

Abstract
Streptococcal pyrogenic exotoxin A (SpeA1) is a bacterial superantigen associated with scarlet fever and streptococcal toxic shock syndrome (STSS). SpeA1 is found in both monomeric and dimeric forms, and previous work suggested that the dimer results from an intermolecular disulfide bond between the cysteines at positions 90 of each monomer. Here, we present the crystal structure of the dimeric form of SpeA1. The toxin crystallizes in the orthorhombic space group P212121, with two dimers in the crystallographic asymmetric unit. The final structure has a crystallographic R-factor of 21.52% for 7248 protein atoms, 136 water molecules, and 4 zinc atoms (one zinc atom per molecule). The implications of SpeA1 dimer on MHC class II and T-cell receptor recognition are discussed.
Keywords: X-ray crystallography, superantigen, streptococcal pyrogenic exotoxin A1, T-cell receptor, MHC class II molecule
The streptococcal and staphylococcal superantigens are exotoxins of ~25,000 Da that induce fever, hypotension, and multiorgan failure in infected individuals. In contrast to conventional antigens, which are presented to the T-cell receptor after they have been processed and bound to the peptide groove of class II MHC (Marrack and Kappler 1990), superantigens act by binding simultaneously to the T-cell receptor and the class II MHC molecule outside of the peptide-binding groove. Superantigens interact with the class II MHC molecules either via a generic MHC binding site that is conserved among members of the bacterial superantigen family, or via a high-affinity zinc site. The presence of this zinc binding site is varied among superantigens: Some have no zinc-binding sites at all (e.g., SEB), some have one zinc site (e.g., SEA), and some have multiple zinc binding sites (e.g., SpeH). In addition, superantigens can act either as monomers (e.g., SEB) or dimers (e.g., SpeC; for a recent review, see Baker and Acharya 2003). Thus, there are numerous strategies used by these toxins to interact with class II MHC molecule.
Streptococcal pyrogenic exotoxin A1 (SpeA1) is produced by Streptococcus pyogenes strains associated with scarlet fever and streptococcal toxin shock, and the toxin has been linked to early events leading to rheumatic fever and guttate psoriasis (Hauser et al. 1991). Many of the biological effects of SpeA1 are attributed to the production of cytokines by the stimulated T-cell population. SpeA1 contains a generic class II MHC binding site. In addition, the toxin contains a zinc-binding site, but the role of this site in class II binding has not been established. Purification of recombinant SpeA1 from Escherichia coli yields both dimeric and monomeric forms of the toxin in approximately equal amounts (Papageorgiou et al. 1999). The crystal structure of monomeric SpeA1 (Papageorgiou et al. 1999; Baker et al. 2001) suggests that the dimer form results from the formation of a disulfide bond between the free cysteines at positions 90 (Cys 90) in the monomers. Cys 90 is located in a flexible loop and, as such, would be relatively free to form a disulfide bond with Cys 90 of a neighboring SpeA1 molecule. The advantage provided by dimerization of SpeA1 is not clearly evident. To further understand the binding interaction between this toxin and class II MHC, and to examine the potential role of zinc in the binding, the crystal structure of the dimeric form of SpeA1 was determined. The structure presented here confirms the role of Cys 90 in dimer formation. Class II MHC binding and the physiological role of SpeA1 dimer in the disease process are discussed.
Results and Discussion
Quality of the structure
The dimer form of SpeA1 crystallizes in the space group P212121, with unit cell dimensions of a = 53.27, b = 127.18, c = 148.04 Å. This differs from the cell dimensions of monomeric SpeA1, which crystallizes in the space group P21212 and has a unit cell of a = 126.9, b = 101.3, c = 82.0 Å. The crystallographic data processing and refinement statistics for dimeric SpeA1 are shown in Table 1.
Table 1.
Data collection and refinement statistics
| Cell dimensions (Å) | a = 53.3, b = 127.2, c = 148.0 |
| Space group | P212121 4 mol/a.u. |
| Resolution (Å) | 40.0–2.6 |
| No. of measurements | 525,004 |
| No. of unique reflections | 31,727 |
| Completeness | 88.8 (83.3)a |
| 1/σ | 16.6 (12.2) |
| Rmerge (%)b | 13.0 (25.3) |
| Refinement | |
| Rcryst (%)c | 21.52 |
| Rfree (%)d | 28.22 |
| No. of protein atoms | 7248 |
| No. of water molecules | 136 |
| Temperature factors (Å2) | |
| Main-chaine | 24.1, 24.5, 25.2, 26.2 |
| Side-chaine | 24.9, 25.8, 26.3, 27.4 |
| Zinc ionse | 33.2, 33.6, 36.0, 56.1 |
| Solvent | 19.8 |
| RMSD in bond lengths (Å) | 0.007 |
| RMSD in bond angles (Å) | 1.37 |
a Outermost shell 2.69 to 2.6 Å.
b Rmerge = ∑(|Ij − 〈I〉|)/∑ 〈I〉, where Ij is the observed intensity of relfection j and 〈I〉 is the average intensity of multiple observations.
c Rcryst = ∑|Fo| − |Fc|/∑|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
d Five percent of the data that were used for the calculation of Rfree were randomly excluded from the refinement.
e Temperature factors for individual molecules quoted.
The final model consists of four molecules, or two dimers, in the asymmetric unit containing 7248 nonhydrogen protein atoms, four zinc ions, and 138 water molecules. Dimer one is composed of molecule one and molecule three, and dimer two of molecule two and four. The root mean square (RMS) deviation between the dimeric structure and the native structure is 0.55 Å (molecule 1), 0.59 Å (molecule 2), 0.46 Å (molecule 3), and 0.48 Å (molecule 4). The regions that deviate most between the two structures include the flexible disulfide loop and the first six residues at the N terminus (Fig. 1C ▶). Exclusion of these areas from the calculation improves the RMS deviation to 0.33 Å (molecule 1), 0.32 Å (molecule 2), 0.38 Å (molecule 3), and 0.37 Å (molecule 4). The Ramachandran plot for all four molecules shows 84% of the residues in allowed regions and none in disallowed regions. The overall electron density allowed all side chains to be modeled. This is in contrast to the structure of monomeric toxin, in which residues 1, 2, and 221 were not modeled due to poor density and residues 5, 88, 112, 115, 179, and 180 in all four molecules and residues 91 and 92 in molecules 2, 3, and 4 were modeled as alanine due to insufficient density.
Figure 1.
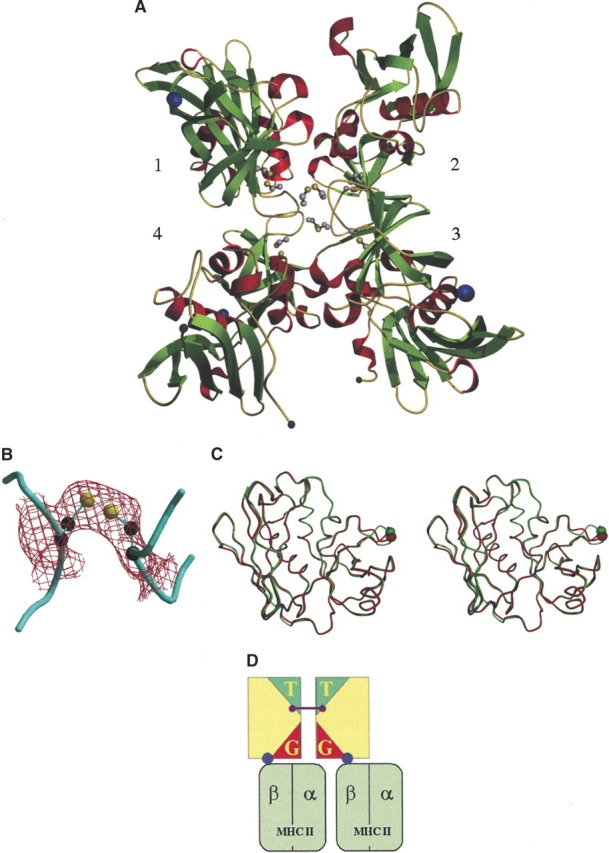
(A) The crystal structure of native SpeA1; the arrangement of the four molecules in the crystallographic asymmetric unit. Inter- and intramolecular disulfide bonds are shown in ball-and-stick representation. (B) 2Fo − Fc electron density map contoured at 1σ clearly shows the density for the intermolecular disulfide bond. (C) Cα trace of molecule 1 from both native (red) and dimeric (green) SpeA1 indicating movement of the disulfide loop. N and C termini are labeled; Cα of Cys 90 is shown as a colored ball. (D) Cartoon representation of SpeA1 dimer possible interactions. The location of the T-cell receptor binding sites (T) in the dimer prevents interaction with TCRs if SpeA1 dimer binds to MHC class II molecules via its zinc site. The location of the generic MHC class II binding sites (G) means that they are unable to interact with MHC II molecules.
The SpeA1 structure is characteristic of the bacterial superantigen family and is comprised of an N- and C-terminal domains separated by a long central α-helix (Papageorgiou and Acharya 1997). The N-terminal tail (residues 1–16) is packed against the β-grasp motif of the C-terminal domain and, as such, is considered part of the C-terminal domain. A single zinc atom is located in the N-terminal domain of the protein and is coordinated by Glu 33, Asp 77, His 106, and His 110. Mutagenesis studies have shown residues Asp 77, His 106, and His 110 to be essential for zinc binding, whereas Glu 33 has a lesser role (Baker et al. 2001).
Although the crystallization conditions for the dimeric form were the same as for the monomeric form of SpeA1, the space group and unit cell dimensions differ for the two forms. This, in addition to the fact that the protein used in the crystallization was purified as a stable disulfide linked form, suggests it is unlikely that the dimer seen here is a crystallization artefact. The dimer comprised of molecule 1 and molecule 3 will be used throughout the discussion.
The disulfide bridge and the dimer interface
The dimeric SpeA1 structure contains three cysteine residues; two of them (Cys 87 and Cys 98) form a disulfide bridge at the top of the N-terminal β-barrel, between strands β4 and β5 (Fig. 1A ▶). The third cysteine (Cys 90) forms an intermolecular disulfide bond with Cys 90 of the second molecule in the dimer. This disulfide bond length is 2.03 Å (Fig. 1B ▶). As in the monomeric form, Cys 90 is solvent exposed and forms part of a flexible disulfide loop. The 10 residues that comprise the disulfide loop of dimeric SpeA1 possess higher temperature factors than the average for the rest of the protein, 52 Å2 (residues 88–93) compared to 25 Å2, indicating that this region retains some of the flexibility seen in the monomeric form. As well as forming an intermolecular disulfide bond, the loop is also involved in crystal packing interactions at the interface between the four molecules. The loop and its surrounding regions show more order than that of the monomeric form, particularly in residues 87 to 88 and 94 to 98. This is reflected in the improved electron density and the ability to model all side chains within this region. In the native structure, residues 88, 91, and 92 were modeled as alanines due to poor density. However, the loop still retains some degree of flexibility, and superposition of the residues within the loop (87–98) shows an RMS deviation of 0.37 Å (molecule 1 to molecule 2), 1.19 Å (molecule 1 to molecule 3), and 1.42 Å (molecule 1 to molecule 4). To form the disulfide bond, the loop moves 1.62b Å/1.96 Å (molecule 1/molecule 2 dimer) from its position in the native structure and 4.24 Å/4.81 Å from its position in the cadmium bound structure (Earhart et al. 2000). Interestingly, in the cadmium bound form of SpeA1 the cadmium interacts with the disulfide loop, especially Cys 90.
The buried surface area at the dimer interface is 544.9 Å2. Apart from the disulfide bond itself, several other residues are in close contact (Table 2). The slightly increased B-factors in this region suggest that the loop still retains some degree of flexibility, and as such, other transient interactions may occur between the two molecules.
Table 2.
Contacts between molecule 1 and molecule 3 in formation of SpeA1 dimer
| Mol1 | Mol3 |
| Leu 42 CD2 | Glu 91 CD OE2 |
| Leu 89 CB | Tyr 88 CZ, OH |
| CD1 | OH |
| O | CE2, CZ, CA |
| Cys 90 CA | Leu 89 O |
| Cys 90 SG | Cys 90 SG |
| Glu 91 CD | Leu 41 CD2 |
| OE1 | Tyr 88 O |
| OE2 | Leu 41 CD2 |
| Leu 89 O, C |
Intermolecular contact cutoff values: C-C 4.1 Å; C-N 3.8 Å; C-O 3.7 Å; O-O 3.3 Å; O-N 3.4 Å; and N-N 3.4 Å
MHC class II recognition
Dimerization of the bacterial superantigens SED (Sundstrom et al. 1996) and SpeC (Roussel et al. 1997) has been suggested as a prerequisite for T-cell activation. The positions of the proposed zinc-MHC binding site and the generic MHC class II binding site within the structure of SpeA1 are similar to the positions of the MHC class II binding sites seen in the structure of enterotoxin SEC2 (Papageorgiou et al. 1995). The role of both the generic (Jardetzky et al. 1994), and zinc sites (Li et al. 2001; Petersson et al. 2001) in complex formation is well established. In all cases, contact with the class II associated antigenic peptide is observed. In the dimeric form, the zinc MHC class II binding sites of both SpeA1 molecules are free to interact with one MHC class II molecule each. In contrast, those residues thought to form a generic MHC II site are buried such that neither of the generic sites in the monomer could bind an MHC class II molecule (Fig. 1D ▶). The conformation of the disulfide loop between Cys87 and Cys98 has been shown to be important for MHC class II binding (Kline and Collins 1996; Roggiani et al. 1997), so even if some degree of flexibility in the dimer interface existed, the fact that it is part of the dimer interface precludes this interaction.
Taking into account the monomeric and dimeric forms of this toxin, it is possible that SpeA1 interacts with MHC class II molecules in three distinct manners: (1) as a monomer via its generic MHC class II binding site (Papageorgiou et al. 1999), (2) as a zinc bound monomer interacting with a single MHC class II molecule via its zinc site, and (3) as a disulfide linked dimer SpeA1 could interact with two MHC class II molecules via the zinc sites of both molecules of the dimer.
Dimerization and the TCR binding site
The prospect that SpeA1 possess several mechanisms for interacting with MHC class II molecules depending on its form opens up the possibility that it may also interact differently with T-cell receptors either in monomeric or in dimeric state. In a monomeric form, SpeA1 bound to a single MHC class II molecule via either its generic or zinc binding site would be able to make direct contact with a TCR in an appropriate manner (Papageorgiou et al. 1999; Baker et al. 2001). However, how it would interact with the TCR as a dimeric toxin is unclear at present.
SpeA1 binds to, and activates, human T cells bearing Vβ12.2, Vβ14.1, and Vβ2.1 (Kline and Collins 1997; Lavoie et al. 1999). It has been shown that SpeA1 uses different residues when binding to TCRs depending on the TCR Vβ chain (Kline and Collins 1997), a mechanism that has become evident from the known superantigen structures so far. The dissociation constant for monomeric SpeA1 with the mouse TCR Vβ8.2 is in the same range as that of SEC2 (6.2 and 7.9 μM, respectively; Malchiodi et al. 1995). Based on the structural similarities of SpeA1 and SEC2, in combination with mutagenesis data, it is proposed that the TCR-binding site encompasses residues from the α2 helix, β2–β3 loop, the disulfide loop, α4–β9 loop, and the α5 helix.
The disulfide loop also appears to be important for interactions with TCRs as well as and MHC class II molecules. In SEC2, four residues from the disulfide loop have the potential to make contacts with the complementarity determining region 1 (CDR1) chain and Gln 72 of Vβ. The disulfide loop of SpeA1 is significantly shorter, and these interactions may not be feasible. However, Cys 90 and Cys 98 from the disulfide loop of SpeA1 are required for stimulation of Vβ 12.2 expressing T cells, with Cys 90 being essential for Vβ12.2 T-cell stimulation, and to a lesser extent for Vβ14.1, whereas it has no observable influence on the stimulation of Vβ2.1 expressing cells (Kline and Collins 1997). Therefore, it is possible that dimerization of SpeA1, although allowing interaction with MHC class II molecules via its zinc binding site, may prohibit the binding to TCR molecules with specific Vβ (Fig. 1D ▶), as the disulfide loop is involved in the dimer interface; as such, dimerization of SpeA1 may serve to further regulate its activity.
How structural data fits with biology
In vitro experiments indicated that monomeric and dimeric SpeA1 are equally active. This might result from reduction of the dimer disulfide bond at the cell surface, or it might reflect a flexibility of the molecule at the dimer interface. This flexibility may allow a different orientation of the generic and zinc-mediated MHC binding sites from those observed in the crystal structure. In combination with the inherent flexibility of its receptors (Davis et al. 2003), that would make it possible for SpeA1 to act directly as a dimer on MHC class II and in turn allow it to bind one or two TCR molecules. It is a formal possibility that the dimer form of SpeA1 is the more potent form in the in vivo conditions. Physiological concentrations of SpeA1 could be extremely low and dimerization might server to increase the local concentration of toxin on the cell surface. A similar model has been postulated for SpeC (Tripp et al. 2003).
Materials and methods
Protein purification and crystallization
Recombinant SpeA1 with an N-terminal His tag was expressed in E. coli BL21 (DE3; Novagen) and purified by immobilized metal ion affinity chromatography as described previously by Papageorgiou et al. (1999). The eluted fusion protein was cleaved with thrombin, and tag and toxin were separated by dialysis against phosphate buffered saline (PBS). Thrombin was removed from the preparation by chromatography over p-amino benziamidine aga-rose (Sigma). Monomeric and dimeric SpeA1 were separated by size exclusion chromatography over a Sephadex G75 column and dialysed against PBS, 0.14 M NaCl, 2.7 mM KCl, 5.4 mM Na2HPO4, and 1.8 mM KH2PO4 before being concentrated with Centricon-10 filter units (Amicon).
Crystals were grown at 16°C by using the hanging drop vapor diffusion method. Small, irregular crystals were produced in 16% to 19% PEG 3350, 0.1 M sodium cacodylate buffer (pH 6.5), and 16% isopropanol. These crystals were then used for microseeding of hanging drops equilibrated against a reservoir solution containing 17% PEG 8000, 0.2 M ammonium sulfate, and 0.1 M sodium cacodylate buffer (pH 6.5). Two distinct crystal morphologies were produced; long needles (as for the monomeric forms of the toxin) and large octagonal plates. The plate octagonal crystals were used for data collection.
Data collection and refinement
X-ray diffraction data to 2.6 Å were collected at 100 K by using the crystallization buffer containing 20% glycerol as a cryoprotectant at Daresbury synchrotron radiation facility on station PX14.2. One hundred fifty images were collected, with an oscillation range of 0.75° per image. Data processing, scaling, and merging were performed by using the HKL program suite (Otwinowski 1993a,b). Phases were determined by using the structure of native SpeA1 (Papageorgiou et al. 1999) with the flexible loop omitted (residues 88 to 93) as a starting model. The initial model was subject to rigid body refinement. Calculation of an (|Fo| − |Fc|) electron density map revealed the presence of a zincion in the zinc binding site of the toxin for all four molecules in the asymmetric unit. The backbone of the flexible loop was easily traceable at this point. Residues 88 to 93 were initially modeled as alanines with side chains being added in subsequent rounds of refinement. The structure was refined by simulated annealing using tight noncrystallographic symmetry restraints and the maximum likelihood target as implemented in the program CNS (Brünger et al. 1998). Rfree and Rcryst were used to follow the progress of refinement (Brünger 1992). At the final stages of refinement, grouped B-factor refinement and release of the noncrystallographic symmetry restraints were performed. SigmaA-weighted electron density maps (|Fo| − |Fc|) and (2|Fo| − |Fc|) were calculated after each cycle of refinement and visualized by using the program O (Jones et al. 1991). Water molecules were added to the model toward the end of refinement with the aid of difference maps and the program water_pick in CNS (Brünger et al. 1998). The final model has a crystallographic R-factor (Rcryst) of 21.52% for all data and an Rfree of 28.22% for 5% of the data omitted (Table 1).
Acknowledgments
We thank the staff at the Synchrotron radiation source (Daresbury, UK) for their help with X-ray data collection.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04826804.
References
- Baker, M.D. and Acharya, K.R. 2003. Superantigens: Structure, function, and diversity. Methods Mol. Biol. 214 1–31. [PubMed] [Google Scholar]
- Baker, M.D., Gutman, D.M., Papageorgiou, A.C., Collins, C.M., and Acharya, K.R. 2001. Structural features of a zinc binding site in the superantigen streptococcal pyrogenic exotoxin A (SpeA1): Implications for MHC class II recognition. Protein Sci. 10 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. The free R-value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–474. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., GrosseKunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Davis, S.J., Ikemizu, S., Evans, E.J., Fugger, L., Bakker, T.R., and van der Merwe, P.A. 2003. The nature of molecular recognition by T cells. Nat. Immunol. 4 217–224. [DOI] [PubMed] [Google Scholar]
- Earhart, C.A., Vath, G.M., Roggiani, M., Schlievert, P.M., and Ohlendorf, D.H. 2000. Structure of streptococcal pyrogenic exotoxin A reveals a novel metal cluster. Protein Sci. 9 1847–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser, A.R., Stevens, D.L., Kaplan, E.L., and Schlievert, P.M. 1991. Molecular analysis of pyrogenic exotoxins from Streptococcus pyogenes isolates associated with toxic shock-like syndrome. J. Clin. Microbiol. 29 1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky, T.S., Brown, J.H., Gorga, J.C., Stern, L.J., Urban, R.G., Chi, Y.I., Stauffacher, C., Strominger, J.L., and Wiley, D.C. 1994. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 368 711–718. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 109–110. [DOI] [PubMed] [Google Scholar]
- Kline, J.B. and Collins, C.M. 1996. Analysis of the superantigenic activity of mutant and allelic forms of streptococcal pyrogenic exotoxin A. Infect. Immun. 64 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997. Analysis of the interaction between the bacterial superantigen streptococcal pyrogenic exotoxin A (SpeA) and the human T-cell receptor. Mol. Microbiol. 24 191–202. [DOI] [PubMed] [Google Scholar]
- Lavoie, P.M., Thibodeau, J., Erard, F., and Sekaly, R.P. 1999. Understanding the mechanism of action of bacterial superantigens from a decade of research. Immunol. Rev. 168 257–269. [DOI] [PubMed] [Google Scholar]
- Li, Y., Li, H., Dimasi, N., McCormick, J.K., Martin, R., Schuck, P., Schlievert, P.M., and Mariuzza, R.A. 2001. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity 14 93–104. [DOI] [PubMed] [Google Scholar]
- Malchiodi, E.L., Eisenstein, E., Fields, B.A., Ohlendorf, D.H., Schlievert, P.M., Karjalainen, K., and Mariuzza, R.A. 1995. Superantigen binding to a T cell receptor β chain of known three-dimensional structure. J. Exp. Med. 182 1833–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack, P. and Kappler, J. 1990. The staphylococcal enterotoxins and their relatives. Science 248 705–711. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. 1993a. DENZO: An oscillation data processing programme for macromolecular crystallography. Yale University, New Haven, CT.
- ———. 1993b. SCALEPACK: Software for the scaling together of integrated intensities measured on a number of separate diffraction images. Yale University, New Haven, CT.
- Papageorgiou, A.C. and Acharya, K.R. 1997. Superantigens as immunomodulators: Recent structural insights. Structure 5 991–996. [DOI] [PubMed] [Google Scholar]
- Papageorgiou, A.C., Acharya, K.R., Shapiro, R., Passalacqua, E.F., Brehm, R.D., and Tranter, H.S. 1995. Crystal structure of the superantigen enterotoxin C2 from Staphylococcus aureus reveals a zinc-binding site. Structure 3 769–779. [DOI] [PubMed] [Google Scholar]
- Papageorgiou, A.C., Collins, C.M., Gutman, D.M., Kline, J.B., O’Brien, S.M., Tranter, H.S., and Acharya, K.R. 1999. Structural basis for the recognition of superantigen streptococcal pyrogenic exotoxin A (SpeA1) by MHC class II molecules and T-cell receptors. EMBO J. 18 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson, K., Hakansson, M., Nilsson, H., Forsberg, G., Svensson, L.A., Liljas, A., and Walse, B. 2001. Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 20 3306–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggiani, M., Stoehr, J.A., Leonard, B.A., and Schlievert, P.M. 1997. Analysis of toxicity of streptococcal pyrogenic exotoxin A mutants. Infect. Immun. 65 2868–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel, A., Anderson, B.F., Baker, H.M., Fraser, J.D., and Baker, E.N. 1997. Crystal structure of the streptococcal superantigen SPE-C: Dimerization and zinc binding suggest a novel mode of interaction with MHC class II molecules. Nat. Struct. Biol. 4 635–643. [DOI] [PubMed] [Google Scholar]
- Sundstrom, M., Abrahmsen, L., Antonsson, P., Mehindate, K., Mourad, W., and Dohlsten, M. 1996. The crystal structure of staphylococcal enterotoxin type D reveals Zn2+-mediated homodimerization. EMBO J. 15 6832–6840. [PMC free article] [PubMed] [Google Scholar]
- Tripp, T.J., McCormick, J.K., Webb, J.M., and Schlievert, P.M. 2003. The zinc-dependent major histocompatibility complex class II binding site of streptococcal pyrogenic exotoxin C is critical for maximal superantigen function and toxic activity. Infect. Immun. 71 1548–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]