 114–121. [DOI] [PubMed] [Google Scholar]
- Bozzi, M., Veglia, G., Paci, M., Sciandra, F., Giardina, B., and Brancaccio, A. 2001a. A synthetic peptide corresponding to the 550–585 region of α-dystroglycan binds β-dystroglycan as revealed by NMR spectroscopy. FEBS Lett. 499 210–214. [DOI] [PubMed] [Google Scholar]
- Bozzi, M., Battistoni, A., Sette, M., Melino, S., Rotilio, G., and Paci, M. 2001b. Unfolding and inactivation of monomeric superoxide dismutase from E. coli by SDS. Int. J. Biol. Macromol. 29 99–105. [DOI] [PubMed] [Google Scholar]
- Bozzi, M., Bianchi, M., Sciandra, F., Paci, M., Giardina, B., Brancaccio, A., and Cicero, O.D. 2003. Structural characterization by NMR of the natively unfolded extracellular domain of β-dystroglycan: Toward the identification of the binding epitope for α-dystroglycan. Biochemistry 42 13717–13723. [DOI] [PubMed] [Google Scholar]
- Cao, W., Henry, M.D., Borrow, P., Yamada, H., Elder, J.H., Ravkov, E.V., Nichol, S.T., Compans, R.W., Campbell, K.P., and Oldstone, M.B. 1998. Identification of α-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 282 2079–2081. [DOI] [PubMed] [Google Scholar]
- Cartaud, A., Coutant, S., Petrucci, T.C., and Cartaud, J.J. 1998. Evidence for in situ and in vitro association between β-dystroglycan and the subsynaptic 43K rapsyn protein. Consequence for acetylcholine receptor clustering at the synapse. J. Biol. Chem. 273 11321–11326. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, K. and Mazumdar, S. 2003. Stabilization of partially folded states of cytochrome C in aqueous surfactant: Effects of ionic and hydrophobic interactions. Biochemistry 42 14606–14613. [DOI] [PubMed] [Google Scholar]
- Cregut, D., Civera, C., Macias, M.J., Wallon, G., and Serrano, L. 1999. A tale of two secondary structure elements: When a β-hairpin becomes an α-helix. J. Mol. Biol. 292 389–401. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Di Stasio, E., Sciandra, F., Maras, B., Di Tommaso, F., Petrucci, T.C., Giardina, B., and Brancaccio, A. 1999. Structural and functional analysis of the N-terminal extracellular region of β-dystroglycan. Biochem. Biophys. Res. Commun. 266 274–277. [DOI] [PubMed] [Google Scholar]
- Di Stasio, E., Bizzarri, P., Misiti, F., Pavoni, E., and Brancaccio, A. 2004. A fast and accurate procedure to collect and analyze unfolding fluorescence signal: The case of dystroglycan domains. Biophys. Chem. 107 197–211. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., Lawson, J.D., Iakoucheva, L.M., and Obradovic, Z. 2002. Intrinsic disorder and protein function. Biochemistry 41 6573–6582. [DOI] [PubMed] [Google Scholar]
- Durbeej, M. and Campbell, K.P. 2002. Muscular dystrophies involving the dystrophin-glycoprotein complex: An overview of current mouse model. Curr. Opin. Genet. Dev. 12 349–361. [DOI] [PubMed] [Google Scholar]
- Durbeej, M., Henry, M.D., Ferletta, M., Campbell, K.P., and Ekblom, P. 1998. Distribution of dystroglycan in normal adult mouse tissues. J. Histochem. Cytochem. 46 449–457. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. 1997. Fluorescence methods for studying equilibrium macromolecule-ligand interaction. Methods Enzymol. 278 221–257. [DOI] [PubMed] [Google Scholar]
- Ervasti, J.M. and Campbell, K.P. 1993. A role for the dystrophinglycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fändrich, M., Fletcher, M.A., and Dobson, C.M. 2001. Amyloid fibrils from muscle myoglobin. Nature 410 165–166. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1993. The importance of not saturating water in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 115 12593–12594. [Google Scholar]
- Gunasekaran, K., Tsai, C.-J., Kumar, S., Zanuy, S., and Nussinov, R. 2003. Extended disordered proteins: Targeting function with less scaffold. Trends Biochem. Sci. 28 81–85. [DOI] [PubMed] [Google Scholar]
- Henry, M.D. and Campbell, K.P. 1999. Dystroglycan inside and out. Curr. Opin. Cell Biol. 11 602–607. [DOI] [PubMed] [Google Scholar]
- Higgins, D., Thompson, J., Gibson, T., Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Jones, M.N. 1996. Protein-surfactant interactions. In Surface activity of the proteins (ed. Shlomomagdassi), pp. 243–284. Dekker, New York.
- Jung, D., Yang, B., Meyer, J., Chamberlain, J.S., and Campbell, K.P. 1995. Identification and characterization of the dystrophin anchoring site on β-dystroglycan. J. Biol. Chem. 270 27305–27310. [DOI] [PubMed] [Google Scholar]
- Kammerer, R.A., Schulthess, T., Landwehr, R., Lustig, A., Fischer, D., and Engel, J. 1998. Tenascin-C hexabrachion assembly is a sequential two-step process initiated by coiled-coil α-helices. J. Biol. Chem. 273 10602–10608. [DOI] [PubMed] [Google Scholar]
- Krueger-Koplin, R.D., Sorgen, P.L., Krueger-Koplin, S.T., Rivera-Torres, I.O., Cahill, S.M., Hicks, D.B., Grinius, L., Krulwich, T.A., and Girvin, M.E. 2004. An evaluation of detergents for NMR structural studies of membrane proteins. J. Biomol. NMR 28 43–57. [DOI] [PubMed] [Google Scholar]
- Lohman, T.M. and Bujalowski, W. 1991. Thermodynamic methods for model-independent determination of equilibrium binding isotherms for protein-DNA interactions: Spectroscopic approaches to monitor binding. Methods Enzymol. 208 258–290. [DOI] [PubMed] [Google Scholar]
- Losasso, C., Di Tommaso, F., Sgambato, A., Ardito, R., Cittadini, A., Giardina, B., Petrucci, T.C., and Brancaccio, A. 2000. Anomalous dystroglycan in carcinoma cell lines. FEBS Lett. 484 194–198. [DOI] [PubMed] [Google Scholar]
- Marley, J., Lu, M., and Bracken, C. 2001. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20 71–75. [DOI] [PubMed] [Google Scholar]
- Minor Jr., D.L. and Kim, P.S. 1996. Context-dependent secondary structure formation of a designed protein sequence. Nature 380 730–734. [DOI] [PubMed] [Google Scholar]
- Sciandra, F., Schneider, M., Giardina, B., Baumgartner, S., Petrucci, T.C., and Brancaccio, A. 2001. Identification of the β-dystroglycan binding epitope within the C-terminal region of α-dystroglycan. Eur. J. Biochem. 268 4590–4597. [DOI] [PubMed] [Google Scholar]
- Spence, H.J., Dhillon, A.S., James, M., and Winder, S.J. 2004. Dystroglycan, a scaffold for the ERK-MAP kinase cascade. EMBO Rep. 5 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent, J., Alvarez-Martinez, M.T., Heitz, F., Liautard, J.P., Balny, C., and Lange, R. 2003. Alternative prion structural changes revealed by high pressure. Biochemistry 42 1318–1325. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. 2002. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 11 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V.N., Li, J., and Fink, A.L. 2001. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular link between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276 44284–44296. [DOI] [PubMed] [Google Scholar]
- White, S.H., Ladokhin, A.S., Jayasinghe, S., and Hristova, K. 2001. How membranes shape protein structure. J. Biol. Chem. 276 32395–32398. [DOI] [PubMed] [Google Scholar]
- Williamson, R.A., Henry, M.D., Daniels, K.J., Hrstka, R.F., Lee, J.C., Sunada, Y., Ibraghimov-Beskrovnaya, O., and Campbell, K.P. 1997. Dystroglycan is essential for early embryonic development: Disruption of Reichert’s membrane in Dag1-null mice. Hum. Mol. Genet. 6 831–841. [DOI] [PubMed] [Google Scholar]
- Winder, S.J. 2001. The complexities of dystroglycan. Trends Biochem. Sci. 26 118–124. [DOI] [PubMed] [Google Scholar]
- Wright, P.E. and Dyson, H.J. 1999. Intrinsically unstructured proteins: Reassessing the protein structure-function paradigm. J. Mol. Biol. 293 321–331. [DOI] [PubMed] [Google Scholar]
- Yang, B., Jung, D., Motto, D., Meyer, J., Koretzky, G., and Campbell, K.P. 1995. SH3 domain-mediated interaction of dystroglycan and Grb2. J. Biol. Chem. 270 11711–11714. [DOI] [PubMed] [Google Scholar]
- Yoshida, T., Pan, Y., Hanada, H., Iwata, Y., and Shigekawa, M. 1998. Bidirectional signaling between sarcoglycans and the integrin adhesion system in cultured L6 myocytes. J. Biol. Chem. 273 1583–1590. [DOI] [PubMed] [Google Scholar]