

Abstract
Decay-accelerating factor (DAF, CD55) is a glycophosphatidyl inositol-anchored glycoprotein that regulates the activity of C3 and C5 convertases. In addition to understanding the mechanism of complement inhibition by DAF through structural studies, there is also an interest in the possible therapeutic potential of the molecule. In this report we describe the cloning, expression in Escherichia coli, isolation and membrane-targeting modification of the four short consensus repeat domains of soluble human DAF with an additional C-terminal cysteine residue to permit site-specific modification. The purified refolded recombinant protein was active against both classical and alternative pathway assays of complement activation and had similar biological activity to soluble human DAF expressed in Pichia pastoris. Modification with a membrane-localizing peptide restored cell binding and gave a large increase in antihemolytic potency. These data suggested that the recombinant DAF was correctly folded and suitable for structural studies as well as being the basis for a DAF-derived therapeutic. Crystals of the E. coli-derived protein were obtained and diffracted to 2.2 Å, thus permitting the first detailed X-ray crystallography studies on a functionally active human complement regulator protein with direct therapeutic potential.
Keywords: complement, decay-acceleration, membrane anchor, crystallography
DAF is a GPI-anchored glycoprotein that regulates complement activation, preventing the destruction of host tissue. It is widely distributed in tissue, especially upon endothelial cells (Nishikawa et al. 1998), but significant variations in expression have been noted within specific organs such as the kidney (Spiller et al. 1999) and in skin (Sayama et al. 1991). DAF plays a role in local control of complement activation, for example, on melanocytes (Venneker et al. 1998) and on the endometrium (Young et al. 2002).
DAF is a member of the regulators of complement activation (RCA) gene family of proteins (Hourcade et al. 1989), which inhibit the C3 and C5 convertases, the central amplification enzymes of the complement cascade. The RCA family includes the fluid phase regulators Factor H and C4b binding protein; and the membrane bound proteins DAF, membrane cofactor protein (MCP or CD46), and complement receptor type 1 (CR1 or CD35). Membrane-associated DAF prevents the assembly of the C3 convertases C4b2a and C3bBb by dissociating C2a and Bb from the complexes (Medof et al. 1984; Fujita et al. 1987) and acts only on convertase complexes generated on the same membrane and not extrinsically on convertases assembled on target cells (Medof et al. 1984). DAF is normally expressed as a glycosylated 70-kDa protein, although an 82-kDa form has been described (Hensel et al. 2001). The functional sites of DAF are contained within four short consensus repeats (SCRs, also known as complement control protein motifs—CCPs—or sushi domains). DAF is therefore structurally related to other complement regulatory proteins such as CR1/CD35 and MCP/CD46, which are also composed of SCR domains; CR1 allotypes have up to 44 such repeats, and MCP, like DAF, has 4. It has been shown that classical and alternative pathway regulatory activity of DAF is associated with SCRs 2 and 3 and SCRs 2, 3, and 4, respectively (Brodbeck et al. 1996). DAF has an N-linked glycan between SCRs 1 and 2, but it has been shown (Brodbeck et al. 2000) that the N glycan is not required for the complement regulatory function of DAF. A membrane-proximal O-linked polysaccharide rich region (S/T region) separates the SCRs from the membrane to which the protein is tethered by a GPI anchor (Medof et al. 1986). It has been suggested that the S/T region may be a functional spacer (Coyne et al. 1992), because deletion mutants (DAFΔS/T), although expressed normally, were nonfunctional. Forms of DAF with the GPI anchor replaced by an encoded transmembrane domain were, however, shown to be active (Lublin and Coyne 1991). Fodor et al. (1995) transfected a murine fibroblast cell line with CD59/DAF chimeric proteins, and found that, unlike CD59, DAF does not have a requirement to be proximal to the membrane. The outline structures of the various forms of DAF used in this study are shown in Figure 1 ▶.
Figure 1.

Summary of the domain structures and modifications made to the DAF forms used in this study. For definitions of the forms, see Abbreviations. Note that the exact composition of the glycoform carbohydrate differs between nDAF and PpDAF.
Moran et al. (1992) engineered soluble and secretory forms of DAF lacking GPI anchors. These anchorless forms of DAF were 50-fold less active than GPI-anchored DAF extracted from CHO cells at inhibiting complement activation on the cell surface in vitro, and they inhibited fluid phase activation where the GPI-anchored DAF did not. The soluble form of DAF was found to inhibit the reversed passive Arthus reaction in guinea pigs.
Encouraged by this and by more recent studies (Harris et al. 2002) on the biological activity of soluble forms of DAF, our aim was to express DAF in Escherichia coli and refold it to produce an active protein for therapeutic applications and structural studies. We have described elsewhere a general strategy for restoring membrane-binding function in recombinant soluble complement regulatory proteins by modification with synthetic peptide derivatives (Smith and Smith 2001; Fraser et al. 2002, 2003; Smith 2002). Such a modification strategy has the advantage that it enables the large-scale production of soluble proteins derived from the extracellular domains of membrane-bound proteins and does not require extraction of such proteins from host cell membranes.
This approach has been shown to permit novel therapeutic applications of complement regulators in animal models of disease (Dong et al. 1999; Linton et al. 2000), and one modified agent derived from CR1 has progressed to human clinical studies (Smith 2002). The purpose of the present studies was therefore to establish expression and refolding conditions for large-scale production of DAF derivatives with therapeutic potential and utility for structural studies. Large-scale production is facilitated by utilizing an E. coli expression system which, in this case, has an added benefit of not introducing glycosylation into the DAF molecule. Glycosylation can complicate crystallization and NMR studies by introducing heterogeneity, and is itself not required for the function of DAF (Brodbeck et al. 2000; Kuttner-Kondo et al. 2001).
Structural studies on SCR-containing proteins have been reported previously (Norman et al. 1991; Barlow et al. 1992; Wiles et al. 1997), but only limited information is available on proteins containing more than two such domains. The pathogen binding domains of DAF (SCRs 3 and 4) have been expressed in the yeast Pichia pastoris and their structure determined to 1.7 Å (Williams et al. 2003). Additionally, Uhrinova et al. (2003) have solved the solution structure of DAF modules 2 and 3 that are necessary and sufficient to accelerate decay of the classical pathway convertase. However, prior to the present work no high-resolution structure of a multi-SCR human protein with intact complement (classical and alternative) regulatory function had been described.
We demonstrate here that it is possible to produce recombinant soluble DAF from E. coli-derived inclusion bodies, that the material produced is active in decay-accleration and hemolytic assays of both the classical and alternative pathways, that it can be modified in a site-specific fashion to restore membrane binding function with an associated increase in potency, and that it can be crystallized.
Results
DNA encoding a DAF protein comprising the four extra-cellular SCR domains with a C-terminal cysteine (Fig. 1 ▶) was cloned into a pET-based vector (see Materials and Methods) and expressed in E. coli. The C-terminal cysteine was added to permit site-specific terminal localization of a membrane-targeting peptide. The product was isolated and solubilized from inclusion body pellets. Using high-pH re-folding conditions similar to those previously employed for multi-SCR constructs derived from CR1 (Dodd et al. 1995; Mossakowska et al. 1999), we obtained a soluble protein that could be isolated in ~80% purity (as assessed by nonreducing SDS-PAGE [Fig. 2 ▶, lane 8]) using simple fractional precipitation and gel filtration methods.
Figure 2.

Expression and refolding of EcDAF. Reducing 4%–12% PAGE gels. Lane 1, MW markers; lane 2, homogenized cell pellet; lane 3, pellet wash (PBS/Tween-80); lane 4, supernatant after solubilization; lane 5, pellet after solubilization; lane 6, pellet after dialysis against 6 M urea, 10 mM HCl; lane 7, supernatant after dialysis; lane 8, soluble product post-refold.
For crystallization, detailed assay studies, and for modification, the EcDAF preparation was further purified by gel filtration yielding fractions with different mobilities on nonreducing SDS-PAGE gels. Only one fraction had the same mobility as PpDAF (data not shown), and this was selected for use in crystallization trials and most assays, because PpDAF is known to be correctly folded from studies with conformationally sensitive antibodies and from NMR studies (Powell et al. 1997). The results were corroborated by MALDI-TOF mass spectrometry (MS), which confirmed that the isolated species had the correct molecular mass as predicted from the amino acid sequence.
To confer membrane binding, EcDAF was site-specifically modified with a synthetic analog of a “myristoyl-electrostatic switch” peptide, APT542 (Smith and Smith 2001).
This resulted in approximately 95% conversion to a singly modified species as observed by a gel shift (Fig. 3 ▶) confirmed by MALDI-TOF MS.
Figure 3.

Conjugation of EcDAF with the membrane-localizing peptide MP (APT542). Nonreducing 4%–12% PAGE gels. Lane 1, MW markers; lane 2; EcDAF reduced with TCEP; lane 3, EcDAF reduced with TCEP and modified with MP.
Decay acceleration assays in which the C3 convertases of each pathway were assembled from purified components (see Materials and Methods) were used to check this key activity in EcDAF. Figure 4 ▶ shows that the concentration of EcDAF required to reduce C3a production by 50% was approximately 30 nM in classical pathway (C1s triggered) activation and <10 nM for the alternative pathway (factor D triggered). This potency was comparable to or better than that of reference standards (e.g., SCRs 1–3 of CR1 which has an IC50 value of ~100 nM against the classical pathway and ~20 nM against the alternative pathway—data not shown). Under the conditions of these fluid-phase assays detergent-solubilized nDAF was only weakly active. Purified DAF derivatives were also assayed for activity in hemolytic assays using the classical and alternative pathways of complement activation. In a classical pathway hemolytic assay the EcDAF preparation was shown to be of comparable potency to soluble PpDAF (Fig. 5A ▶).
Figure 4.

Decay-accelerating activity of EcDAF in fluid-phase assays: Inhibition of C3a release from the classical pathway (open circles) and alternative pathway (filled squares) convertases. Means ± SD of two separate experiments.
Figure 5.

(A) Comparison of the activity of EcDAF (filled circles) and PpDAF (open triangles) in a classical pathway antihemolytic assay. (B) Comparison of EcDAF (open circles) with EcDAF-MP (open squares) and nDAF (open diamonds) in the classical pathway antihemolytic assay. (C) Comparison of EcDAF (open circles) with EcDAF-MP (open squares) in the alternative pathway hemolytic assay at 1 in 8 serum dilution, and (D) 1 in 16 serum dilution.
Figure 5B ▶ compares purified EcDAF, GPI-anchored nDAF, and EcDAF-MP in a classical pathway antihemolytic assay. The IC50 of EcDAF-MP was ~0.2 nM in this assay, approximately 100-fold more potent than the unmodified protein and significantly more active than detergent-solubilized nDAF. The detergent CHAPS was required to ensure the solubility of nDAF, and the possibility cannot be ruled out that CHAPS inhibits incorporation of GPI-anchored proteins into cell membranes and thus causes the potency of nDAF in this assay to be underestimated.
The effect of CHAPS on EcDAF-MP activity was investigated and found to be negligible except at a concentration of <0.5 nM where a slight potentiating effect was observed (data not shown). In an alternative pathway hemolytic assay using a 1 in 8 final dilution of serum, the IC50 of EcDAF was approximately 800 nM (Fig. 5C ▶) and using 1 in 16 final serum dilution it was lowered to 30–40 nM (Fig. 5D ▶). Under both alternative pathway assay conditions, there was no significant difference in potency between modified and unmodified forms of EcDAF. When this assay was repeated in a “wash” format (i.e., with the target erythrocytes exposed to agents in the absence of serum, followed by buffer washing and exposure to diluted serum), EcDAF-MP was not able to protect the target cells (data not shown). However, it was active if present in the assay mixture continuously (see Fig. 5C, D ▶).
Binding of EcDAF-MP to cells could be demonstrated directly under other conditions. Figure 6, A and B ▶, compares Raji cells exposed to EcDAF-MP with untreated controls using immunofluorescent detection of DAF with murine antihuman DAF mAB (MBC1) followed by an Alexa-Fluor-488 conjugated goat antimouse IgG. Binding of the modified protein was clearly evident following washing of the cells, whereas controls showed much lower background fluorescence. Figure 6, C and D ▶, compares binding of EcDAF-MP and unmodified EcDAF to guinea pig erythrocytes (which, unlike Raji cells, lack human DAF). A punctate pattern of distribution suggestive of localization in lipid rafts (Simons and Ikonen 1997) was observed for EcDAF-MP but not for the unmodified protein.
Figure 6.

EcDAF binding to cells, detected with murine anti-hDAF mAB and AlexaFluor 488 labeled anti-mIgG. Magnification ×40. (A) Raji cells following exposure to 1 μM EcDAF-MP; (B) Raji cells + control (no agent); (C) guinea pig erythrocytes + EcDAF-MP; (D) guinea pig erythrocytes + EcDAF.
Initial crystallization screening was performed using commercially available screens, and resulted in small crystals in condition 43 of Crystal Screen 1 (Molecular Dimensions Ltd. [20% mPEG4K, 0.2 M ammonium sulphate]). However, several optimization steps were required before diffraction quality crystals could be obtained. First, it was observed that minor changes to the chemical composition of the mother liquor as well as growth at low temperatures (2°C) favored crystal formation. Second, growth conditions had to be altered to allow cryoprotection (Garman 1999) of the crystals. A number of cryoprotection protocols were investigated, but the most successful approach was to incorporate glycerol cryoprotectant into the mother liquor and grow crystals under these conditions. However, high glycerol concentrations (>10%) drastically reduced the number of crystals obtained. Third, the method of seeding (Stura 1991) allowed for an increase in the frequency of crystal appearance as well as limited control over the size of crystals grown (see Fig. 7 ▶). The crystals belonged to the space group P1 and diffracted to a maximum resolution of 2.2 Å. Table 1 gives unit cell parameters for three typical crystals as well as data collection and processing statistics for three typical data sets.
Figure 7.
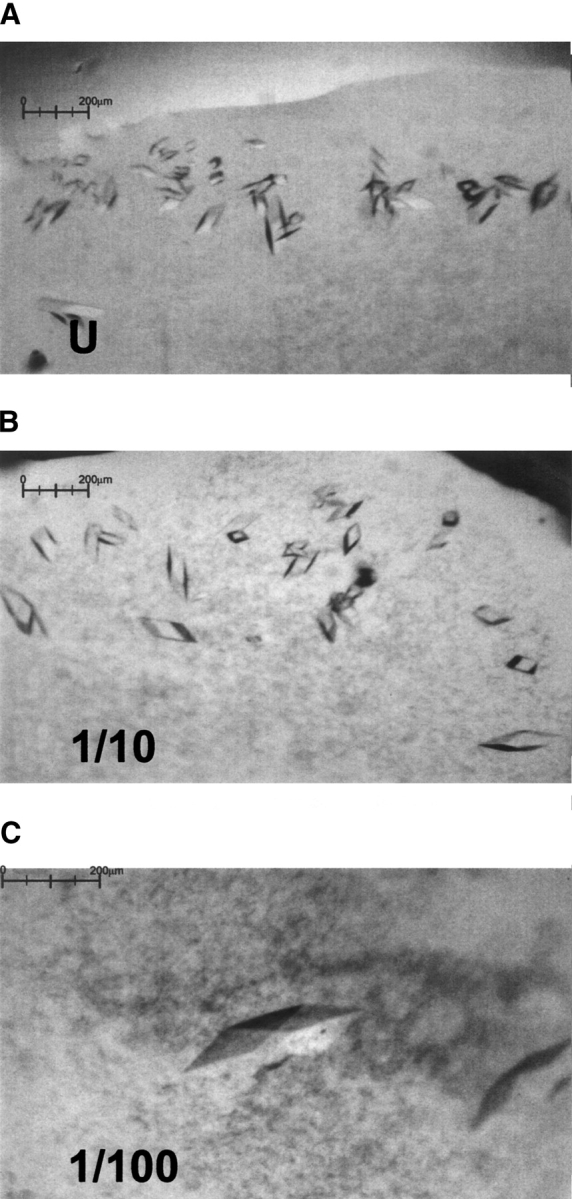
Effect of seeding on DAF crystal growth: (A) crystals grown from an undiluted seeding stock, (B) 10-fold diluted stock, (C) 100-fold diluted stock. The largest crystal at center of insert (C) is 210 μm in the longest dimension.
Table 1.
Data collection and processing parameters for DAF crystals
| Data set 1 | Data set 2 | Data set 3 | |
| Collection parameters | |||
| Detector | ADSC Q4 CCD | ADSC | MAR345 |
| Source | ESRF beamline ID14-2 | Daresbury beamline 9.6 | Inhouse Rigaku RU200H rotating |
| Oscillation angle (°) | 1 | 1.5 | 1 |
| Crystal to detector | 220 | 200 | 260/170 |
| Exposure time (sec) | 45 | 90 | 600/1200 |
| Wavelength (Å) | 0.933 | 0.870 | 1.542 |
| Processing parameters | |||
| Unit cell parametersa | 53.1, 51.9, 64.1 | 46.7, 55.1, 61.8, 88.8 | 46.7, 55.1, 61.6, 88,8 |
| Space group | P1 | P1 | P1 |
| Resolution range (outer) | 39.5–2.8 (2.9–2.8) | 16.0–2.2 (2.32–2.2) | 17–2.3 (2.42–2.3) |
| Observed reflections | 232,001 | 543,567 | 722,760 |
| Unique Reflections | 14,573 | 26,342 | 23,375 |
| Completeness (outer) | 99.0 (98.4) | 93.5 (93.1) | 94.4 (92.0) |
| Rmergeb (outer shell) | 13.5 (38.8) | 9.6 (36.3) | 8.3 (16.4) |
| I/σI (outer shell) | 15.4 (3.5) | 6.3 (1.9) | 6.1 (4.0) |
a Dimensions a, b, c given in Å, and α, β, γ given in degrees.
b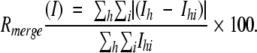
Ih is the weighted mean measured intensity of the observations Ihi in which the intensities of the symmetry related reflections, which should be the same, are compared. Rmerge (I) gives an estimate of their disagreement.
Discussion
We have cloned and expressed a recombinant form of the four SCRs of human DAF as inclusion bodies in E. coli and then isolated, refolded, and purified it to produce a functional protein that inhibits complement activation in vitro at a potency comparable with soluble DAF expressed in a eukaryotic system (P. pastoris). The ability to produce biologically active multi-SCR domains in bacteria was first demonstrated for a 3-SCR fragment of human CR1 (Dodd et al. 1995) and then extended to a 4-SCR unit from the same protein (Mossakowska et al. 1999) and a 4-SCR form of rat Crry (Fraser et al. 2002). The refolding protocol employed may well be general for multi-SCR proteins but the practical limit on the number of SCRs in such constructs has not yet been determined. In the case of human DAF, the recombinant material described here (EcDAF) was also able to form regular crystals that diffracted to 2.2 Å, implying that EcDAF is a homogenous species and is likely to be correctly folded.
Our results are consistent with previous conclusions regarding the roles of both the S/T rich membrane-proximal region (Fodor et al. 1995) and the N-glycan (Brodbeck et al. 2000), neither of which appear to be essential for the complement-regulatory function of nonmembrane bound DAF. EcDAF was active in both classical and alternative pathway decay-acceleration and antihemolytic assays. In the case of the hemolytic assays, the apparent potency of the protein was lower against the alternative pathway than the classical pathway, whereas in the decay accleration assay, the reverse was true. This can be attributed to the strong dependence of the hemolytic assays on serum concentration (see Fig. 5C, D ▶).
We have also modified EcDAF by site-specific attachment of a membrane localizing peptide at the C-terminus and EcDAF-MP also bound to cell surfaces in vitro under conditions where EcDAF itself did not bind. Under the assay conditions used, the soluble product (EcDAF-MP) had a classical pathway antihemolytic potency apparently greater than that of native GPI-anchored DAF, although this comparison may be affected by the use of detergent to solubilize nDAF. DAF is thus added to a growing list of proteins (including complement regulatory proteins such as CR1 fragments (Smith and Smith 2001), rat Crry (Fraser et al. 2002), and CD59 (Fraser et al. 2003), to which GPI anchor-like membrane localizing properties can be conferred by an efficient posttranslational modification process using synthetic peptides. We observed that although the attachment of the membrane-localizing peptide produced a large (>100-fold) increase in potency against the classical pathway (Fig. 5 ▶), no such effect was seen against the alternative pathway. To establish if this was due to the higher concentrations of serum used in the alternative pathway assays preventing membrane binding, “wash” experiments were performed in which rabbit or guinea pig erythrocytes were loaded with EcDAF-MP in the absence of serum, washed, and then challenged. The results suggested that under the conditions of the alternative pathway assay (unlike those of the classical pathway assay), EcDAF-MP was not able to bind sufficiently strongly to the target erythrocytes to confer intrinsic protection against challenge by a relatively high concentration of serum. Other studies have suggested that the effect of serum is largely mediated by albumin and can be circumvented by altering the targeting tail structure (D. Esser, L. Fiedler, J.R. Betley, I. Dodd, P.J.E. Rowling, G.P. Smith, R.G. Oldroyd, L. Affleck, S. Ridley, and R.A.G. Smith, unpubl.). The effect of albumin does not appear to abrogate the activity of membrane-targeted complement regulators delivered to sites of disease in vivo (Linton et al. 2000; Pratt et al. 2003).
DAF has significant potential as a therapeutic agent (Moran et al. 1992), and recent studies have demonstrated therapeutic utility for DAF fusions with immunoglobulin Fc domains (Harris et al. 2002; Yanagawa et al. 2003). However, except in transgenic systems (Fecke et al. 2002), the potential for an intrinsically acting membrane bound form of DAF has not been realized. In part, this is because of the difficulties in obtaining sufficient quantities of nDAF for evaluation, and in part because of the relatively low potency of non-GPI-anchored forms (Moran et al. 1992; Walter et al. 1992). The strategy adopted here circumvents these problems first by providing for a potentially large-scale and low-cost production system, and second, because of the increased potency of the modified soluble derivatives. In addition, myristoylated peptide modifications permit transfer of modified materials from the plasma phase to vascular sites in vivo (Smith 2002) and the distribution of such modified proteins in vivo can be manipulated by varying the structure of the peptide “tail” (D. Esser, L. Fiedler, J.R. Betley, I. Dodd, P.J.E. Rowling, G.P. Smith, R.G. Oldroyd, L. Affleck, S. Ridley, and R.A.G. Smith, unpubl.).
The crystallization of a biologically active four-domain form of DAF suggests that this material is correctly folded, and the collection of high-quality and reproducible diffraction data (Table 1) has permitted the determination of an atomic structure for this protein. This has been extended to the modeling of the complete extracellular region of the molecule as well as its interaction with Factor B (Lukacik et al. 2004). The complete structural description also sheds light on the function of the MP targeting peptides. Despite being much smaller than the combined GPI anchor and S/T region, the MP peptide appears to be capable of localizing the SCRs of DAF in a functionally efficient orientation on the outer cell membrane. Figure 8 ▶ compares full-length DAF (based on the atomic structure including the S/T-rich region) with EcDAF-MP, in which that region is replaced by the MP peptide. Clearly, the region of DAF implicated in regulation of the classical pathway convertases (SCRs 2 and 3; Brodbeck et al. 1996) is spaced very differently from the membrane surface in the two cases. Although the S/T rich region may be assumed to be flexible, the small size of the MP peptide (at least half of which is probably in direct contact with the membrane), provides a constraint on the distance of the C2a component of the classical pathway C3 convertase from cell surfaces.
Figure 8.

A comparison of full-length DAF (right) with EcDAF-MP (left) based on the atomic structure including the modeled S/T- rich region (Lukacik et al. 2004). In EcDAF-MP, the S/T region is replaced by the MP peptide. The structures are modeled upon a simulated phospholipid bilayer. Both S/T and MP peptides are depicted as maximally extended (the latter is proposed to be in contact with the bilayer through the myristoyl group and at least the first three lysine residues).
In conclusion, the atomic structure of DAF, when combined with the use of membrane-orienting probes, significantly extends our knowledge of structure–function relationships that have previously been based solely on partial structures of soluble fragments (e.g., Uhrinova et al. 2003; Williams et al. 2003). Derivatives of the EcDAF-MP type also provide powerful tools for probing the biological roles and therapeutic potential of DAF.
Materials and methods
Proteins and chemicals
GPI-anchored DAF (nDAF) was prepared as previously described (Morgan 2000). PpDAF was prepared by the method of Powell et al. (1997). SCRs(1–3) of CR1 were prepared as described by Mossakowska et al. (1999).
Chemicals and reagents were supplied by Sigma-Aldrich unless otherwise stated.
Phosphate-buffered saline (Dulbecco A) was from Oxoid Ltd. Sodium chloride and Tween 20 were from Merck; EDTA and urea were from Prolabo; Novex 4%–12% Bis Tris Nupage gels, and Mark 12 protein molecular weight standards were from Invitrogen.
N-myristoyl GlySerSerLysSerProSerLysLysLysLysLysLysPro-GlyAspCys-(S-2-thiopyridyl) carboxamide (APT542) was prepared as previously described (Smith et al. 1998; Smith and Smith 2001). Tris-(2-carboxyethyl) phosphine (TCEP) was supplied by Pierce.
Alexa488 conjugated goat antimouse IgG was from Molecular Probes.
Cells and serum
Rabbit antibody sensitized sheep erythrocytes were purchased from Diamedix. Rabbit and guinea pig erythrocytes in Alsever’s buffer were purchased from TCS Biologicals. Normal human serum was prepared from a pool of volunteers by standard methods.
Cloning and expression
The gene encoding the entire human DAF sequence was obtained by RT-PCR from a human brain total RNA preparation (Origene Inc.) using primers: (GCATATGACCGTCGCGCGGCCGAGC) DAF-F and (GGAATTCTAAGTCAGCAAGCCCATGGTTACT) DAF-R.
The product of the PCR was T-cloned into the plasmid pUC57/T (MBI Fermentas) and authenticated by nucleotide sequence analysis.
A fragment of the full-length gene encompassing the four SCRs of DAF, along with an N-terminal Met-Gln addition and a C-terminal Cys (MQD35-G285C) was generated from this plasmid template by PCR using primers DAF1–4F (GCATATGCAGGAC TGTGGCCTGCCGCCGGACGTT) and DAF4R (GGAATTC TATCAGCATCCTCTGCATTCAGGTGGTGG) that incorporated NdeI and EcoRI sites for subsequent subcloning steps, and the product T-cloned. The DAF1–4 DNA fragment was inserted, and excised from this plasmid using the NdeI and EcoRI restriction sites incorporated into the primers. This was followed by ligation into the pET-26b(+) vector downstream of the T7 RNA polymer-ase promoter (Novagen) for expression in E. coli (see below).
Expression was carried out in E. coli UT5600(DE3). A 25-mL culture in LB-kanamycin was grown overnight at 37°C. Ten milliliters of the overnight culture was then transferred into 1 liter of EZMix (Sigma)-kanamycin in a 5l-culture flask. The culture was grown for 75 min (OD600 = 0.6) and then induced by adding 1 mL of 1 M IPTG. The induced culture was grown for a further 3 h, after which it was centrifuged (9000g for 30 min) to obtain bacterial pellets.
Isolation of inclusion bodies, refolding and purification
Frozen cell pellets were resuspended in 20 mM Tris•HCl, 1 mM EDTA (pH 8.0 [40 mL]), and passed through a homogenizer twice (Emulsiflex-C5, Avestin, 12 kpsi). The homogenate was centrifuged (10.000g/4°C/30 min) and the supernatant discarded. The pellet containing the inclusion bodies was washed twice in PBS/0.05% (w/v) Tween 20, centrifuged (10,000g/4°C/30 min) and the supernatants discarded. The pellet was resuspended in 8 M urea, 25 mM dithiothreitol, 1 mM EDTA, 0.1 M Tris (pH 8.0 [10 mL]) to facilitate solubilization. After 2 h (room temperature/gentle stirring) the pH of the solution was adjusted to 3.0–4.0 with HCl, and the solution was centrifuged (10,000g/4°C/30 min) before dialysis in 6 M urea, 10 mM HCl (pH 3.0–4.0). After dialysis, the major contaminant (OmpA) precipitated and was removed by centrifugation (10,000g/4°C/30 min).
Folding of solubilized protein was carried out in 0.02 M ethanolamine, 1 mM EDTA, 0.5 M arginine, 1 mM cysteine, 2 mM cystine (pH 11.0) using rapid dilution (1:50 v/v) with three additions of the same volume over a 32-h period. Additions were drop-wise with stirring, and then the solution, which remained clear, was left static at 4°C. This material was ultrafiltered using an Amicon YM10 membrane (Millipore), the retentate (20 mL) was centrifuged (20,000g/4°C/20 min). Soluble EcDAF was buffer-exchanged into PBS using a G25 Sephadex column (G25 Sephadex medium grade, Amersham Biosciences) by monitoring the A280 of the eluate and collecting the Vo fraction. The resulting EcDAF preparation was approximately 80% pure, as determined by SDS-PAGE using staining with Coumassie brilliant blue R250 (Fig. 2 ▶, lane 8).
For crystallization studies, the EcDAF preparation was buffer exchanged into 50 mM Tris•HCl (pH 7.5), 150 mM NaCl and further purified by gel filtration on Hi Load 26/60 Superdex S75 (Amersham Biosciences) run at 18°C.
The resultant preparation was >95% pure as determined by SDS-PAGE/CBBR250.
Protein concentrations
Protein concentrations were determined by quantitative SDS-PAGE/CBBR250 analysis (UVP Gel Documentation System GDS8000) and analyzed using GelWorks 1D Analysis software using an in-house protein standard and/or by A280 readings using a calculated molar extinction coefficient of 38,880 M−cm− for EcDAF (Protean) or 36,840 M− cm− (ProtParam; Gill and von Hippel 1989). Concentration of nDAF was determined by dye binding assay using BSA as a standard (Bradford 1976).
Modification with APT542
EcDAF at 2 mg/mL (71 μM) in PBS Dulbeccos A was treated with threefold molar excess of TCEP and incubated overnight at room temperature to cleave the exo-disulfide bonded between C-terminal protein cysteine and cysteine derived from the refold buffer. DAF was then incubated with fivefold molar excess of the myristoylated peptide APT542 for 2 h at room temperature. In certain experiments, residual APT542 was removed by gel filtration using PD-10 G25 Sephadex columns as recommended by the manufacturer (Amersham Biosciences).
Assays
For studies of classical pathway activation, sensitized sheep erythrocytes (100 μL) were incubated with DAF proteins (50 μL) and 1 in 100 diluted normal human serum (50 μL) added for 1 h at 37°C. Cells were pelleted at 430 rcf for 3 min and lysis was determined by measuring the OD of the supernatant at 410 nm. The diluent for this assay was: 0.1 M HEPES, 0.15 M NaCl, 0.1% gelatin (pH 7.4). CHAPS (3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate) (0.05% w/v) was used in the diluent for assay of nDAF to retain solubility. For studies of alternative pathway activation, rabbit erythrocytes in Alsever’s buffer were washed and diluted 1 in 50 (~5 × 108cells/mL), 50 μL of cells were incubated with DAF proteins (50 μL), and 1 in 8 normal human serum (100 μL) for 1 h at 37°C. Cells were pelleted at 430 rcf for 3 min and lysis was determined by measuring optical density at 410 nm of the supernatant. The alternative pathway diluent was 0.3 M HEPES, 0.15 M NaCl, 8 mM EGTA, 5 mM MgCl2, 0.1% gelatin (pH 7.4). The “wash” variant of this assay (Fraser et al. 2002) was carried out by preincubation of the cells with varying concentrations of an agent in the absence of serum, pelleting, and resuspension three times in the above buffer, followed by serum challenge.
Antihemolytic activity in both assays was expressed as the concentration of the agent giving 50% of the total cell lysis (IC50).
Fluid-phase assays of decay accleration were performed as described by Nickells and Atkinson (1997) using purified components (Advanced Research Technologies) and C3a immunoassay (Quidel).
Crystallization and diffraction data collection
A number of crystallization conditions were initially screened by the sitting drop vapor diffusion method in Linbro plates.
After several rounds of optimization the following protocol was devised to reliably obtain diffraction quality crystals: 0.5 μL of well solution and 0.5 μL (9.6 mg/mL) of protein were mixed on a polypropylene microbridge (Harlos 1992). The well solution contained 0.2 M ammonium sulfate, 20% w/v methoxy polyethyleneglycol 5000, 0.1 M sodium acetate (pH 4.6), 10% glycerol. The 1 μL drop was allowed to preequilibrate at 2°C in a sealed well of a Linbro plate for 12 h before seeding (Stura 1991). Seeding was performed by transferring dilutions of a crushed crystal stock into preequilibrated drops. Approximately 15 DAF crystals in a 1-μL drop were crushed, and the drop contents were diluted 10-fold and 100-fold. An animal hair fiber was used to transfer the diluted stocks into a preequilibrated drop. This method increased the frequency of crystal appearance and allowed for growth of larger crystals. The largest crystals could be obtained by using more dilute stocks (e.g., 100-fold). Diffraction quality crystals were obtained by this method in approximately 6 days.
Immediately prior to the diffraction experiment, crystals grown in the presence of 10% glycerol were transferred to a solution replicating mother liquor conditions but with a higher (16%) glycerol concentration. This glycerol concentration was just sufficient to achieve full cryoprotection of the crystal. The crystals were then flash frozen in a cold stream of nitrogen gas at 100 K. X-ray diffraction data were collected on an ADSC Q4 CCD-based detector at beamline ID14-2 European Synchrotron Radiation Facility (ESRF), Grenoble, for data set 1; on an ADSC detector at beamline 9.6, Synchrotron Radiation Source (SRS), Daresbury, UK for data set 2, and on a MAR345 detector utilizing a Rigaku RU200H rotating anode X-ray generator for data set 3 (University of Oxford).
A number of data sets were collected from these crystals. Data sets were integrated and scaled using either the HKL package (Otwinowski and Minor 1997) or programs MOSFLM and SCALA available as part of the CCP4 suite of programs (Collaborative Computational Project 1994).
Acknowledgments
We gratefully acknowledge the contributions of Katie Thurston and Louise Mansfield to several aspects of this work. We also thank Ian Dodd and Seán Gallagher for advice and assistance in protein refolding and isolation and Andrew Heinrich for fermentation support. P.L. was funded by a Cooperative Award in Science and Engineering of the Medical Research Council (UK) to S.M.L. and R.S. We also thank the staff of the ESRF Grenoble, SRS Daresbury, Elspeth Garman, and Ed Lowe for assistance with in-house and synchrotron data collection, and Simon Ridley for critical review of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
GPI, glycophosphatidyl inositol
PpDAF, human DAF1–4 expressed in Pichia pastoris, N glycosylated and with an oligohistidine tag
EcDAF, nonglycosylated human DAF 1–4 expressed in Escherichia coli
nDAF, human native glycosylated (GPI-anchored) DAF from erythrocytes
EcDAF-MP, soluble E. coli human DAF linked through a C-terminal cysteine to the myristoylated peptide APT542
PCR, polymerase chain reaction
SCR, short consensus repeat
TCEP, Tris-(2-carboxyethyl) phosphine
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03455604.
References
- Barlow, P.N., Norman, D.G., Steinkasserer, A., Horne, T.J., Pearce, J., Driscoll, P.C., Sim, R.B., and Campbell, I.D. 1992. Solution structure of the fifth repeat of factor H: A second example of the complement control protein module. Biochemistry 31 3626–3634. [DOI] [PubMed] [Google Scholar]
- Bradford, M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brodbeck, W.G., Liu, D., Sperry, J., Mold, C., and Medof, M.E. 1996. Localization of classical and alternative pathway regulatory activity with the decay-accelerating factor. J. Immunol. 156 2528–2533. [PubMed] [Google Scholar]
- Brodbeck, W.G., Kuttner-Kondo, L., Mold, C., and Medof, M.E. 2000. Structure/function studies of human decay-accelerating factor. Immunology 101 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project. 1994. The CCP Suite: Programs for protein crystallography. Acta Crystallogr. D. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Coyne, K.E., Hall, S.E., Thompson, S., Arce, M.A., Kinoshita, T., Fujita, T., Anstee, D.J., Rosse, W., and Lublin, D.M. 1992. Mapping of epitopes, glycosylation sites, and complement regulatory domains in human decay accelerating factor. J. Immunol. 149 2906–2913. [PubMed] [Google Scholar]
- Dodd, I., Mossakowska, D.E., Camilleri, P., Haran, M., Hensley, P., Lawlor, E.J., McBay, D.L., Pindar, W., and Smith, R.A. 1995. Overexpression in Escherichia coli , folding, purification and characterisation of the first three short consensus repeat modules of human complement receptor type 1. Protein Exp. Purif. 5 727–736. [DOI] [PubMed] [Google Scholar]
- Dong, J., Pratt, J.R., Smith, R.A., Dodd, I., and Sacks, S.H. 1999. Strategies for targeting complement inhibitors in ischaemia/reperfusion injury. Mol. Immunol. 36 957–963. [DOI] [PubMed] [Google Scholar]
- Fecke, W., Long, J., Richards, A., and Harrison, R. 2002. Protection of hDAF-transgenic porcine endothelial cells against activation by human complement: Role of the membrane attack complex. Xenotransplantation 9 97–105. [DOI] [PubMed] [Google Scholar]
- Fodor, W.L., Rollins, S.A., Guilmette, E.R., Setter, E., and Squinto, S.P. 1995. A novel bifunctional chimeric complement inhibitor that regulates C3 convertase and formation of membrane attack complex. J. Immunol. 155 4135–4138. [PubMed] [Google Scholar]
- Fraser, D.A., Harris, C.L., Smith, R.A, and Morgan, B.P. 2002. Bacterial expression and membrane targeting of the rat complement regulator Crry: A new model anticomplement therapeutic. Protein Sci. 11 2512–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, D.A., Harris, C.L., Williams, A.S., Mizuno, M., Gallagher, S., Smith, R.A., and Morgan, B.P. 2003. Generation of a recombinant membrane-targeted form of the complement regulator CD59: Activity in vitro and in vivo. J. Biol. Chem. 278 48921–48927. [DOI] [PubMed] [Google Scholar]
- Fujita, T., Inoue, T., Ogawa, K., Idia, K., and Tamura, N. 1987. The mechanism of action of decay-accelerating factor (DAF). DAF inhibits the assembly of C3 convertase by dissociating C2a and Bb. J. Exp. Med. 166 1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garman, E. 1999. Cool data: Quantity and quality. Acta Crystallogr. D Biol. Crystallogr. 55 1641–1653. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Harlos, K. 1992. Micro-bridges for sitting drop crystallizations. J. Appl. Crystallogr. 25 536–538. [Google Scholar]
- Harris, C.L., Williams, A.S., Linton, S.M, and Morgan, B.P. 2002. Coupling complement regulators to immunoglobulin domains generates effective anti-complement reagents with extended half-life in vivo. Clin. Exp. Immunol. 129 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel, F., Hermann, R., Brandlein, S., Krenn, V., Schmausser, B., Geis, S., Muller-Hermelink, H.K., and Vollmers, H.P. 2001. Regulation of the new coexpressed CD55 (decay-accelerating factor) receptor on stomach carcinoma cells involved in antibody SC-1-induced apoptosis. Lab Invest. 81 1553–1563. [DOI] [PubMed] [Google Scholar]
- Hourcade, D., Holers, V.M., and Atkinson, J.P. 1989. The regulators of complement activation (RCA) gene cluster. Adv. Immunol. 45 381–416. [DOI] [PubMed] [Google Scholar]
- Kuttner-Kondo, L.A., Mitchell, L., Hourcade, D.E., and Medof, M.E. 2001. Characterization of the active sites in decay-accelerating factor. J. Immunol. 167 2164–2171. [DOI] [PubMed] [Google Scholar]
- Linton, S.M., Williams, A.S., Dodd, I., Smith, R., Williams, B.D., and Morgan, B.P. 2000. Therapeutic efficacy of a novel membrane-targeted complement regulator in antigen-induced arthritis in the rat. Arthritis Rheum. 43 2590–2597. [DOI] [PubMed] [Google Scholar]
- Lublin, D. and Coyne, K.E. 1991. Phospholipid-anchored and transmembrane versions of either decay-accelerating factor of membrane cofactor protein show equal efficiency in protection from complement-mediated cell damage. J. Exp. Med. 174 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacik, P., Roversi, P., White, J., Esser, D., Smith, G.P., Billington, J., Williams, P.A., Rudd, P.A., Wormald, M.R., Harvey, D.J., et al. 2004. Complement regulation at the molecular level: The structure of decay-accelerating factor. Proc. Natl. Acad. Sci. 101 1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof, M.E., Kinoshita, T., and Nussenzweig, V. 1984. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J. Exp. Med. 160 1558–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof, M.E, Walter, E.I., Roberts, W.L., Haas, R., and Rosenberry, T.L. 1986. Decay accelerating factor of complement is anchored to cells by a C-terminal glycolipid. Biochemistry 25 6740–6747. [DOI] [PubMed] [Google Scholar]
- Moran, P., Beasley, H., Gorrell, A., Martin, E., Gribling, P., Fuchs, H., Gillett, N., Burton, L.E., and Caras, I.W. 1992. Human recombinant soluble decay accelerating factor inhibits complement activation in vitro and in vivo. J. Immunol. 149 1736–1743. [PubMed] [Google Scholar]
- Morgan, B.P. 2000. Immunoaffinity methods for purification of complement components and regulators. Methods Mol. Biol. 150 53–60. [DOI] [PubMed] [Google Scholar]
- Mossakowska, D., Dodd, I., Pindar, W., and Smith, R.A. 1999. Structure–activity relationships within the N-terminal short consensus repeats (SCR) of human CR1 (C3b/C4b receptor, CD35): SCR3 plays a critical role in inhibition of the classical and alternative pathways of complement activation. Eur. J. Immunol. 29 1955–1965. [DOI] [PubMed] [Google Scholar]
- Nickells, M.W. and Atkinson, J.P. 1997. Identification, functional assessment and quantitation of receptors and membrane regulatory proteins whose ligands are C3 and C4. In Complement: A practical approach (ed. A.A. Dodds and R.B. Sim), pp. 69–91. Oxford University Press, Oxford.
- Nishikawa, K., Matsuo, S., Tamai, H., Okada, N., and Okada, H. 1998. Tissue distribution of the guinea-pig decay-accelerating factor. Immunology 95 302–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, D.G., Barlow, P.N., Baron, M., Day, A.J., Sim, R.B., and Campbell, I.D. 1991. Three-dimensional structure of a complement control protein module in solution. J. Mol. Biol. 219 717–725. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Academic Press, New York. [DOI] [PubMed]
- Powell, R.M., Ward, T., Evans, D.J., and Almond, J.W. 1997. Interaction between echovirus 7 and its receptor, decay-accelerating factor (CD55): Evidence for a secondary cellular factor in A-particle formation. J. Virol. 71 9306–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt, J.R., Jones, M.E., Dong, J., Zhou, W., Chowdhury, P., Smith, R.A., and Sacks, S.H. 2003. Nontransgenic hyperexpression of a complement regulator in donor kidney modulates transplant ischemia/reperfusion damage, acute rejection, and chronic nephropathy. Am. J. Pathol. 163 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayama, K., Shiraishi, S., Shirakata, Y., Kobayashi, Y., and Miki, Y. 1991. Characterization of decay-accelerating factor (DAF) in human skin. J. Invest. Dermatol. 96 61–64. [DOI] [PubMed] [Google Scholar]
- Simons, K. and Ikonen, E. 1997. Functional rafts in cell membranes. Nature 387 569–572. [DOI] [PubMed] [Google Scholar]
- Smith, R.A.G. 2002. Targeting anticomplement agents. Biochem. Soc. Trans. 30 1037–1041. [DOI] [PubMed] [Google Scholar]
- Smith, G.P. and Smith, R.A.G. 2001. Membrane-targeted complement inhibitors. Mol. Immunol. 38 249–255. [DOI] [PubMed] [Google Scholar]
- Smith, R.A.G., Dodd, I., and Mossakowska, D.E. 1998. Conjugates of soluble peptide compounds with membrane binding agents. Int. Patent Pub. WO 98/02454.
- Spiller, O.B., Hanna, S.M., and Morgan, B.P. 1999. Tissue distribution of the rat analogue of decay-accelerating factor. Immunology 97 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stura, E.A. 1991. Application of the streak-seeding technique in protein crystallization. J. Cryst. Growth 110 270–282. [Google Scholar]
- Uhrinova, S., Lin, F., Ball, G., Bromek, K., Uhrin, D., Medof, M.E., and Barlow, P.N. 2003. Solution structure of a functionally active fragment of decay-accelerating factor. Proc. Natl. Acad. Sci. 100 4718–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneker, G.T., Vodegel, R.M., Okada, N., Westerhof, W., Bos, J.D., and Asghar, S.S. 1998. Relative contributions of decay accelerating factor (DAF), membrane cofactor protein (MCP) and CD59 in the protection of melanocytes from homologous complement. Immunobiology 198 476–484. [DOI] [PubMed] [Google Scholar]
- Walter, E.I., Ratnoff, W.D., Long, K.E., Kazura, W., and Medof, M.E. 1992. Effect of glycoinositolphospholipid anchor lipid groups on functional properties of decay-accelerating factor protein in cells. J. Biol. Chem. 267 1245–1252. [PubMed] [Google Scholar]
- Williams, P., Chaudhry, Y., Goodfellow, I.G., Billington, J., Powell, R., Spiller, O.B., Evans, D.J., and Lea, S. 2003. Mapping CD55 function. The structure of two pathogen binding domains at 1.7 Å. J. Biol. Chem. 278 10691–10696. [DOI] [PubMed] [Google Scholar]
- Wiles, A.P., Shaw, G., Bright, J., Perczel, A., Campbell, I.D., and Barlow, P.N. 1997. NMR studies of a viral protein that mimics the regulators of complement activation. J. Mol. Biol. 272 253–265. [DOI] [PubMed] [Google Scholar]
- Yanagawa, B., Spiller, O.B., Choy, J., Luo, H., Cheung, P., Zhang, H.M., Goodfellow, I.G., Evans, D.J., Suarez, A., Yang, D., et al. 2003. Coxsackievirus B3-associated myocardial pathology and viral load reduced by recombinant soluble human decay-accelerating factor in mice. Lab. Invest. 83 75–85. [DOI] [PubMed] [Google Scholar]
- Young, S.L., Lessey, B.A., Fritz, M.A., Meyer, W.R., Murray, M.J., Speckman, P.L., and Nowicki, B.J. 2002. In vivo and in vitro evidence suggest that HB-EGF regulates endometrial expression of human decay-accelerating factor. J. Clin. Endocrinol. Metab. 87 1368–1375. [DOI] [PubMed] [Google Scholar]