

Abstract
E. coli Par10 is a peptidyl-prolyl cis/trans isomerase (PPIase) from Escherichia coli catalyzing the isomerization of Xaa-Pro bonds in oligopeptides with a broad substrate specificity. The structure of E. coli Par10 has been determined by multidimensional solution-state NMR spectroscopy based on 1207 conformational constraints (1067 NOE-derived distances, 42 vicinal coupling-constant restraints, 30 hydrogen-bond restraints, and 68 φ/ψ restraints derived from the Chemical Shift Index). Simulated-annealing calculations with the program ARIA and subsequent refinement with XPLOR yielded a set of 18 convergent structures with an average backbone RMSD from mean atomic coordinates of 0.50 Å within the well-defined secondary structure elements. E. coli Par10 is the smallest known PPIase so far, with a high catalytic efficiency comparable to that of FKBPs and cyclophilins. The secondary structure of E. coli Par10 consists of four helical regions and a four-stranded antiparallel β-sheet. The N terminus forms a β-strand, followed by a large stretch comprising three α-helices. A loop region containing a short β-strand separates these helices from a fourth α-helix. The C terminus consists of two more β-strands completing the four-stranded anti-parallel β-sheet with strand order 2143. Interestingly, the third β-strand includes a Gly-Pro cis peptide bond. The curved β-strand forms a hydrophobic binding pocket together with α-helix 4, which also contains a number of highly conserved residues. The three-dimensional structure of Par10 closely resembles that of the human proteins hPin1 and hPar14 and the plant protein Pin1At, belonging to the same family of highly homologous proteins.
Keywords: protein structure, NMR, parvulin, PPIase, cis peptide bond
Parvulins represent the most recently discovered third protein family within the enzyme class of peptidyl prolyl cis/trans isomerases (PPIases, EC 5.2.1.8). These enzymes accelerate the adjustment of the cis/trans equilibrium of peptidyl-prolyl bonds in peptides and proteins (Fischer 2000). The Escherichia coli Par10 embodies the archetype of the homonymous protein family, and defines the minimal catalytic domain of the whole body of parvulin-like enzymes. Par10 was originally identified by its enzymatic activity in a screen for unknown PPIases in the cytoplasm of E. coli (Rahfeld et al. 1994b).
Parvulins are ubiquitously found in pro- and eukaryotes. Homologous proteins that have been described and predicted from available genome sequence databases so far possess molecular masses between 10 and 68 kDa. With 92 amino acid residues, E. coli Par10 consists of the minimal number of amino acid residues facilitating the cis/trans isomerization of peptidyl-prolyl bonds. Generally, parvulins consist of one, rarely two parvulin boxes and additional N-and C-terminal extensions. Three signature sequences occur within this parvulin box: H-[ILV]-[LVQ], G-G-[DYILR-SKE]-[LIM]-[GSEN]-[WEKAFPY]-[FMVAIL], and G-[YVILWF]-[HEA]-[IVL]-[ILV] (Fig. 1 ▶).
Figure 1.

Sequence homology of selected members of the parvulin family with respect to E. coli Par10 (PPIC_ECOLI, Swiss-Prot entry P39159). Homologous sequences were taken from the genomes of Salmonella typhimurium (SALMONELLA, Q9L6S3), Cenarchaeum symbiosum (CENARCHEUM, O74049), Arabidopsis thaliana PPIase (ARABIDOPSIS, O23727), human Par14 (PAR14, Q9Y237), human Pin1 (PIN1_HUMAN, Q13526), Drosophila melanogaster Par18 (DOD_DROMEO, P54353), and Saccharomyces cerevisiae Par19 (ESS1_YEAST, P22696). The letters on black background denote identical amino acids; the ones on gray background, similar amino acid types. On top the secondary structure of E. coli Par10 is shown for easier orientation. Alignment based on FASTA (version 3.2t09) (Pearson and Lipman 1988; Pearson 1990) with manual corrections, graphical output, and consensus by BOXSHADE (version 3.21) (Hofmann and Baron).
Based on the catalytic capacity of their Par10-like domains, eukaryotic parvulins such as mammalian Pin1 (Par18) and Saccharomyces cerevisiae Ess1 (Par19) were found to be involved in a novel type of regulation of phosphorylation dependent cell signaling. The PPIase activity-mediated conformational changes of Pin1 substrates, for example, they can have profound effects in transcriptional regulation (Arevalo-Rodriguez et al. 2000; Wu et al. 2000), mitotic cell cycle control (Lu et al. 1996; Winkler et al. 2000) and phosphorylation-dependent signal transduction pathways (Ryo et al. 2003). By promotion of protein phosphatase-catalyzed dephosphorylation Pin1 activity can restore the conformation and function of phosphorylated tau protein. Consequently, Pin1 knockout in mice causes progressive age-dependent neuropathy characterized by motor and behavioral deficits, tau hyperphosphorylation, tau filament formation, and neuronal degeneration (Liou et al. 2003). Parvulins participating in these processes share a unique substrate specificity, with pSer/pThr-Pro motifs in the targeted substrates representing the prolyl bonds to be isomerized. (Yaffe et al. 1997; Hani et al. 1999; Zhou et al. 2000).
In prokaryotes parvulins have been shown to assist the maturation of proteins and to assure their molecular function. The Bacillus subtilis SurA (Par32.5) is necessary for secretion of active α-amylase into the periplasm (Kontinen and Sarvas 1993), whereas the Lactococcus lactis PrtM (Par33) is essential for the major cell envelope associated serine protease PrtP to achieve its full proteolytic activity (Vos et al. 1989). In nitrogen assimilating bacteria, such as Klebsiella pneumoniae and Azotobacter vinelandii, the nifM gene products (e.g., K. pneumoniae Par31) ensure the enzymatic activity of the nitrogenase reductase (Howard et al. 1986). In E. coli cells the combined knockout of the two periplasmic parvulins E. coli SurA (Par47) and E. coli Ybau (Par68) results in a lethal phenotype. Both proteins were shown to be responsible for the assembly of outer membrane proteins (Dartigalongue and Raina 1998).
In the X-ray structure of human Pin1 (hPin1) three basic residues located within a loop are supposed to form the binding pocket for the phosphate group of the specific substrates (Ranganathan et al. 1997), explaining why these parvulins reach their full catalytic efficiency only on negatively charged substrates.
Besides the parvulin-like catalytic core of hPin1 (supplemented with a WW domain), a second parvulin, hPar14, was identified within the human genome (Rulten et al. 1999; Uchida et al. 1999). hPar14 is an N-terminally extended form of a parvulin-like catalytic core, where a basic, glycine-rich DNA recognition motif within the 34-residue extension targets the PPIase to the nucleus (Sekerina et al. 2000; Surmacz et al. 2002; Reimer et al. 2003).
The solution structure of hPar14 was solved by NMR spectroscopy (Sekerina et al. 2000; Terada et al. 2001). A comparison with the X-ray crystal structure of hPin1 (Ranganathan et al. 1997) revealed an identical topological arrangement of the secondary structure elements of the PPIase domains. In contrast to hPin1 and the other pSer/pThr-Pro specific parvulins, hPar14 is suggested to be involved in chromatin architecture and/or chromatin remodeling (Uchida et al. 1999). It may also play a role in regulating entry or offset of M-phase transition or transcription. Therefore, the function of hPar14 might be closely related to hPin1 (Surmacz et al. 2002; Uchida et al. 2003). The solution structure of the single-domain parvulin Pin1At of A. thaliana gave first evidence for a modified surface charge of the catalytic core in domain-supplemented parvulins (Landrieu et al. 2002). Of the two parvulin-like domains in E. coli SurA, the domain with the actual PPIase activity is located in a satellite position tethered about 30 Å from the core module in the crystal structure of SurA (Bitto and McKay 2002).
The object of this investigation, E. coli Par10, is characterized by a broad substrate specificity combined with high enzymatic activity (Rahfeld et al. 1994a). The monomeric protein represents the fully catalytically active enzyme. The architecture of Par10 should provide direct evidence for the minimal number of interacting sites needed for the formation of the active cleft of parvulins. Here we report the high resolution structure of E. coli Par10, as determined by NMR spectroscopy in aqueous solution.
Results
Resonance assignment
Sequential assignment of the backbone resonances of E. coli Par10 was performed with sequential information derived from a set of triple-resonance experiments [HNCACB, CBCA(CO)NH, HNCO, HNHA, HA(CACO)NH]. Evaluation was aided by the automatic assignment program PASTA (Leutner et al. 1998). The sequential assignment procedure yielded complete 1HN and 15N (except for the five prolines, P41, P59, P69, P73, and P76), Cα, Hα, Cβ and C′ assignments for 87 residues.
Assignment of the aliphatic side-chain resonances was completed to about 93% from a combination of 3D HCCH-TOCSY and HCCH-COSY experiments, together with information from the triple-resonance sequential assignment experiments. Assignment of the aromatic 1H resonances was achieved from Cβ-Hδ and Cβ-Hɛ correlation experiments (Yamazaki et al. 1993). Only a small number of aliphatic and aromatic side-chain signals remained unassigned, mainly few Hγ and Hδ shifts of lysine, isoleucine, and leucine residues as well as all Hζ protons of aromatic side chains.
A table of all assigned chemical shifts of E. coli Par10 has been deposited in the BMRB database (accession number 5225).
Secondary structure determination
The comparison of Cα, Cβ, C′ and Hα chemical shifts with random coil values was utilized to locate secondary structure elements using the program CSI (Wishart et al. 1992; Wishart and Sykes 1994). Thus, the four β-strands and three α helices expected for the parvulin fold could be clearly identified. In Par10, helices 1 and 2 extend from E14 to G27 and from F30 to S38, respectively. The β-strands 1 and 2 extend from residues A4–V11 and E51–R53. Concluding from chemical shift data, helix 4 (A60 to F66) is separated from the surrounding β-strands by two loop regions. The β-strands 3 and 4 (L77–T79 and G82–L89, respectively) were also clearly determined. In addition, evidence of another α-helix, helix 3, comprising residues G43–G47, was found in the CSI pattern. Confirmation of its existence and exact localization could be derived from the NOE data. All secondary-structure elements were confirmed by typical NOE connectivity patterns between backbone protons and 3JHNHα coupling constants (Fig. 2 ▶), as well as amide proton exchange rates estimated from MEXICO experiments (Gemmecker et al. 1993). The topology of the β-sheet was determined from NOE contacts between the β-strands showing an antiparallel arrangement with strand order 2143 (Fig. 3 ▶).
Figure 2.

Secondary structure indicators for E. coli Par10. Existence and intensity of typical NOEs are symbolized by the corresponding black rectangles, likewise the size of the 3J coupling. The last row shows the results of the Chemical Shift Index, CSI (Wishart et al. 1992; Wishart and Sykes 1994), with the letters b and h denoting β-sheets and helices. For comparison, the secondary structure element locations of the final structural ensemble are given on top of the sequence.
Figure 3.

Topology of E. coli Par10. Filled arrows symbolize experimental Hα-Hα NOE contacts; open arrows reflect observed HN-HN NOEs within the β-sheet.
Tertiary structure
From the experimental data (NOE derived distances, 3JHNHα coupling constants, chemical shifts, and exchange rates) an ensemble of 50 structures was calculated with the programs ARIA and XPLOR (for details see Materials and methods). The 18 best structures were selected and further refined with XPLOR to yield the final structural ensemble representing the result of the NMR structure determination (PDB code 1JNS; minimized average structure 1JNT). The structural quality in terms of restraint violation and deviation from ideal geometry (Table 1) was checked with the program PROCHECK (version 3.4, Laskowski et al. 1993). The RMSD values shown in Table 2 demonstrate the global fold to be well-defined. Figure 4A ▶ shows the superimposition of the 18 converged structures.
Table 1.
Structural statistics for E. coli Par10
| SA (20 Structures) | <SA>r | |
| Restraint RMSD | ||
| Distance restraints [Å] | ||
| All (1097) | 0.023 | 0.018 |
| Intra-Residue NOEs (494) | 0.017 | 0.011 |
| Sequential NOEs (272) | 0.034 | 0.026 |
| Medium-range NOEs (133) | 0.023 | 0.023 |
| Long-range NOEs (168) | 0.019 | 0.015 |
| H-bonds (30) | 0.006 | 0.012 |
| Dihedral restaints ([°], 68) | 0.098 | 0.096 |
| 3J(HNHα) restraints ([Hz], 42) | 0.906 | 0.817 |
| Deviations from ideal covalent geometry | ||
| Bonds [Å] | 0.002 | 0.002 |
| Angles [°] | 0.561 | 0.532 |
| Structure quality indicatorsa | ||
| Ramachandran map regions [%] | 82.8/9.6/4.9/2.6 | 82.9/13.2/2.6/1.3 |
SA: the final set of 20 simulated annealing structures; <SA>r: the structure obtained by regularizing the mean structure of SA under experimental restraints. The numbers in brackets represent the number of restraints of each type.
a Structural quality indicators were determined with the program PROCHECK (version 3.4, Laskowski et al. 1993). Percentages are shown for residues in most favored/additionally allowed/generously allowed/disallowed regions of the Ramachandran map.
Table 2.
Atomic coordinate RMSD [Å] of E. coli Par10
| SA vs. <SA> | SA vs. <SA>r | |||
| Backbone | All | Backbone | All | |
| All residues | 0.85 ± 0.14 | 1.39 ± 0.16 | 1.17 ± 0.20 | 1.87 ± 0.25 |
| All secondary structure | 0.58 ± 0.11 | 1.15 ± 0.14 | 0.87 ± 0.09 | 1.64 ± 0.16 |
| β-sheet, α1, α2, α4 | 0.50 ± 0.06 | 1.05 ± 0.13 | 0.79 ± 0.07 | 1.56 ± 0.15 |
SA: the final set of 18 simulated annealing structures; <SA>: the mean structure calculated by averaging the coordinates of the SA ensemble after fitting over secondary structure elements, exluding helix three; <SA>r: the structure obtained by regularizing <SA> under experimental restraints. Total secondary structure was defined as: β-sheet: 4–11, 51–53, 77–79, 82–89; α-helices: 14–27 (helix 1), 30–38 (helix 2), 43–47 (helix 3), 60–66 (helix 4).
Figure 4.
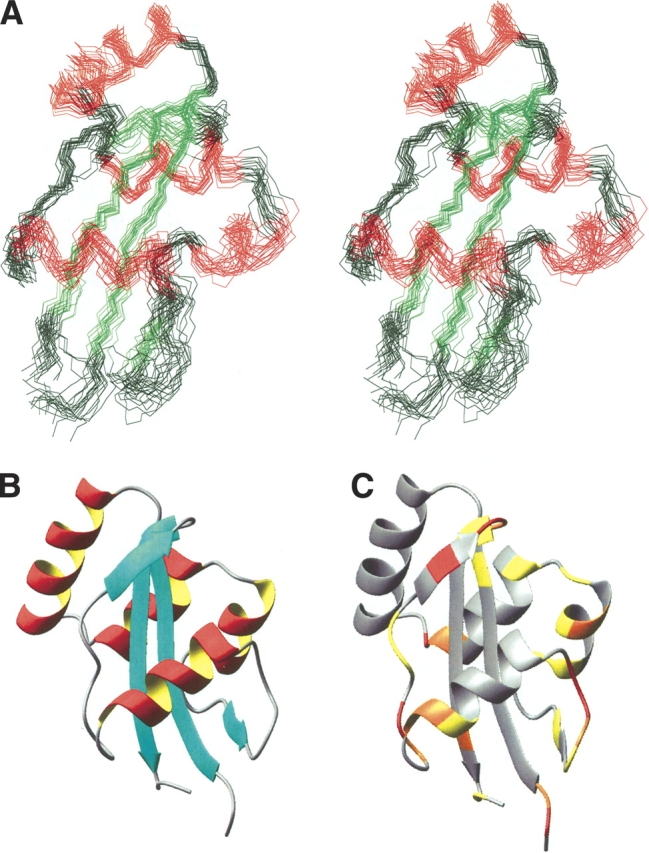
Three-dimensional structure of E. coli Par10. (A) Stereoview of the ensemble of the 18 best structures calculated from NMR data. The structures are superimposed with respect to the backbone atoms of secondary structure elements (excluding helix 3). For easier orientation, the antiparallel β-sheet is colored in green, the four helices in red. (B) Cartoon representation of the E. coli Par10 structure with secondary structure elements. (C) Tertiary structure of E. coli Par10 color-coded according to the MEXICO data: red, fast exchange; orange, medium exchange; yellow, weak exchange of the NH protons on a 100-ms time scale. Figure produced with MOLMOL, version 2k.1 (Koradi et al. 1996).
The experimentally determined three-dimensional structure of E.coli Par10 bears a strong similarity to the crystal structures of the hPin1 PPIase domain (Ranganathan et al. 1997) and the P2 domain of E. coli SurA (Bitto and McKay 2002), as well as to the solution NMR structures of hPar14 (Sekerina et al. 2000; Terada et al. 2001) and Pin1At (Landrieu et al. 2002). The four β-strands form a curved β-sheet, which is enclosed between α helices 1 and 2 on its convex side and α-helix 4 on the concave side, resulting in an α β α-sandwich structure with helix 4 being partially enclosed by the curved β-sheet (Fig. 4B ▶).
Dynamic nature of the Par10 structure
As expected, the loop regions are less well-defined in the structural calculations, especially the region I39–G50 (surprisingly also including α-helix 3). Contradictory NOE distance data and averaged 3JHNHα coupling constants indicate high flexibility in the loop regions Q54–P59 and S67–P73.
Unfortunately, no good-quality relaxation data could be obtained due to the low sample concentration and limited stability. However, the dynamic nature of helix 3 is clearly visible in the data derived from a MEXICO experiment (Fig. 4A ▶), in which the extent of exchange of HN protons with water is evaluated on a millisecond to second timescale (Gemmecker et al. 1993). Amide protons without visible exchange peaks can be assumed to be either sterically shielded from the solvent or to be involved in stable intra-molecular hydrogen bonds.
Within the β-sheet of E. coli Par10, no HN–water proton exchange was observed, except for the amide protons of E51 (weak), H78, and V88. This is in agreement with the three-dimensional structure, showing the localization of the amide protons of these residues to be on the edge of the β-sheets. As expected, also residues Q80 and F81—located in positions i + 1 and i + 2 of a βI-turn—show rather intense exchange peaks.
From the lack of MEXICO signals of the HN atoms in helices 1, 2, and 4, the existence of intramolecular H-bonds in a fairly rigid secondary structure can be concluded. In contrast, helix 3 shows a significant exchange of its backbone amide protons. Although the number of experimental restraints per residue is comparable to the average, the NOE data concerning helix 3 do not establish an equally well-defined secondary structure as for helices 1, 2, and 4. While the CSI gives a clear evidence for the existence of a helical core region near positions 43–47, this helix 3 is clearly less rigid than the others. Interestingly, in the highly homologous hPar14 and hPin1 structures there is no equivalent to helix 3, but merely a loop region without a well-defined secondary structure. On the other hand, helix 3 does indeed exist in Pin1At, where it seems to be even more stable than in Par10.
As stated above, the loop regions around helix 4 are also less well-defined. In Figure 5 ▶, the local RMSD values and the number of NOEs are compared. Although the number of NOEs per residue is more or less constant for all residues, the local RMSD values increase in the region around helix 3 and the loop between strand 2 and helix 4, a clear indication for increased structural flexibility in these parts. Hypothetically, this flexibility in the anchor loops around helix 4 could be interpreted in terms of the binding mechanism, with helix 4 moving away from the β-sheet, thereby opening the postulated active center for substrate access.
Figure 5.

Local RMSD values and number of NOEs per residue: although the NOE density is almost evenly distributed over the length of the sequence, significant differences can be seen in the local RMSD (black line). While most secondary structure elements (indicated in the top row) are well-defined, helix α3 and the two following loop regions show a high local RMSD that can be attributed to local flexibility.
Discussion
Comparison of the homologous parvulin domains of hPin1 and hPar14 with Par10
Interestingly, in hPar14 and Par10 the β-strand 3 includes a bulge containing a proline residue, which is responsible for the pronounced kink in the β-sheet. In E. coli Par10 this position is occupied by Pro76. While the chemical shift of its Cβ atom indicates that Pro76 is cis configured, the Cγ shift is more typical for a trans conformation. Nevertheless, characteristic NOEs in this region show unambiguously that the peptide bond Gly75-Pro76 adopts the cis conformation: in the 3D CCH-NOESY the NOE signals from the Gly75 Hα protons to Pro76 Cδ are missing, but the NOEs to Pro76 Cα are present. A peak is also visible for Pro76 Hα to Gly75 Cα, whereas the peaks from Pro76 Hδ1 and Hδ1 to Gly75 Cα are absent.
In contrast to E. coli Par10, the previously published structures of hPar14 and hPin1 do not contain a cis peptide bond at this location; for hPar14 a cis peptide bond was discussed (Sekerina et al. 2000), but could not be proven. Surprisingly, a close look at the three-dimensional structures reveals that the cis bond of E. coli Par10 allows the formation of the full β-sheet H-bond pattern between strands 3 and 4, even in the presence of the bulge (Fig. 6A ▶). The complexity of refolding of urea-denatured Par10 and the low rates had indeed suggested that a cis prolyl bond might exist in native Par10 (Scholz et al. 1997). Thus, the Gly75-Pro76 motif has now been identified as the site responsible for the strongly autocatalytic behavior in the re-folding of Par10.
Figure 6.
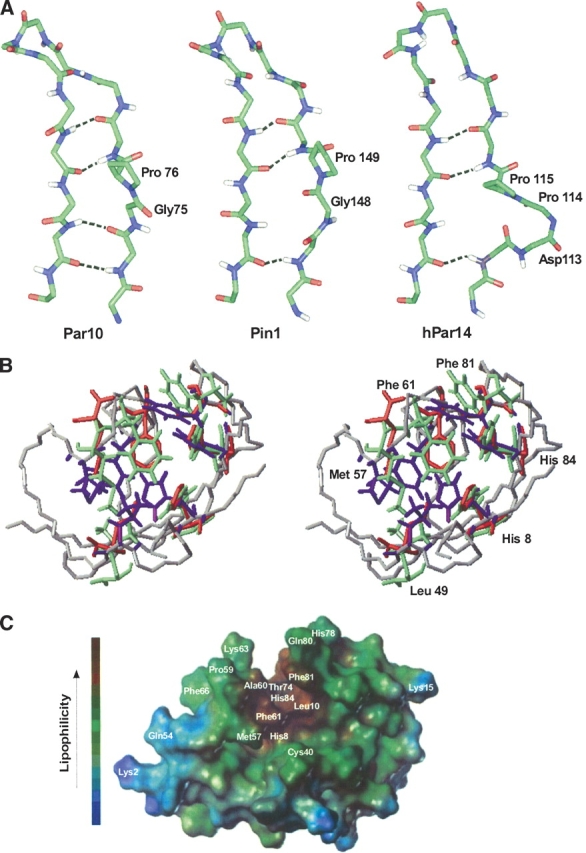
Orientation of the H-bonds in the β-sheet and putative substrate binding site of different Parvulins. (A) Arrangement of the G75-P76 cis amide bond in E. coli Par10, compared with the two homologous structures hPin1 and hPar14. In spite of the bulge resulting from the cis peptide bond, the β-sheet of E. coli Par10 can form all four H-bonds whereas the structures of hPar14 and hPin1 with trans-Pro are only stabilized by three H-bonds in this region (figure produced with Insight II, MSI Inc.). (B) Comparison of the putative binding pockets of hPar14 (PDB code 1eq3, green residues) and hPin1 (PDB code 1pin; red residues) overlaid on the E. coli Par10 structure (PDB code 1jnt; blue residues, grey backbone). The labeled residues are highly conserved in all parvulins and may be involved in the catalytic activity of these parvulins (figure produced with MOLMOL, version 2k.1; Koradi et al. 1996). (C) Connolly surface of E. coli Par10. Helix 4 (left) and the curved β-sheet form a lipophilic gap (brown), enabling a lipophilic oligopeptide substrate to bind (figure produced with the MOLCAD module of the program Sybyl, version 6.3; Tripos AG).
Putative substrate binding pocket of Par10
When compared to the putative binding pockets of hPar14 and Pin1, the corresponding residues in E. coli Par10 are highly conserved. This fact and the orientation of these residues in the binding pocket substantiate the hypothesis that these amino acids are substantial for enzyme function (Fig. 6B ▶); however, the exact nature of the catalytic mechanism is still unclear. For hPin1 (Pin1) a hypothetical mechanism has been suggested (Ranganathan et al. 1997), with residues L49 (E. coli Par10)/L122 (hPin1)/L82 (hPar14), M57 (E. coli Par10)/M130 (hPin1)/M90 (hPar14) and F61 (E. coli Par10)/F134 (hPin1)/F94 (hPar14) being assumed to form the substrate binding site, while residues H8 (E. coli Par10)/H59 (hPin1)/H42 (hPar14), H84 (E. coli Par10)/H157 (hPin1)/H123 (hPar14), C40 (E. coli Par10)/C113 (hPin1)/D74 (hPar14) and F81 (E. coli Par10)/S154 (hPin1)/F120 (hPar14) are distributed around the putative bond rotation axis and supposed to work as a catalytic cascade. However, residues C113 and S154 of hPin1 are not conserved in other parvulin homologs.
The putative substrate binding pocket contains a large aggregation of lipophilic side chains (Fig. 6C ▶). In combination with helix 4, the curved β-sheet forms a lipophilic gap on the protein surface, enabling a lipophilic oligopeptide to bind. Although the substrate specificities of subfamilies of the members of the parvulin family differs, the structure concerning this region is highly conserved. Only the residues that are necessary for the pSer/pThr and Arg side-chain-specific substrate recognition in hPin1 and hPar14, respectively, are missing in E. coli Par10.
In contrast to hPar14 and hPin1, E. coli Par10 does not possess any additional N-terminal domain. E. coli Par10 is located in the cytosol of E. coli where it seems to perform its cellular function. Although Par10 knockout in E. coli is followed by increased sensitivity toward oxidative stress (B. Hernandez-Alvarez, in prep.), the physiological role of the enzyme still remains to be elucidated.
Materials and methods
Cloning and overexpression of recombinant E. coli Par10
The parA gene was amplified by PCR using pSEP38 as a template (Rahfeld et al. 1994b). The amplified DNA fragment encoding E. coli Par10 was cloned into the NcoI, BamHI restriction sites of pQE60 (Qiagen), yielding the new plasmid pSEP612. After transformation in competent E. coli M15[pREP4] cells the clone was used for overexpression of recombinant E. coli Par10 protein. Expression cultures were grown in selective 2xYT-medium (16 g/L peptone, 10 g/L yeast extract, 5 g/L NaCl) by inoculation of 1 L with 20 mL of an overnight culture of the pSEP612 containing E. coli M15 [pREP4] cells. After induction of protein expression at A600 = 0.7 with 1 mM IPTG, cells were shaken for further 5 h at 37°C. Cells were harvested and treated as described (Rahfeld et al. 1994a).
Overexpression and purification of 13C, 15N-labeled E. coli Par10
The cells of a 50 mL overnight culture were harvested and resuspended into 50 mL of [U-13C,15N] MARTEK 9-CN medium (Martek). One liter of MARTEK 9-CN medium was inoculated with these adapted cells. After induction of protein expression at A600 = 0.3 with 1 mM IPTG, cells were shaken for further 10 h at 37°C. Cells were harvested at A600 = 0.8 by centrifugation at 4°C for 15 min at 6000 g in a Beckmann J2-HC centrifuge (Beckman Instruments). Cells were ruptured using a SLM Aminco French pressure cell (SLM Aminco). The cell lysate was stirred with 0.1% (v/v) Benzonase for 15 min and ultracentrifuged in a Beckmann L8 60M centrifuge at 100,000 g for 30 min at 4°C. The supernatant was applied to a Fractogel EMD DEAE-650(M) column (2.5 × 20 cm), equilibrated with 2 mM Tris buffer (pH 8.0). E. coli Par10 passed the column and was applied to a Fractogel TSK AF-Blue column (1 × 6 cm) equilibrated with 2 mM Tris buffer (pH 8.0). E. coli Par10 was eluted by running a linear gradient from 0 to 3 M KCl in 60 ml, 2 mM Tris buffer (pH 8.0). PPIase containing fractions were pooled and concentrated with a Filtron OMEGACELL (Pall-Filtron) with an exclusion size of 3,000 Da. Samples of 1 ml were applied to a Superdex 75 gel filtration column (1.6 × 60 cm; Pharmacia LKB), equilibrated with 10 mM phosphate buffer (pH 6.0), containing 100 mM KCl, 1 mM EDTA, and 1 mM DTE. Fractions containing E. coli Par10 were pooled, concentrated as described and used for NMR investigations.
NMR spectroscopy
Samples of [U-13C, 15N] and [U-15N] parvulin (0.8 mM) were prepared in 10 mM phosphate buffer (pH 6.0) containing 100 mM KCl and 1 mM EDTA, 1 mM DTE and 10% D2O. The number of signals observed in initial 1H,15N-HSQC spectra of both samples was consistent with that expected from the primary sequence of the protein. The spectra also showed good dispersion and lineshape characteristics, indicating that the solution contained a folded, monomeric protein. However, samples degraded slowly at room temperature within 2–3 wk, as could be seen from 2D 15N-HSQC spectra.
All spectra were recorded at 298 K at 600 or 750 MHz 1H resonance frequency on Bruker DMX600 and DMX750 spectrometers, respectively (Bruker Biospin GmbH). All spectra were processed with the Bruker program package XWINNMR, for spectra evaluation the TRIAD module of the software package SYBYL was used (version 6.6; Tripos Inc.).
Sequential assignment was achieved using information on the intraresidue and sequential Cα, Cβ, and Hα chemical shifts taken from 3D HNCACB (Stonehouse et al. 1995), 3D CBCA(CO)NH (Grzesiek and Bax 1992) and 3D HA(CACO)NH experiments (Boucher et al. 1992). Additionally, carbonyl carbon assignments were available from a 3D HNCO experiment (Kay et al. 1990). A 3D HNHA experiment performed on the 15N-labeled sample was used to extract 3JHNHα coupling constants (Vuister and Bax 1993). Side-chain assignment experiments included 3D HCCH-COSY (Ikura et al. 1991), 3D HCCH-TOCSY (Kay et al. 1993), 3D TOCSY-HSQC (Fesik and Zuiderweg 1988; Kay et al. 1989; Marion et al. 1989) and a 3D (H)CC(CO)NH experiment (Farmer and Venters 1995). 2D experiments correlating 13Cβ with 1Harδ/ɛ were used to assign most of the aromatic δ and ɛ protons (Yamazaki et al. 1993).
Two MEXICO spectra (Gemmecker et al. 1993) with mixing times of 50 and 200 ms were acquired to determine exchange rates of the amide protons with water. Amide protons not showing any significant MEXICO cross-peaks were considered to be potentially involved in hydrogen bonds. This was the case for all amide protons in the β-sheet (except for the peripheral amide protons of E51, H78, and V88) and in the central parts of helices α1, α2, and α4. In contrast, helix α3 displayed several significant exchange peaks indicating this helix to be less rigid (Fig. 4C ▶).
Distance data was derived from four different 3D NOESY spectra. A 3D 15N-HSQC-NOESY was recorded with the 15N-labeled sample, and 3D 13C-HSQC-NOESY, 3D CCH-NOESY, and 3D CNH-NOESY spectra with the 13C, 15N-labeled sample. The NOE mixing times of all experiments were set to 80 ms.
Structure calculations and analysis
Initial NOE assignment was performed with the program ARIA (Ambiguous Restraints in Iterative Assignment, version 0.53; Nilges 1997; Nilges and O’Donoghue 1998; Linge and Nilges 1999) in combination with CNS (Crystallography and NMR System, version 0.5; Brunger et al. 1998). All programs were run on SGI (Silicon Graphics Inc.) Origin 200 computers equipped with four R10000 processors, running under IRIX 6.4. ARIA input parameters included the chemical shift table with all backbone and most of the sidechain shifts assigned, as well as 42 3JHNHα coupling constants. A single ARIA run consisted of eight iterations, with 20 structures per iteration being calculated, using the standard ARIA/CNS simulated annealing protocol. After convergence of the ARIA runs, further calculations were performed in XPLOR (version 3.851, Brunger 1992), utilizing only unambiguous data derived from the ARIA calculations (1067 NOEs), as well as 30 H-bond restraints (according to secondary structure locations from Chemical Shift Index, CSI, and solvent exposure data derived from MEXICO experiments). Furthermore, 68 dihedral restraints for φ and ψ angles along the CSI predicted α-helical segments were used (φ = −65 ± 40°; ψ = −40 ± 40°). With standard simulated annealing protocols, a set of 50 structures was calculated, and 17 structures of this ensemble discarded due to high Ramachandran energy. From the remaining 33 structures, a subset SA was chosen with no NOE restraint being violated by more than 0.17 Å, and no dihedral restraint violated by more than 3°. For the NOE selection criterion, NOEs in flexible regions (residues 48–50 and 54–59, as well as flexible side chains, as observed by contradictory NOEs) were not taken into consideration. Final refinement of this ensemble, containing 18 structures, was performed utilizing a conformational database potential function in the nonbonded potential (Kuszewski et al. 1996, 1997). To obtain a single representative structure, an averaged structure <SA> was calculated from the SA ensemble, and regularized under experimental restraints, yielding <SA>r . Fitting was performed over all secondary structure elements, excluding helix three (residues 43–47). Structure improvement in all stages was controlled using the program PROCHECK (Laskowski et al. 1993).
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, the Sonderforschungsbereich 369, the Fonds der Chemischen Industrie and the Dr.-Ing. Leonhard-Lorenz-Stiftung. We also thank Murray Coles for help with the structure calculations and Jens Liermann for his support with the PASTA program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ARIA, ambiguous restraints in iterative assignment
CNS, crystallography and NMR system
CSI, chemical shift index
DTE, dithioerythrol
EDTA, ethylendiamine tetraacetate
HSQC, heteronuclear single-quantum correlation
isopropyl ß-D-thiogalactopyranoside (IPTG)
MEXICO, measurement of exchange in isotopically labelled compounds
RMSD, root mean square deviation
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04756704.
References
- Arevalo-Rodriguez, M., Cordenas, M.E., Wu, X., Hanes, S.D., and Heitman, J. 2000. Cyclophilin A and Ess1 interact with and regulate silencing by the Sin3-Rpd3 histone deacetylase. EMBO J. 19 3739–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto, E. and McKay, D.B. 2002. Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10 1489–1498. [DOI] [PubMed] [Google Scholar]
- Boucher, W., Laue, E.D., Campbell-Burk, S.L., and Domaille, P.J. 1992. Improved 4D experiments for the assignment of backbone nuclei in 13C/15N labelled proteins. J. Biomol. NMR 2 631–637. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T. 1992. X-PLOR, Version 3.1. A system for X-ray crystallography and NMR. Yale University Press, New Haven, CT.
- Brunger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software system for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Dartigalongue, C. and Raina, S. 1998. A new heat-shock gene, ppiD, encodes a peptidyl-prolyl isomerase required for folding of outer membrane proteins in Escherichia coli. EMBO J. 17 3968–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer II, B. and Venters, R. 1995. Assignment of side chain 13C resonances in perdeuterated proteins. J. Am. Chem. Soc. 117 4187–4188. [Google Scholar]
- Fesik, S.W. and Zuiderweg, E.R.P. 1988. Heteronuclear three-dimensional NMR spectroscopy. A strategy for the simplification of homonuclear two-dimensional NMR-spectra. J. Magn. Reson. 78 588–593. [Google Scholar]
- Fischer, G. 2000. Chemical aspects of peptide bond isomerisation. Chem. Soc. Rev. 29 119–127. [Google Scholar]
- Gemmecker, G., Jahnke, W., and Kessler, H. 1993. Measurement of fast proton exchange rates in isotopically labelled compounds. J. Am. Chem. Soc. 115 11620–11621. [Google Scholar]
- Grzesiek, S. and Bax, A. 1992. Correlating backbone amide and side-chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114 6291–6293. [Google Scholar]
- Hani, J., Schelbert, B., Bernhardt, A., Domdey, H., Fischer, G., Wiebauer, K., and Rahfeld, J.-U. 1999. Mutations in a peptidylprolyl cis/trans isomerase gene lead to a defect in 3′-end formation of a pre-mRNA in Saccharomyces cerevisiae. J. Biol. Chem. 274 108–116. [DOI] [PubMed] [Google Scholar]
- Howard, K.S., McLean, P.A., Hansen, F.B., Lemley, P.V., Koblan, K.S., and Orme-Johnson, W.H. 1986. Klebsiella pneumoniae nifM gene product is required for stabilization and activation of nitrogenase iron protein in Escherichia coli. Biol. Chem. 261 772–778. [PubMed] [Google Scholar]
- Ikura, M., Kay, L.E., and Bax, A. 1991. Improved three-dimensional 1H-13C-1H correlation spectroscopy of a 13C-labeled protein using constant-time evolution. J. Biomol. NMR 1 299–304. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Marion, D., and Bax, A. 1989. Practical aspects of 3D heteronuclear NMR of proteins. J. Magn. Reson. 84 72–84. [Google Scholar]
- Kay, L.E., Ikura, M., Tschudin, R., and Bax, A. 1990. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson. 89 496–514. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Xu, G.-Y., Singer, A., Muhandiram, D.R., and Forman-Kay, J.D. 1993. A gradient-enhanced HCCH-TOCSY experiment for recording sidechain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. B 101 333–337. [Google Scholar]
- Kontinen, V.P. and Sarvas, M. 1993. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol. Microbiol. 8 727–737. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Kuszewski, J., Gronenborn, A.M., and Clore, G.M. 1996. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 5 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J. Magn. Reson. 125 171–177. [DOI] [PubMed] [Google Scholar]
- Landrieu, I., Wieruszeski, J.-M., Wintjens, R., Inzé, D., and Lippens, G. 2002. Solution structure of the single-domain prolyl cis/trans isomerase PIN1At from Arabidopsis thaliana. J. Mol. Biol. 320 321–332. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–289. [Google Scholar]
- Leutner, M., Gschwind, R.M., Liermann, J., Schwarz, C., Gemmecker, G., and Kessler, H. 1998. Automated backbone assignment of labelled proteins using the threshold accepting algorithm. J. Biomol. NMR 11 31–43. [DOI] [PubMed] [Google Scholar]
- Linge, J. and Nilges, M. 1999. Influence of non-bonded parameters on the quality of NMR structures: A new force-field for NMR structure calculation. J. Biomol. NMR 13 51–59. [DOI] [PubMed] [Google Scholar]
- Liou, Y.C., Sun, A., Ryo, A., Zhou, X.Z., Yu, Z.X., Huang, H.K., Uchida, T., Bronson, R., Bing, G.Y., Li, X.J., et al. 2003. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424 556–561. [DOI] [PubMed] [Google Scholar]
- Lu, K.P., Hanes, S.D., and Hunter, T. 1996. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380 544–547. [DOI] [PubMed] [Google Scholar]
- Marion, D., Kay, L.E., Sparks, S.W., Torchia, D.A., and Bax, A. 1989. Three-dimensional heteronuclear NMR of 15N-labeled proteins. J. Am. Chem. Soc. 111 1515–1517. [Google Scholar]
- Nilges, M. 1997. Ambiguous distance data in the calculation of NMR structures. Fold. Des. 2 53–57. [DOI] [PubMed] [Google Scholar]
- Nilges, M. and O’Donoghue, S. 1998. Ambiguous NOEs and automated NOE assignment. Prog. NMR Spect. 32 107–139. [Google Scholar]
- Pearson, W.R. 1990. Rapid and sensitive sequence comparison with FASTP and FASTA. In Molecular evolution, computer analysis of protein and nucleic acid sequences (ed. R.F. Doolittle), Methods in Enzymology 183: pp. 63–98. Academic Press, San Diego, CA. [DOI] [PubMed]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence analysis. Proc. Natl. Acad. Sci. 85 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahfeld, J.-U., Schierhorn, A., Mann, K., and Fischer, G. 1994a. A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 343 65–69. [DOI] [PubMed] [Google Scholar]
- Rahfeld, J.-U., Rücknagel, K.P., Schelbert, B., Ludwig, B., Hacker, J., Mann, K., and Fischer, G. 1994b. Confirmation of the existence of a third family among peptidyl prolyl cis/trans isomerases. Amino acid sequence and recombinant production of parvulin. FEBS Lett. 352 180–184. [DOI] [PubMed] [Google Scholar]
- Ranganathan, R., Lu, K.P., Hunter, T., and Noel, J.P. 1997. Structural and functional analysis of the mitotic rotamase Pin1 suggest substrate recognition is phosphorylation dependent. Cell 89 875–886. [DOI] [PubMed] [Google Scholar]
- Reimer, T., Weiwad, M., Schierhorn, A., Ruecknagel, P.K., Rahfeld, J.U., Bayer, P., and Fischer, G. 2003. Phosphorylation of the N-terminal domain regulates subcellular localization and DNA binding properties of the peptidyl-prolyl cis/trans isomerase hPar14. J. Mol. Biol. 330 955–966. [DOI] [PubMed] [Google Scholar]
- Rulten, S., Thorpe, J., and Kay, J. 1999. Identification of eukaryotic parvulin homologues: A new subfamily of peptidylprolyl cis/trans isomerases. Biochem. Biophys. Res. Commun. 259 557–562. [DOI] [PubMed] [Google Scholar]
- Ryo A, Liou, Y.-C., Lu, K.P., and Wulf, G. 2003. Prolyl isomerase Pin1: a catalyst for oncogenesis and a potential therapeutic target in cancer. J. Cell. Sci. 116 773–783. [DOI] [PubMed] [Google Scholar]
- Scholz, C., Rahfeld, J., Fischer, G., and Schmid, F.X. 1997. Catalysis of protein folding by parvulin. J. Mol. Biol. 273 752–762. [DOI] [PubMed] [Google Scholar]
- Sekerina, E., Rahfeld, J.-U., Müller, J., Rascher, C., Fischer, G., and Bayer, P. 2000. NMR solution structure of hPar14 reveals similarity to the peptidyl prolyl cis/trans isomerase domain of the mitotic regulator hPin1 but indicates a different functionality of the protein. J. Mol. Biol. 301 1003–1017. [DOI] [PubMed] [Google Scholar]
- Stonehouse, J., Clowes, R., Shaw, G., Keeler, J., and Laue, E. 1995. Minimization of sensitivity losses due to the use of gradient pulses in triple-resonance NMR of proteins. J. Biomol. NMR 5 226–232. [DOI] [PubMed] [Google Scholar]
- Surmacz, T., Bayer, E., Rahfeld, J.-U., Fischer, G., and Bayer, P. 2002. The N-terminal basic domain of human parvulin hPar14 is responsible for the entry to the nucleus and high affinity binding. J. Mol. Biol. 321 235–247. [DOI] [PubMed] [Google Scholar]
- Terada, T., Shirouzu, M., Fukumori, Y., Fujimori, F., Ito, Y., Kigawa, T., Yokoyama, S., and Uchida, T. 2001. Solution structure of the human parvulin-like peptidyl prolyl cis/trans isomerase hPar14. J. Mol. Biol. 305 917–926. [DOI] [PubMed] [Google Scholar]
- Uchida, T., Fujimori, F., Tradler, T., Fischer, G., and Rahfeld, J.-U. 1999. Identification and characterization of a 14 kDa human protein as a novel parvulin-like peptidyl-prolyl cis/trans isomerase. FEBS Lett. 446 278–282. [DOI] [PubMed] [Google Scholar]
- Uchida, T., Takamiya, M., Takahashi, M., Miyashita, H., Ikeda, H., Terada, T., Matsuo, Y., Shirouzu, M., Yokoyama, S., Fujimori, F., et al. 2003. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem. Biol. 10 15–24. [DOI] [PubMed] [Google Scholar]
- Vos, P., van Asseldonk, M., van Jeveren, F., Siezen, R., Simons, G., and de Vos, W.M. 1989. A maturation protein is essential for production of active forms of Lactococcus lactis SK11 serine proteinase located in or secreted from the cell envelope. J. Bacteriol. 171 2795–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuister, G. and Bax, A. 1993. Quantitative J correlation: A new approach for measuring homonuclear three-bond J(HNHα) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 115 7772–7777. [Google Scholar]
- Winkler, K.E., Swenson, K.I., Kornbluth, S., and Means, A.R. 2000. Requirement of the prolyl isomerase Pin1 for the replication checkpoint. Science 287 1644–1647. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994. The 13C Chemical-Shift Index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4 171–180. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1992. The Chemical Shift Index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31 1647–1651. [DOI] [PubMed] [Google Scholar]
- Wu, X., Wilcox, C.B., Devasahayam, G., Hackett, R.L., Arevalo-Rodriguez, M., Cardenas, M.E., Heitman, J., and Hanes, S.D. 2000. The Ess1 prolyl isomerase is linked to the chromatin remodeling complexes and the general transcription machinery. EMBO J. 19 3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe, M.B., Schutkowski, M., Shen, M., Zhou, X.Z., Stukenberg, P.T., Rahfeld, J. U., Xu, J., Kuang, J., Kirschner, M.W., Fischer, G., et al. 1997. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278 1957–1960. [DOI] [PubMed] [Google Scholar]
- Yamazaki,T., Forman-Kay, J., and Kay, L. 1993. Two-dimensional NMR experiments for correlating 13Cβ and 1Hδ/ɛ chemical shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J. Am. Chem. Soc. 115 11054. [Google Scholar]
- Zhou, X.Z., Kops, O., Werner, A., Lu, P., Shen, M., Stoller, G., Kullertz, G., Stark, M., Fischer, G., and Lu, K.P. 2000. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell 6 873–883. [DOI] [PubMed] [Google Scholar]