

Abstract
Pit viper venoms contain a number of serine proteinases that exhibit one or more thrombin-like activities on fibrinogen and platelets, this being the case for the kinin-releasing and fibrinogen-clotting KN-BJ from the venom of Bothrops jararaca. A three-dimensional structural model of the KN-BJ2 serine proteinase was built by homology modeling using the snake venom plasminogen activator TSV-PA as a major template and porcine kallikrein as additional structural support. A set of intrinsic buried waters was included in the model and its behavior under dynamic conditions was molecular dynamics simulated, revealing a most interesting similarity pattern to kallikrein. The benzamidine-based thrombin inhibitors α-NAPAP, 3-TAPAP, and 4-TAPAP were docked into the refined model, allowing for a more insightful functional characterization of the enzyme and a better understanding of the reported comparatively low affinity of KN-BJ2 toward those inhibitors.
Keywords: venom serine proteinase, thrombin, benzamidine-based inhibitors, homology modeling
Arterial and venous thromboses are major causes of human morbidity and mortality. Strategies to inhibit arterial thrombogenesis focus mainly on drugs that block platelet function, but they often include anticoagulants to prevent fibrin deposition, an approach also used in the prevention and treatment of venous thrombosis because venous thrombi are composed mainly of fibrin and red blood cells (Weitz and Hirsh 2001). Fibrin results from the hydrolysis of fibrinogen, a process catalyzed by thrombin, a procoagulant serine protease.
Snake venoms are complex mixtures containing many different proteins, a number of them interacting with components of the human hemostatic system and playing key roles in hemorrhagia and blood-clotting disorders (Markland 1998). The serine proteinase ones often display fibrinogen-clotting (thrombin-like) and kinin-releasing (kallikrein-like) activities and have been the focus of many studies for their clinical and biochemical applications (Watanabe et al. 2002). Unlike thrombin, these enzymes are incapable of activating factor XIII; thus, they hydrolyze fibrinogen only to produce a soft clot (non-crosslinked fibrins) that is different from the crosslinked hard clots formed by thrombin. Paradoxically, despite their observed thrombin-like coagulant action in vitro, such purified venombins have anticoagulant action in vivo, inducing a benign state of defibrinogenation for which the abnormal soft fibrin clot is thought to be crucial (Markland 1998). Ancrod (from the Calloselasma rhodostoma venom) and Batroxobin (from the Bothrops atrox, moojeni venom) are commercially produced for clinical use as defibrinogenating (anticoagulant) agents to prevent thrombi and improve blood circulation. Other related defibrinogenating peptidases with similar properties have been reported in a number of snake species, this being the case for the kinin-releasing and blood-clotting KN-BJ2 serine proteinase from the Bothrops Jararaca venom, which shares a 69% sequence identity with Batroxobin (ClustalW [Higgins et al. 1994] default alignment) and has already been experimentally characterized for its primary sequence, enzyme specificity, and comparatively low affinity toward a number of benzamidine-based thrombin inhibitors (Serrano et al. 1998).
The functional differences between venombin-like enzymes and other serine proteinases, namely, thrombin, would be best understood if a representative three-dimensional (3D) structure of the former were available. At present, the only experimental structure for a snake venom serine proteinase is the X-ray structure of the Trimeresurus stejnejeri venom plasminogen activator (TSV-PA; Parry et al. 1998), an enzyme with no fibrinogen-clotting activity. Still, KN-BJ2—the target of the present study—shares a 64% sequence identity with TSV-PA (ClustalW default alignment), allowing for a homology modeling of its structure using that plasminogen activator as template. Considering that, during evolution, the 3D structures among homologous proteins have been much better preserved than their primary sequences, comparative modeling is a reliable method to predict a protein’s 3D structure when sequence identity between the target and the template(s) is high, with accuracy comparable to a low-resolution X-ray structure (Martí-Renom et al. 2000; Ramos et al. 2001).
The homology-built 3D computational model of KN-BJ2 proposed in the present contribution has been generated using the TSV-PA structure as major template and a set of five other trypsin-like serine proteinases for local modeling of a few critical loops. The model was further improved by including a set of 22 buried waters, which was considered to be part of the structure by analogy with the pattern of secluded water molecules found in other members of the same enzyme family (Henriques et al. 1997, 2001). This conserved water matrix ensured a structurally preserved protein environment, a good indicator that the model is most probably a good representative structure of the enzyme, and imposed a new type of structural constraint that otherwise would be impossible to define in any subsequent force-field simulation.
In an effort to understand the observed affinity differences between KN-BJ2 and thrombin toward some active-site directed benzamidine-based inhibitors, a docking exercise of three Nα-substituted amidinophenylalanine piperidines into the model structure(s) has been performed, the inhibitors being α-NAPAP (Nα-(2-naphthylsulfonyl-glycyl)-4-amidinophenylalanine piperidine), 3-TAPAP (Nα-tosyl-3-amidinophenylalanine piperidine), and 4-TA-PAP (Nα-tosyl-4-amidinophenylalanine piperidine). The subject of van der Waals (steric) and electrostatic complementarity has been also addressed by analysis of the enzyme interaction (contact) surfaces mapped with the electrostatic potential (ESP). The modeling/docking results have been analyzed and compared with the available crystallographic structures of those three inhibitors complexed with thrombin and/or trypsin (Banner and Hadváry 1991; Turk et al. 1991), allowing for a more critical assessment of the KN-BJ2 model and providing a first explanation for the reduced affinity of this venombin-like enzyme toward the inhibitors in question.
Materials and methods and Results
Multiple sequence alignment
The sequence of KN-BJ2 was obtained from the Swiss-Prot database (Boeckmann et al. 2003; entry name VSP2_BOTJA). The PSI-BLAST (Altschul et al. 1997) engine was used to search for those sequence homologs of the enzyme’s catalytic domain with available X-ray structures. The structures for a few selected sequences producing significant alignments were extracted from the Protein Data Bank (Berman et al. 2000) and relevant information about them is listed in Table 1. The percentage of sequence identity was obtained from a default ClustalW (Higgins et al. 1994) automatic sequence alignment. The alignment was then manually corrected against the secondary structure superposition analysis of the potential templates, within the QUANTA (QUANTA 2000, Molecular Simulations Inc.) Protein Design application.
Table 1.
Serine proteinase structures selected as possible templates for the KN-BJ2 model
| Ramachandran plot statistics (%)a | |||||||||
| Name | Species | In complex with | PDB ID | Resolution (Å) | Core | Allow | Gen/dis | 3D profile scoreb | % of sequence identity |
| TSV-PA | Trimeresurus stejnejeri | CMK-derivative | 1BQY | 2.5 | 83.7 | 16.3 | N/N | 78.1 | 64 |
| 84.2 | 15.3 | Y/N | 80.4 | ||||||
| Trypsin | Bos taurus | Diisopropylphosphoryl | 4PTP | 1.34 | 88.3 | 11.7 | N/N | 82.4 | 41 |
| α-NAPAP | 1PPC | 1.8 | 86.2 | 13.8 | N/N | 78.5 | |||
| 3-TAPAP | 1PPH | 1.9 | 85.6 | 14.4 | N/N | 77.4 | |||
| Kallikrein | Sus scrofa | Hirustasin | 1HIA | 2.4 | 83.2 | 16.3 | Y/N | 79.1 | 40 |
| 84.7 | 15.3 | N/N | 81.7 | ||||||
| BPTI | 2KAI | 2.5 | 81.2 | 17.7 | N/Y | 73.2 | |||
| t-PA | Homo sapiens | Benzyldiamine | 1RTF | 2.3 | 84.6 | 15.0 | Y/N | 84.8 | 31 |
| Factor D | Homo sapiens | Isatoic anhydride inhibitor | 1BIO | 1.5 | 86.3 | 12.1 | Y/N | 81.0 | 29 |
| Thrombin | Bos taurus | Fibrinopeptide A | 1UCY | 2.2 | 80.9 | 18.4 | Y/N | 85.2 | 27 |
| 82.0 | 16.0 | Y/Y | 89.5 | ||||||
| 78.7 | 19.7 | Y/Y | 90.5 | ||||||
| α-NAPAP | 1ETS | 2.3 | 78.7 | 20.3 | Y/N | 84.6 | |||
| 4-TAPAP | 1ETT | 2.5 | 76.7 | 22.3 | Y/N | 85.7 | |||
| Thrombin | Homo sapiens | NAPAP-derivative | 1QUR | 2.0 | 84.7 | 15.3 | N/N | 86.0 | 27 |
| Fibrinopeptide A | 1FPH | 2.5 | 84.2 | 15.8 | N/N | 81.5 | |||
| α-NAPAP | 1DWD | 3.0 | 82.8 | 17.2 | N/N | 71.4 | |||
PDB entries 1BQY, 1HIA, and 1UCY have crystal units with multiple structures and were assessed accordingly. Ramachandran plot statistics were obtained with PROCHECK (Laskowski et al. 1993); 3D profile scores are Luthy/Eisenberg’s (Gribskov et al. 1990); and sequence identity percentages are for a ClustalW (Higgins et al. 1994) default alignment (more details in the text).
a Residues in most favored (core), additionally allowed (allow), generously allowed (gen), and disallowed (dis) regions of the φ-ψ Ramachandran plot
(Y = yes, N = no).
b Analysis performed for the catalytic domains only, with no hydrogens nor heteroatoms.
The final sequence alignment of the most promising templates is presented in Figure 1 ▶. As expected, TSV-PA is, by far, the best candidate for major template: it has the highest percentage identity with the fewest number of gaps/insertions in the alignment, differing in sequence length by a single residue only, and exhibiting the seven-residue C-terminal extension that is highly conserved among snake venom serine proteinases (Parry et al. 1998) and present in KN-BJ2 as well. As for the remaining less homologous sequences, each is representative of a particular subspecificity within the trypsin S1 family. The selected trypsin, kallikrein, and thrombin structures were resolved in the complex form with an inhibitor/substrate that is also an inhibitor/substrate of KN-BJ2. Besides, unlike TSV-PA, KN-BJ2 has blood-clotting and kinin-releasing activities (Serrano et al. 1998), prompting the inclusion of thrombin and kallikrein structures to guide the modeling. As for the tissue-type plasminogen activator t-PA, its resistance to Kunitz-type inhibitors (e.g., bovine pancreatic trypsin inhibitor, BPTI) is also common to snake venom serine proteinases (Braud et al. 2000), a characteristic that KN-BJ2 might predictably exhibit and therefore should be considered during the modeling.
Figure 1.
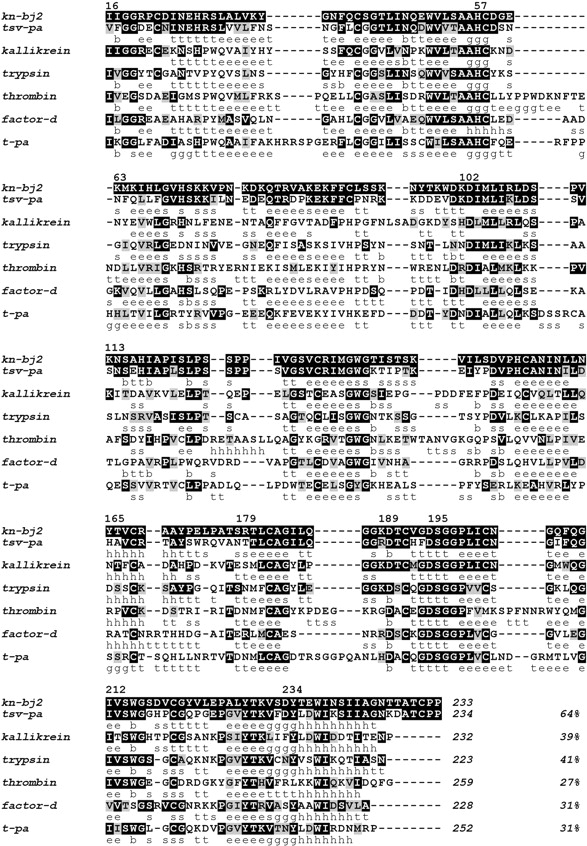
Multiple sequence alignment used in the KN-BJ2 modeling. Sequences for the templates TSV-PA (1BQY.pdb), porcine pancreatic kallikrein (1HIA.pdb), bovine trypsin (1PPH), human thrombin (1DWD.pdb), human factor-D (1BIO.pdb), and human t-PA (1RTF.pdb) were aligned according to their topological equivalence. The residue numbering refers to chymotrypsinogen. Shaded residues denote identity (black) or close similarity (gray) to KN-BJ2. Sequence lengths are displayed in italic after the C terminus as well as the calculated percentage of identity with KN-BJ2. The secondary structure elements indicated below each sequence is the Kabsch and Sander (1983) assignment provided in the Protein Data Bank online information; the assignments are h = helix; b = residue in isolated β bridge; e = extended β strand; g = 310 helix; t = hydrogen bonded turn; s = bend.
Crude model
The sequence alignment in Figure 1 ▶ was used to derive the 3D model structures of KN-BJ2. A first set of three crude structures was automatically generated with MODELLER (Sali and Blundell 1993) using the default options; for templates 1BQY.pdb and 1HIA.pdb, the structure with the best (higher) 3D profile score was used (see Table 1). The quality of the generated structures was evaluated with PROCHECK (Laskowski et al. 1993), and Luthy/Eisenberg (Gribskov et al. 1990) profiles were calculated with QUANTA, the results being summarized in Table 2. Secondary structural elements were also assigned, those of models 1 and 3 being practically equivalent to the TSV-PA motifs, and model 2 displaying a slightly less characterized folding. Because model 3 combines a higher 3D profile score with the smaller number of bad contacts and highlighted residues, this was the structure chosen for further refinement.
Table 2.
Quality parameters for the MODELLER (Sali and Blundell 1993)-generated KN-BJ2 structural models and for the refined model 3 and corresponding MD average structure
| Ramachandran plots | |||||||
| Overall (%)a | Individual | Chi1/Chi2 plot | |||||
| Core | Allow | Gen/dis | Residues highlightedb | Number of bad contacts | 3D profile score | ||
| Model 1 | 87.9 | 11.6 | Y/N | 7 | 5 | 5 | 70.2 |
| Model 2 | 86.4 | 12.6 | Y/Y | 8 | 5 | 7 | 61.8 |
| Model 3 | 86.9 | 12.6 | Y/N | 6 | 1 | 1 | 70.1 |
| Refined model 3 | 89.4 | 10.6 | N/N | 4 | 0 | 0 | 70.4 |
| MD average | 88.4 | 11.6 | N/N | 1 | 3 | 0 | 61.1 |
a Residues in most favored (core), additionally allowed (allow), generously allowed (gen), and disallowed (dis) regions of the φ-ψ Ramachandran plot
(Y = yes, N = no).
b Number of residues deviating by more than 2.0 S.D. from ideal in individual (by residue type) Ramachandran and Chi1/Chi2 plots.
Six disulfide bonds were assigned to this initial model as predicted by homology with TSV-PA. Five of them match the kallikrein pattern, the sixth one being between Cys 91 (chymotrypsinogen numbering) and Cys 245e from the C-terminal extension, which is strictly conserved among snake venom serine proteinases (Parry et al. 1998). Henceforth, the chymotrypsinogen numbering system will be used unless stated otherwise, the model(s) numbering matching the TSV-PA numbering.
Refinement
One first refinement to crude model 3 was the inclusion of a set of inner water molecules that are known to be preserved in proteinases sharing the primary specificity of trypsin (Sreenivasan and Axelsen 1992), with positions predetermined by structural homology. This strategy has been proved valuable in other homology prediction studies (Henriques et al. 1997, 2001), for one such water matrix imposes additional structural constraints to guide the modeling. A comparative analysis of the two TSV-PA molecules in 1BQY.pdb revealed that only 2 of the expected 22 conserved buried waters (Henriques et al. 1997, 2001) are absent in both molecules, those assigned to position identifiers 2 and 15 (cf. Sreenivasan and Axelsen 1992). Yet, the cavities to accommodate them are present in the TSV-PA structure(s) and with a residue environment highly homologous to the equivalent cavities in KN-BJ2 model 3. The remaining templates all have a water molecule in position 2, the striking feature in its environment being the bulky Trp 29 or Tyr 29 that these mammalian enzymes possess (see alignment in Fig. 1 ▶), in contrast with the much smaller, although also polar, Ser 29 of the two snake proteinases. This fact results in a much wider, solvent-accessible cavity in the venom enzymes, thus allowing for a water molecule to reside there but exchange with the bulk solvent, and therefore the corresponding position 2 in the KN-BJ2 model has been occupied with one such water residue. As for position 15, the cavity environment features residue 165, a His in TSV-PA and a Tyr in KN-BJ2, with no structural parallel in any of the other chosen templates (for which a water molecule is found in that position). However, within the serine proteinase family, the 1NES.pdb structure of porcine pancreatic elastase—which has a tyrosine as residue 165—bares a solvent molecule in the equivalent structural site 15. Consequently, a complete set of 22 conserved waters was added to the KN-BJ2 model, copied by homology from the relevant template structures as identified in Table 3.
Table 3.
Matrix of conserved water molecules included in the KN-BJ2 model by homology
| Site identifiers | Water | Site identifiers | Water |
| 1 | 1BQY:ESOL:479 | 12 | 1BQY:ESOL:474 |
| 2 | 1PPH:DSOL:250 | 13 | 1BQY:ESOL:481 |
| 3 | 1BQY:ESOL:488 | 14 | 1BQY:ESOL:572 |
| 4 | 1BQY:ESOL:555 | 15 | 1HIA:GSOL:94 |
| 5 | 1BQY:ESOL:501 | 16 | 1BQY:ESOL:470 |
| 6 | 1BQY:ESOL:498 | 17 | 1BQY:ESOL:924 |
| 7 | 1BQY:ESOL:475 | 18 | 1BQY:ESOL:477 |
| 8 | 1BQY:ESOL:478 | 19 | 1BQY:ESOL:574 |
| 9 | 1BQY:ESOL:545 | 20 | 1BQY:ESOL:550 |
| 10 | 1BQY:ESOL:513 | 21 | 1BQY:ESOL:947 |
| 11 | 1BQY:ESOL:492 | 22 | 1BQY:ESOL:482 |
Site identifiers conform with Henriques et al. (1997). Waters are identified by the PDB ID of the protein from which they were copied and by the segment and residue number.
With the conserved waters matrix already “in place”, a process of regularization of the structure was carried out in order to further improve the quality of the model. All energy calculations were performed using the CHARMM22 (McKerell et al. 1998) force field implementation within CHARMm (Accelyris Inc.). First, a series of local short energy optimizations without nonbonding energy terms were performed, aimed at correcting the main chain structural deviations detected with PROCHECK; in a few cases, a manual redesign, over a particular template, was also accomplished. Next, side chains were explored and corrected for optimal rotamers, favoring those from the existing templates as far as it made sense, while removing undesirable bad contacts.
The modeling refinement of residue Arg 179 is worth describing in more detail: only t-PA has an identical residue in this position (see alignment in Fig. 1 ▶), with a side-chain extended conformation that cannot be transposed to KN-BJ2 because it would clash with its C-terminal extension, as evidenced in Figure 2 ▶. Rough model 3 (and models 1 and 2, in fact) offers a very unusual buried conformation for this arginine-charged side chain, one that obstructs the conserved water site 4. This conserved water is hydrogen bonded to Tyr 234 in the matching templates; thus, it is valid to assume the same must be true for KN-BJ2 and therefore Arg 179 should adopt a different conformation. And, as can be seen in Figure 2 ▶, the alternative rotamer found for Arg 179 not only frees water site 4, but also allows the arginine guanidinium group to form a surface-exposed intramolecular salt bridge with the terminal Pro 245g.
Figure 2.

Cα-trace superposition of the neighboring regions of Arg 179 in factor-D (white) and crude (gray) and refined (black) model 3. Side chains of relevant residues are displayed in ball and stick. Notice that conserved site 4 is occupied with a water molecule (oxygen only) in the refined model and factor-D structures and that the C-terminal extension is not a feature of the latter.
The regularized KN-BJ2 model was finally subjected to an energy optimization with harmonic constraints on the protein backbone (100.0 kcal mole−1 Å−2) and water oxygens (25.0 kcal mole−1 Å−2); the system went through enough minimization steps for the structure to relax and clear out any remaining bad contacts. No significant displacements of the conserved water positions or side-chain distortions were observed. The overall Ramachandran plot of the resulting model is presented in Figure 3 ▶, and its quality check parameters are also listed in Table 2 for comparison. Figure 4 ▶ depicts the secondary structure and estimated accessibility (PROCHECK results) of the refined model and its major template, TSV-PA.
Figure 3.

Ramachandran map and corresponding plot statistics for the KN-BJ2 refined model.
Figure 4.

Secondary structure and estimated solvent accessibility for the KN-BJ2 refined model (a), its subsequent MD average structure (b), see text for details), and TSV-PA.
Model dynamics
Considering 3D structures of proteins change rapidly over time under physiological conditions, a more realistic representation of the KN-BJ2 could be a dynamic trajectory of its model, solvated to some extent. Although the feasible length of a molecular dynamics (MD) simulation samples only a small fraction of a protein’s conformational space, one such approach could further assess the consistency of the refined model and account for solvation effects such as flexibility increase of solvent-accessible residues.
Using the QUANTA/CHARMm package, we then soaked the refined KN-BJ2 model with a 12-Å-thick shell of equilibrated water molecules and submitted it to 500 psec of MD under NVT conditions at 300K, with an integration time step of 1 fsec and SHAKE on all bonds involving hydrogens. A 12.0 Å cutoff distance with a switch smoothing function at 11.25 Å was used for the nonbonding interactions, and a harmonic constraint of 0.60 kcal mole−1 Å−2 applied on the water oxygens of the solvent outer-layer (2 Å thick) because no periodic boundary conditions were used. Trajectories were saved every 0.02 psec.
Figure 5 ▶ displays the potential energies as a function of the simulation time, for both the entire solvated system and the protein alone. The correspondent root mean square deviations (RMSDs) of the protein coordinates (heavy atoms) relative to the initial structure—overall and backbone only—are displayed in Figure 6 ▶. Temperature was well conserved throughout the simulation, as intended. The system was considered to be equilibrated after 300 psec of simulation and an average structure was calculated for the last 200 psec (10,000 frames) of production. The resulting structure was finally relaxed for the internal parameters to regularize, and it displays a best-fit backbone RMSD of 1.28 Å relative to the initial refined model. Other relevant structural quality parameters have been included in Table 2 and Figure 4 ▶.
Figure 5.

Potential energy plotted against the simulation time for the entire solvated system (thin line) and the protein alone (thick line). Energy values have been averaged every 50 saved conformations.
Figure 6.

Root mean square deviations (RMSDs) of the heavy atom coordinates relative to the initial structure plotted against the simulation time, for the overall (thick line) and backbone (thin line) KN-BJ2 model.
For the earlier mentioned best-fit backbone superposition, the average KN-BJ2 structure exhibits all but one conserved water site occupied by a water molecule, with a 1.58 Å overall RMSD for the water oxygens (average vs. initial equivalent positions). Table 4 summarizes the buried waters rearrangement in the averaged MD structure. Three buried water molecules in peripheral sites have been replaced by bulk solvent residues, a predicted situation under ordinary solvent conditions (Sreenivasan and Axelsen 1992). Shuffling has occurred between buried waters in the conjugated sites 9 to 13 and eventually one water molecule (assigned position 13) has been lost. Interestingly enough, kallikreins have been reported either to have positions 9 and 13 alternatively occupied by His 27 side chain or to have water 9 present but not effectively buried (Sreenivasan and Axelsen 1992), depending on the structure. In fact, this is also the case for the more recently reported 1HIA.pdb kallikrein structure (Mittl et al. 1997), one of the KN-BJ2 templates, which is devoid of buried water 13 and has water 9 on a more exposed position compared with the templates without a histidine residue 27, as exemplified in Figure 7 ▶. Both KN-BJ2 and TSV-PA preserve His 27, and the latter also exhibits a most appropriate “kallikrein-like” water 9 position, the one copied to the model.
Table 4.
Rearrangement and deviation from initial positions of the conserved water molecules in the KN-BJ2 MD average structure
| Identifiers | Identifiers | ||||
| Site | Water | Deviation from initial (Å) | Site | Water | Deviation from initial (Å) |
| 1 | 1 | 1.818 | 12 | 11 | 1.085 |
| 2 | 2 | 0.962 | 13 | Missing | — |
| 3 | Bulk water | 0.859 | 14 | Bulk water | 0.708 |
| 4 | 4 | 2.005 | 15 | 15 | 0.897 |
| 5 | 5 | 2.896 | 16 | 16 | 0.955 |
| 6 | 6 | 1.404 | 17 | 17 | 1.888 |
| 7 | 7 | 1.327 | 18 | 18 | 2.667 |
| 8 | 8 | 0.513 | 19 | 19 | 2.188 |
| 9 | 12 | 0.783 | 20 | 20 | 0.359 |
| 10 | 10 | 1.120 | 21 | 21 | 0.929 |
| 11 | Bulk water | 2.723 | 22 | 22 | 1.269 |
Waters are identified according to the sites they occupied in the model before dynamics; “bulk water” means the original molecule has been replaced by a bulk solvent residue.
Figure 7.

Cα-trace superposition of the neighboring region of assigned waters 9 and 13 in trypsin (gray), kallikrein (black), and the KN-BJ2 MD-average structure (white). Residue 27 side chain(s) is displayed in ball and stick. Notice how the occurrence of a histidine as residue 27 induces a more exposed (peripheral) position for water 9 and allows for water 13 to be absent.
Docking
The refined KN-BJ2 model was used in the docking procedures because its side-chain rotamers closely match those in the analogous residues of the inhibitor:enzyme template structures. The structures of the three inhibitors to dock, α-NAPAP, 3-TAPAP, and 4-TAPAP, are represented in Figure 8 ▶. The KN-BJ2:inhibitor starting structures were generated by rigid-body fitting the inhibitors into the model, having their conformations and relative orientations taken from the crystal structures of the corresponding enzyme: inhibitor guiding templates, that is, PDB entries 1DWD (Banner and Hadváry 1991), 1PPH (Turk et al. 1991), and 1ETT (Brandstetter et al. 1992), respectively, for α-NAPAP, 3-TAPAP, and 4-TAPAP; the other available α-NAPAP-bound conformations (1PPC.pdb [Bode et al. 1990], 1ETS.pdb [Brandstetter et al. 1992]) were found to be equivalent to the chosen one.
Figure 8.
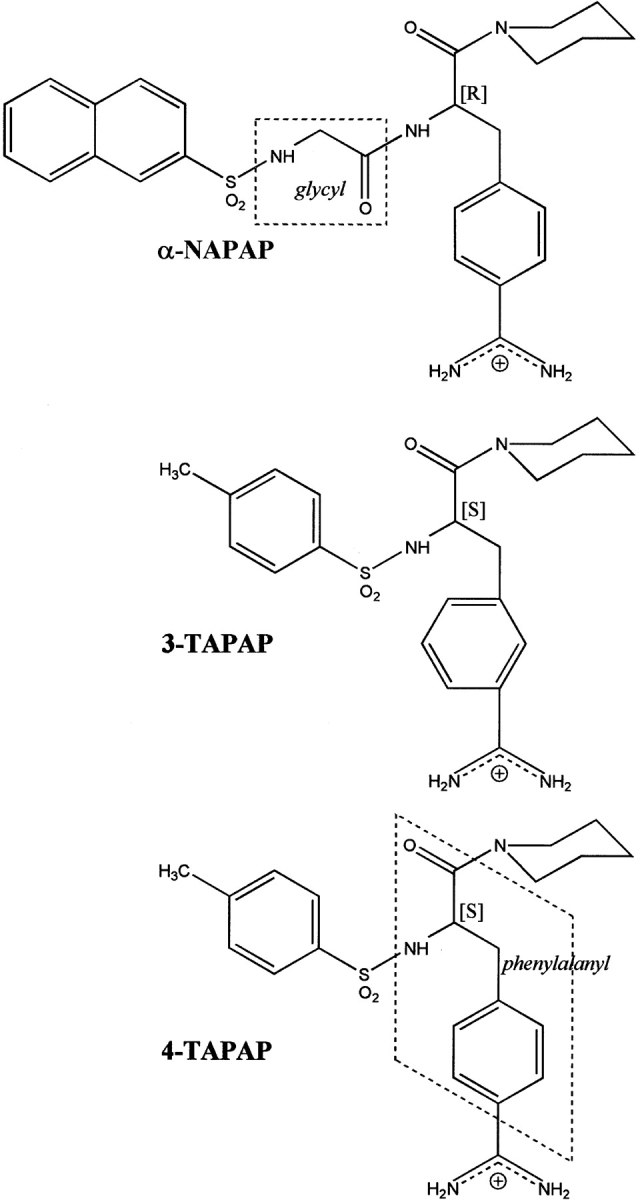
Chemical formulas of the inhibitors α-NAPAP, 3-TAPAP, and 4-TAPAP. Peptidic-like residue frames have been emphasized with dashed boxes.
The preliminary fitting exercise revealed a potential problem, because severe clashes occurred between the piperidyl and naphthyl/tosyl groups of the inhibitors and residue Trp 99 of KN-BJ2, which could invalidate any subsequent reasonable docking. None of the template structures has a bulky tryptophan in this position, but kallikrein (and t-PA, see alignment in Fig. 1 ▶) has a tyrosine residue 99 in a backbone conformation that could possibly accommodate those inhibitors groups. KN-BJ2 being a kallikrein-like enzyme, it was reasonable for its residue 99 region to adopt an alternative kallikrein-derived conformation, this being accomplished without significant changes to the overall quality of the model.
A more refined docking study required the necessary set of parameters for the inhibitors of interest to be derived in conformity with the CHARMM22 force field. Geometric parameters were taken from the available X-ray structures (Bode et al. 1990; Banner and Hadváry 1991; Turk et al. 1991; Brandstetter et al. 1992), and the corresponding force constants were derived by analogy to existing parameters. For the net atomic charges, single-point energy calculations were performed at the density functional theory level with the Becke3-Lee-Yang-Parr (B3LYP) functional and the 6-31G* basis set, using the Gaussian 98 (Frisch et al. 1998) package. Charges were fit to reproduce the inhibitors ESP in their crystallographic bound conformations to thrombin and trypsin. Details of the parameterization will be reported elsewhere (E.S. Henriques, M.A.C. Nascimento, and M.J. Ramos, in prep.).
Next a multisegment Hybrid Boltzmann Jump Conformational Search was performed within QUANTA (QUANTA 2000). The procedure explores conformational space by performing docking rigid body manipulations via a random perturbation method (using a Metropolis selection criterion) while sampling the selected inhibitor’s torsional degrees of freedom using a rotational isomeric state model random search. For each inhibitor, 100 accepted sample conformations were generated and the best-fitting results were clustered and analyzed. These results proved the docking trend of the amidinophenylalanine and piperidine moieties of each inhibitor to be very similar to the corresponding one(s) in the complexes with thrombin and trypsin, but were inconclusive on the fitting of the naphthyl/tosyl groups. Attempts to overcome this situation using the GOLD genetic algorithm for ligand docking (Jones et al. 1997) yield no consensus either (see Kontoyianni et al. 2004 for an evaluation on this and other docking algorithms). The most promising results were then selected for a CHARMm minimization guided fit, performed with a number of plausible hydrogen bonding (H-bond) distance constraints intended to preserve the already established fitting conformations of the amidinophenylalanine piperidine frame. The constraints are consistent with the reported pattern of H-bonds most likely to be found for these inhibitors bound to trypsin-like serine proteinases (Bode et al. 1990; Banner and Hadváry 1991; Turk et al. 1991; Brandstetter et al. 1992), which are between (1) the inhibitor amidino nitrogens and the Asp 198 positioned at the bottom of the S1 specificity pocket, in a typical “symmetrical” twin N—twin O manner for α-NAPAP and 3-TAPAP and in a “lateral” single N—twin O manner for 4-TAPAP and (2) the carbonyl (and possibly amino) group(s) of the inhibitor “main-chain residue” glycyl (α-NAPAP) or phenylalanyl (3– and 4-TAPAP) and the important Gly 216 recognition site.
The constraints’ target distance was 3.0 Å with upper and lower bound limits of 3.5 Å and 1.9 Å and a force constant of 50.0 kcal mole−1 Å−2 for the steepness of the potential outside those limits for all but the distance between the Gly 216 carbonyl group and the inhibitor’s “main-chain” amino group, which was assigned half the value (25.0 kcal mole−1 Å−2). The target distance was chosen on the basis of the analogous crystallographic values, which vary between 2.7 Å and 3.3 Å (Brandstetter et al. 1992); the assignment of a looser distance constraint for the “main-chain” amino group was determined by the one-residue insertion in the KN-BJ2 loop 217–220 relative to thrombin (and trypsin), which protrudes into the interaction region of the referred amino group, thus possibly forcing it to a more twisted distal position. The reader is referred to Figures 8 ▶ and 9 ▶ for a better understanding of these structural/geometric considerations. The enzyme was kept rigid during the minimization fitting procedure, which consisted of a Conjugated Gradient optimization to an energy gradient tolerance of 0.01 kcal mole−1 Å−1, with a distance dependent dielectric constant set to 4 (usual for proteins) and the same cutoff parameters used in the model dynamics, followed by a regularization of the internal parameters of the inhibitor within the context of the cavity van der Waals interactions.
Figure 9.

Depiction of the optimized docking configurations of (A) KN-BJ2:α-NAPAP (black) superposed to thrombin:α-NAPAP (gray, 1DWD.pdb) and (B) the three inhibitors—α-NAPAP (dark gray), 3-TAPAP (light gray), and 4-TAPAP (black)—superposed onto each other inside the model. The inhibitors are displayed in ball and stick and dashed lines represent H-bond interactions.
The optimized docking configurations for the three inhibitors are illustrated in Figure 9 ▶, superposed onto each other inside the model and the KN-BJ2:α-NAPAP one superposed to the thrombin:α-NAPAP crystal structure. Table 5 lists some relevant H-bond distances.
Table 5.
Relevant hydrogen bonding distances (Å) for the modeled KN-BJ2:inhibitor complexes (this work) and the related experimentally resolved complexes (identified by pdb code)
| Enzyme | KN-BJ2 | 1DWD | KN-BJ2 | 1PPH | KN-BJ2 | 1ETT | ||
| Residue | Atom | Inhibitor atom | NAPAP | 3-TAPAP | 4-TAPAP | |||
| Asp 189 | OD1 | NG1 | 3.3 | 3.1 | 3.3 | 3.0 | 3.2b | 3.2b |
| OD2 | NG2 | 3.3 | 2.5 | 3.5 | 2.9 | 3.4 | 3.3 | |
| Gly 216 | N | O | 3.0 | 2.9 | 3.2 | 3.1 | 3.4 | 3.2 |
| O | N | 2.8 | 2.3 | 3.1 | 3.1 | 3.5 | 2.8 | |
| da | 3.0 | 3.1 | 2.7 | 3.1 | 2.9 | 3.0 | ||
a Minimum enzyme–inhibitor contact distance for heavy atoms not involved in specific interactions.
b Values for NG2 also, the 4-TAPAP nitrogen interacting in a single N–twin O manner.
Considering that a comparison of the inhibition constants (Ki) for the inhibition of KN-BJ, human thrombin, and porcine pancreatic kallikrein by the inhibitors of interest (Table 6; Serrano et al. 1998) indicates a closer similarity in affinity between the venom enzyme and kallikrein than to thrombin, an analogous docking exercise has been performed for kallikrein also, using its complex structure with BPTI (Chen and Bode 1983) to further assist the repositioning of the inhibitors. Figure 10 ▶ illustrates the docking complementarity between the binding cavities of those three enzymes and the different best fit conformations/orientations of the inhibitors, which, as discussed next, provides an insightful clarification of the reported different affinity trends.
Table 6.
Inhibition constants (Ki, μM) of human thrombin, bovine trypsin, porcine tissue kallikrein, and snake KN-BJ (1 and 2) as reported in literature
| Inhibitor | Thrombin (Stürzebecher et al. 1986) | Trypsin (Stürzebecher et al. 1984) | Kallikrein (Serrano et al. 1998) | KN-BJ (Serrano et al. 1998) |
| α-NAPAP | 0.006 | 0.69 | 93 | 30 |
| 3-TAPAP | 0.34 | 1.2 | 590 | 263 |
| 4-TAPAP | 2.3 | 64.0 | 38 | 576 |
Figure 10.

Solid-surface representation of the binding cavities for thrombin complexed with α-NAPAP (1DWD.pdb), KN-BJ2 with α-NAPAP best-fit docking conformation, kallikrein with α-NAPAP best-fit docking conformation, and kallikrein with 4-TAPAP best-fit docking conformation. Contour level surface is van der Waals radius +0.9 Å, and coloring is according to the electrostatic potential at the surface calculated with the CHARMm standard atomic charges, color ramping being from intense blue (−50 kcal mole−1) to intense red (−50 kcal mole−1).
Discussion
Overall structure
A structural model of the KN-BJ2 snake venom serine proteinase has been homology built on the basis of the alignment of Figure 1 ▶, TSV-PA being the major (most similar) template by genetic evolutionary proximity and tissue kallikrein being taken as an important auxiliary template for both its topological similarity with TSV-PA (Parry et al. 1998; see secondary structure elements in Fig. 1 ▶) and the reported kinin-releasing (kallikrein-like) activity of KN-BJ2. Despite the 64% sequence identity between KN-BJ2 and TSV-PA and the single one-residue insertion in their alignment, TSV-PA does not display the fibrinogen-clotting activity that prompts us to study KN-BJ2; thus, it was considered advisable to include a number of other relevant serine proteinases, namely, thrombin, to further guide the modeling.
The refined KN-BJ2 model structure exhibits the typical fold of an S1 serine proteinase, in close similarity with TSV-PA topology and solvent accessibility (see Fig. 4 ▶). It has hydrophilic residues His 57, Asp 102 (of the catalytic triad), Asp 189 (S1 pocket), and Asp 194 (in a highly preserved internal salt bridge with the N-terminal residue) buried as expected, and an arrangement of surface-exposed intramolecular salt bridges matching the one in TSV-PA (Parry et al. 1998), except for bridges 143–149 and 97–174, which have no analogy in KN-BJ2. Other charged side chains are exposed to the bulk solvent and one can assume an extra salt bridge between Arg 179 and the C-terminal residue. A set of 22 buried waters known to be preserved in S1 serine proteinases was incorporated into the model, and it fit the structure without any significant adjustments. The overall quality of the model is good and comparable to the quality of its major template structure (TSV-PA, Table 1), as made clear by the results in Table 2 and Figure 3 ▶.
Compared with the refined model, the ensuing MD averaged structure suffered a minor quality loss (see Table 2), most certainly a consequence of the limited sampling and the rather simplified force-field formulation, in particular for the nonbonding interactions, which introduces many very small structural errors; one should recall that secondary structure elements such as β-sheets and α-helices depend on H-bond (i.e., nonbonding) interactions. Thus, the average structure is more relaxed but less well packed as perceived by the slight decrease in the ratio of hydrophilic/hydrophobic solvent-accessible surface area (0.52 versus the initial 0.59 value). Nonetheless, under dynamic conditions, the model rapidly reached equilibrium both energetic (Fig. 5 ▶) and structural (Fig. 6 ▶), and it seems reasonable to assume that such a fast convergence is also a good indicator of the quality of the starting model.
Very interesting MD results arise for the conserved water molecules (see Table 4), their general behavior throughout the simulation being consistent with the available experimental data: the five water molecules assigned to enzymes sharing the primary specificity of trypsin (sites 17–21; Sreenivasan and Axelsen 1992) conserved their positions, and for the three peripheral water sites that have been reoccupied by bulk water molecules, the fact remains that a particular water is bound to reside in each of these sites for sufficiently long periods of time (the average structure covers 200 psec). Of particular interest is the simulation result for waters 9 and 13, which seems to reproduce an already observed pattern for kallikreins in what concerns the presence (or absence) of these two buried waters in a region that might be alternatively occupied by His 27 side chain, as depicted in Figure 7 ▶. Not only does the simulation portray a dynamic solution for that pattern, but it also emphasizes the kallikrein-like character exhibited by the KN-BJ2 venom enzyme, thus contributing to the validation of the model.
Inhibitor binding
The most striking difference between the binding sites of thrombin and KN-BJ2, as well as other trypsin-like serine proteinases, is the specific 60-insertion loop of the former (60.A-D, see alignment in Fig. 1 ▶), which creates with Trp 215, Leu 99, and Ile 174 a double hydrophobic pocket subdivided into a smaller proximal (P) pocket to the His 57 side and a larger distal (D) pocket to the Trp 215 side (Banner and Hadváry 1991). Aromatic moieties such as phenyl or naphthyl rings are well accommodated in this binding pocket, this being the case for the three inhibitors α-NAPAP, 3-TAPAP, and 4-TAPAP. KN-BJ2 (and kallikrein), lacking this peripheral (outer face) rim of the pocket, leave it wide open to the bulk solvent, which could account for much of its observed comparatively low affinity toward those thrombin inhibitors (see Table 6); yet trypsin does not have the 60.A-D loop also and its corresponding affinities are of a much closer order of magnitude to the thrombin ones. The docking exercises were intended to further rationalize these experimental data.
The Hybrid Boltzmann Jump search docked the amidinophenylalanine and piperidine moieties of each inhibitor in a predictable way, that is, equivalent to the corresponding one(s) in the resolved complexes with thrombin and trypsin. As can be seen in Figure 9 ▶, the amidinophenylalanyl group packs favorably inside the S1 binding pocket, forming the desirable interactions between the nitrogens and the carboxylate group of Asp 189, and the piperidine ring is squeezed between the His 57 imidazole side chain and the indole moiety of Trp 99; however, unlike Trp 60.D of thrombin, Trp 99 of KN-BJ2 is in the “inner face” of the P-pocket and pushes the piperidyl group slightly away from the enzyme surface, because it is much bulkier than the corresponding Leu 99 in thrombin (and trypsin). A comparable situation is found for the docking in kallikrein, its residue 99 being (an also bulky) tyrosine.
A close inspection of the binding cavity surfaces represented in Figure 10 ▶ makes it understandable as to why the docking of the naphthyl/tosyl groups was not “straightforward”: the KN-BJ2 and kallikrein cavities lack the striking shape complementarity thrombin has, prompting a looser (ambiguous) fitting of those groups and making it necessary to reduce the conformational freedom of the inhibitors’ “main chain”. This was achieved by imposing a number of plausible distance constraints: those involving the amidinocarboxylate interactions are obvious and were already “in place” after the first docking run, and it follows from experimental evidence that, for all serine proteinases, substrates and inhibitors must establish H-bonds with the non-specific peptide recognition site Gly 216 (Klebe 2000); hence, the second set of constraints.
The calculated KN-BJ2-inhibitor interaction energies for the final docking configurations (−68.1, −55.5, and −49.6 kcal mole−1 for α-NAPAP, 3-TAPAP, and 4-TAPAP, respectively) display the same decreasing tendency of the reported affinities, and although potential energies are not directly comparable with inhibition constants, they are still a good indicator of the reliability of the docked structures. As can be seen in Figure 10 ▶, the docking of α-NAPAP in KN-BJ2 (and kallikrein) induces an extended conformation for this inhibitor, with the naphthyl group being pushed away from the piperidyl ring by the bulky residue 99 and by the rotation around the O2S-NH bond, this latter occurring to avoid the clash of the sulfonyl group with residue 218, which protrudes unfavorably into the binding region in consequence of the previously mentioned one-residue insertion in loop 217–220; plus, the KN-BJ2 structure creates a rather negative ESP around the active-site cleft (as, in fact, does TSV-PA; Parry et al. 1998), facing the negatively polarized SO2 group of the inhibitor. Notice, however, that the extended conformation still allows for the naphthyl group to interact favorably with Trp 215 at the bottom of the latent D-pocket. Similar considerations can be made for the docking of the other two inhibitors, 3- and 4-TAPAP, which also adopt extended conformations (see Fig. 9 ▶) in order to achieve the best possible steric/electrostatic complementarity with the KN-BJ2 binding cavity, yet lead to the systematic lengthening, that is, destabilization, of the crucial interactions, patent in Table 5. One can also infer an increase in steric hindrance by the general shortening of close contacts represented by parameter d in Table 5.
In effect, these discussed modeling results are in excellent agreement with a few consensual ideas regarding the binding of the studied inhibitors (Banner and Hadváry 1991; Brandstetter et al. 1992), which attribute their large affinity to thrombin, mainly as a result of (1) the notched double pocket shaped by the unique 60-insertion loop, which is much more prominent and hydrophobic and therefore better suited to bury the inhibitors hydrophobic moieties than it is possible in trypsin, KN-BJ2, and kallikrein and (2) the good internal packing of the inhibitors themselves, in a compact—close to their “naturally” most stable—conformation (see Fig. 10 ▶), which is also preserved when binding to trypsin, but appears to be inappropriate to dock in the differently shaped KN-BJ2 and kallikrein binding regions, as suggested by the predominance of extended docking conformations obtained in the present modeling study. One particular result might be interpreted in light of this latter topic and illustrates its importance: kallikrein, which induces the most extended conformations of α-NAPAP and 3-TAPAP, has the lowest affinities for these two inhibitors (see Table 6), whereas its off-trend higher affinity to 4-TAPAP concurs with the best-fit compact docking conformation of the latter, as depicted in Figure 10 ▶. Compared with KN-BJ2, kallikrein displays a D-pocket less cramped at position 99 (tyrosine is not as bulky as tryptophan) but somewhat shrunken at the rim of its one-residue shorter loop 172–175, creating a most appropriate concavity to accommodate a tosyl group. In the case of 4-TAPAP, this has been achieved with the inhibitor in a compact docking conformation much similar to the one found in the thrombin: 4-TAPAP complex and with similar anchor interactions to Asp 189 and Gly 216; the different geometry of the 3-TAPAP analog did not promote an equivalent situation.
From the discussion beforehand, it is reasonable to assume that the considerably reduced affinity of KN-BJ2 toward the studied thrombin inhibitors must be largely determined by a significant distortion of their compact stabilizing conformations on docking, mainly induced by the different shape and orientation of the cavities where the two hydrophobic groups of those inhibitors can be accommodated (latent P- and D-pockets), combined with a rather poor complementarity between the referred groups and their host pockets. The bulky residue Trp 99 flanking these pockets must be a key determinant of the KN-BJ2 subspecificity, such as Tyr 99 is to kallikrein (Chen and Bode 1983); incidentally, batroxobin, which possesses a small threonine at position 99, has Ki values for α-NAPAP of the same order of magnitude of trypsin (Leu 99). The KN-BJ2 affinity trend α-NAPAP>3-TAPAP>4-TAPAP is presumably the result of the naphthyl group being more suited (better sterical fit) to occupy the host pockets than the smaller tosyl group, as the docking superposition in Figure 9B ▶ suggests, thus displaying more favorable hydrophobic contacts (e.g., with Trp 215) and a tighter binding; 4-TAPAP has the additional “disadvantage” of anchoring the amidinophenyl group to Asp 189 via a single nitrogen atom. Similar arguments have been used to explain that same trend for the thrombin and trypsin affinity pattern (Bode et al. 1990; Banner and Hadváry 1991; Turk et al. 1991; Brandstetter et al. 1992).
Hydrophobic interactions being a major determinant and discriminating factor in the binding of the inhibitors under study, no attempt has been made to further relax the modeled complex structures by means of full-energy minimizations, for the so-called induced fit the protein must undergo on their binding is unlikely to be correctly reproduced by a simple molecular mechanics force field.
Conclusion
On the basis of the modeling results herein obtained and their comparison with the available experimental data, it seems reasonable to conclude that the structural model we propose for KN-BJ2 is most likely a good representative of this venom enzyme. The model allowed us to tackle a number of interesting aspects that emphasize the kallikrein-like pattern of this proteinase, including how the behavior of its intrinsic set of buried waters under dynamic conditions can be related to a kallikrein motif. The information obtained from the docking procedures proved useful for the interpretation of some reported Ki values for the enzyme toward a number of thrombin inhibitors, convincingly explaining the relative KN-BJ2 affinities and the affinity differences to thrombin and trypsin, and making it possible to rationalize its similarities with kallikrein. Succinctly, both KN-BJ2 and kallikrein feature a distinctive one-residue insertion in loop 217–220 following the Gly 216 recognition site and a bulky aromatic residue 99 protruding into the S2 binding region, two characteristics often associated with kallikrein-like enzymes and absent from thrombin and trypsin. Combined with an ESP around the active site that is markedly more negative than in thrombin and the lack of a thrombin-like 60-insertion loop, the overall result is a more loose-fitting and solvent-exposed binding region and an ill-defined D-pocket that induces extended docking conformations of the inhibitors, exposing their hydrophobic surfaces and not promoting enough cooperatively acting contacts to stabilize the complexes. The fact that enzymes such as trypsin and batroxobin, which also lack the 60-insertion loop but have a smaller aliphatic residue 99 merged in the binding surface, display much higher affinities for the inhibitors in question than KN-BJ2 and kallikrein, underlines the relevance of position 99 for the kallikrein-like affinity. The proposed model provides an insightful clarification of the KN-BJ2 activity and is a valuable solid starting point for a better functional characterization of other related venom proteinases with thrombin- and kallikrein-like activities, which may either reside separately in different enzymes or be found together in a single one (Serrano et al. 1998).
Coordinates for all of the modeled structures are available directly from the authors on request.
Acknowledgments
Financial support from Fundação para a Ciência e Tecnologia (FCT) is gratefully acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04746804.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne P.E. 2000. The distribution and query systems of the RCSB Protein Data Bank. Nucleic Acids Res. 28 D235–D242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode, W., Turk, D., and Sturzebecher, J. 1990. Geometry of binding of the benzamidine- and arginine-based inhibitors N α-(2-naphthyl-sulphonyl-glycyl)-DL-p-amidinophenylalanyl-pipe ridine (NAPAP) and (2R,4R)-4-methyl-1–[N α-(3-methyl-1,2,3,4–tetrahydro-8–quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid (MQPA) to human α-thrombin. X-ray crystallographic determination of the NAPAP-trypsin complex and modeling of NAPAP-thrombin and MQPA-thrombin. Eur. J. Biochem. 193 175–182. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.-C., Estreicher, A., Gasteiger, E., Martin, M.J., Michoud, K., O’Donovan, C., Phan, I., et al. 2003. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandstetter, H., Turk, D., Hoeffken, H.W., Grosse, D., Sturzebecher, J., Martin, P.D., Edwards, B.F., and Bode, W. 1992. Refined 2.3 Å X-ray crystal structure of bovine thrombin complexes formed with the benzamidine and arginine-based thrombin inhibitors NAPAP, 4-TAPAP and MQPA. A starting point for improving antithrombotics. J. Mol. Biol. 226 1085–1099. [DOI] [PubMed] [Google Scholar]
- Braud, S., Parry, M.A., Maroun, R., Bon, C., and Wisner, A. 2000. The contribution of residues 192 and 193 to the specificity of snake venom serine proteinases. J. Biol. Chem. 275 1823–1828. [DOI] [PubMed] [Google Scholar]
- Chen, Z. and Bode, W. 1983. Refined 2.5 Å X-ray crystal structure of the complex formed by porcine kallikrein A and the bovine pancreatic trypsin inhibitor. J. Mol. Biol. 164 283–311. [DOI] [PubMed] [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Zakrzewski, V.G., Montgomery Jr., J.A., Stratmann, R.E., Burant, J.C., et al. 1998. Gaussian 98 (Revision A.9). Gaussian, Inc., Pittsburgh, PA.
- Gribskov, M., Luthy, R., and Eisenberg, D. 1990. Profile analysis. Methods Enzymol. 183 146–159. [DOI] [PubMed] [Google Scholar]
- Henriques, E.S., Ramos, M.J., and Reynolds, C.A. 1997. Inclusion of conserved buried water molecules in the model structure of rat submaxillary kallikrein. J. Comput. Aided Mol. Des. 11 547–556. [DOI] [PubMed] [Google Scholar]
- Henriques, E.S., Floriano, W.B., Reuter, N., Melo, A., Brown, D., Gomes, J.A.N.F., Maigret, B., Nascimento, M.A.C., and Ramos, M.J. 2001. The search for a new model structure of β-Factor XIIa. J. Comput. Aided Mol. Des. 15 309–322. [DOI] [PubMed] [Google Scholar]
- Higgins, D., Thompson, J., Gibson, T., Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, G., Willett, P., Glen, R.C., Leach, A.R., and Taylor, R. 1997. Development and validation of a genetic algorithm to flexible docking. J. Mol. Biol. 267 727–748. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Klebe, G. 2000. Recent developments in structure-based drug design. J. Mol. Med. 78 269–281. [DOI] [PubMed] [Google Scholar]
- Kontoyianni, M., McClellan, L.M., and Sokol, G.S. 2004. Evaluation of docking performance: Comparative data on docking algorithms. J. Med. Chem. 47 558–565. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Markland, F.S. 1998. Snake venoms and the hemostatic system. Toxicon 36 1749–1800. [DOI] [PubMed] [Google Scholar]
- Martí-Renom, M A., Stuart, A., Fiser, A., Sánchez, R., Melo, F., and Sali., A. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29 291–325. [DOI] [PubMed] [Google Scholar]
- McKerell Jr., A.D., Bashford, D., Bellot, M., Dunbrack Jr., R.L., Evanseck, J.D., Field, M.J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102 3586–3616. [DOI] [PubMed] [Google Scholar]
- Mittl, P.R., Di Marco, S., Fendrich, G., Pohlig, G., Heim, J., Sommerhoff, C., Fritz, H., Priestle, J.P., and Grutter, M.G. 1997. A new structural class of serine protease inhibitors revealed by the structure of the hirustasin-kallikrein complex. Structure 5 253–264. [DOI] [PubMed] [Google Scholar]
- Parry, M.A.A., Jacob, U., Huber, R., Wisner, A., Bon, C., and Bode, W., 1998. The crystal structure of the novel snake venom plasminogen activator TSV-PA: A prototype structure for snake venom serine proteinases. Structure 6 1195–1206. [DOI] [PubMed] [Google Scholar]
- Ramos, M.J., Melo, A., and Henriques, E.S. 2001. Modelling enzyme-ligand interactions. In Theoretical biochemistry—Processes and properties of biological systems. Theoretical and computational chemistry (ed. L.A. Eriksson), Vol. 9, pp. 539–595. Elsevier, Amsterdam.
- Sali, A. and Blundell, T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Serrano, S.M.T., Hagiwara, Y., Murayama, N., Highuchi, S., Mentele, R., Sampaio, C.A.M., Camargo, A.C.M., and Fink, E. 1998. Purification and characterization of a kinin-releasing and fibrinogen-clotting serine proteinase (KN-BJ) from the venom of Bothrops jararaca, and molecular cloning and sequence analysis of its cDNA. Eur. J. Biochem. 251 845–853. [DOI] [PubMed] [Google Scholar]
- Sreenivasan, U. and Axelsen, P.H. 1992. Buried water in homologous serine proteases. Biochemistry 31 12785–12791. [DOI] [PubMed] [Google Scholar]
- Stürzebecher, J., Walsmann, P., Voigt, B., and Wagner, G. 1984. Inhibition of bovine and human thrombins by derivatives of benzamidine. Thromb. Res. 36 457–467. [DOI] [PubMed] [Google Scholar]
- Stürzebecher, J., Stürzebecher, U., and Markwardt, F. 1986. Inhibition of batroxobin, a serine proteinase from Bothrops snake venom, by derivatives of benzamidine. Toxicon 24 585–595. [DOI] [PubMed] [Google Scholar]
- Turk, D., Stürzebecher, J., and Bode, W. 1991. Geometry of binding of the N α-tosylated piperidides of m-amidino-, p-amidino- and p-guanidino phenylalanine to thrombin and trypsin. X-ray crystal structures of their trypsin complexes and modeling of their thrombin complexes. FEBS Lett. 287 133–138. [DOI] [PubMed] [Google Scholar]
- Watanabe, L., Vieira, D.F., Bortoleto, R.K., and Arni, R.K. 2002. Crystallization of bothrombin, a fibrinogen-converting serine protease isolated from the venom of Bothrops jararaca. Acta Crystallogr. D58 1036–1038. [DOI] [PubMed] [Google Scholar]
- Weitz, J.I. and Hirsh, J. 2001. New anticoagulant drugs. Chest (Suppl. 1) 11995S–107S. [DOI] [PubMed] [Google Scholar]