

Abstract
SecB, a small tetrameric cytosolic chaperone in Escherichia coli, facilitates the export of precursor poly-peptides by maintaining them in a nonnative conformation and passing them to SecA, which is a peripheral member of the membrane-bound translocation apparatus. It has been proposed by several laboratories that as SecA interacts with various components along the export pathway, it undergoes conformational changes that are crucial to its function. Here we report details of molecular interactions between SecA and SecB, which may serve as conformational switches. One site of interaction involves the final C-terminal 21 amino acids of SecA, which are positively charged and contain zinc. The C terminus of each subunit of the SecA dimer makes contact with the flat β-sheet that is formed by each dimer of the SecB tetramer. Here we demonstrate that a second interaction exists between the extreme C-terminal α-helix of SecB and a site on SecA, as yet undefined but different from the C terminus of SecA. We investigated the energetics of the interactions by titration calorimetry and characterized the hydrodynamic properties of complexes stabilized by both interactions or each interaction singly using sedimentation velocity centrifugation.
Keywords: chaperone, SecB, SecA, calorimetry, sedimentation velocity centrifugation, protein interactions
SecB, a homotetrameric protein of molecular weight 68,000, plays a crucial role in protein export in Escherichia coli by modulating the partitioning of newly synthesized precursors between folding and delivery to the membrane-bound translocation apparatus (Randall and Hardy 2000, 2002). SecB acts as a chaperone and, in this capacity, binds many different precursors with high affinity before they acquire stable tertiary structure. In addition to this promiscuous binding, SecB also exhibits specific binding to SecA, which is a peripheral component of the membrane-bound translocase. SecA is a homodimer with subunits of 102,000 molecular weight (Schmidt et al. 1988; Akita et al. 1991; Driessen 1993). In solution, the interaction between SecA and SecB has a dissociation constant (Kd) of ~1 to 2 μM (den Blaauwen et al. 1997), whereas if SecA is associated with the translocase at the membrane the affinity is higher (Kd ~30 nM; Hartl et al. 1990). Fekkes et al. (1997) proposed that at the membrane, the ligand is transferred from SecB to SecA when SecA binds ATP and undergoes a change in conformation. SecA then acts in concert with SecYEG to translocate the polypeptide through the membrane to the periplasm (for a recent review, see Economou 2002).
The first clues concerning the sites of interaction between SecB and SecA were provided by mutant strains isolated by Gannon and Kumamoto (1993). These strains, which were defective in export in vivo, produced species of SecB with single amino acid substitutions at four positions: Asp 20, Glu 24, Leu 75, and Glu 77. It was subsequently reported (Fekkes et al. 1998) that these altered species of SecB were incapable of binding membrane-associated SecA. The interacting partner within SecA was shown to be the last 21 residues, which contain a bound zinc (Breukink et al. 1995; Fekkes et al. 1999). The X-ray structure of SecB (Xu et al. 2000) shows that the residues on SecB altered by these mutations lie in a ring on the negatively charged flat surface formed by β-sheets from each monomer of a SecB dimer. Because SecB is a tetramer arranged as a dimer of dimers, there is one binding site on each side of the tetramer that could interact with the positively charged C terminus of each subunit in the homodimeric SecA protein (Fig. 1 ▶). Woodbury et al. (2000) demonstrated that although this contact between the C terminus of SecA and the β-sheet of SecB is necessary for a SecA–SecB complex to be active in binding the membrane, in the complete absence of this interaction there are additional sites of contact that provide sufficient binding energy to allow isolation of a SecA–SecB complex in solution. Here we define one additional site of contact on SecB as the C-terminal α-helix. The energy of stabilization derived from the separate sites was determined by titration calorimetry, and the various complexes stabilized by one or both interactions were characterized hydrodynamically by sedimentation velocity centrifugation.
Figure 1.
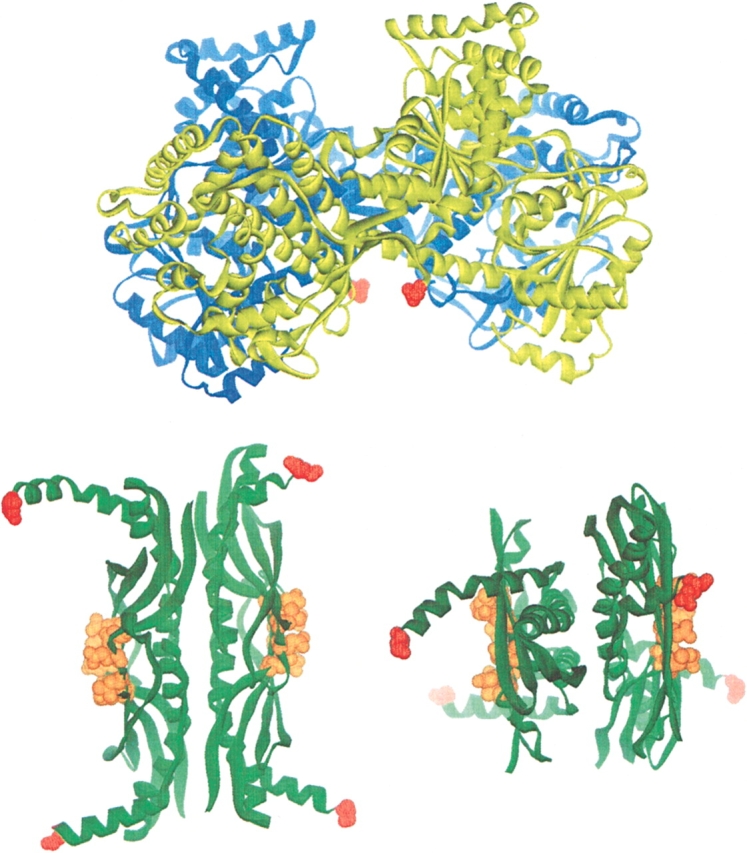
Structures of SecA and SecB. Ribbon representations are shown of the SecA dimer (blue and yellow) from Bacillus subtilis (Hunt et al. 2002) and the SecB tetramer (green) from Hemophilus influenzae. The view of SecB on the right is rotated 90 degrees toward the viewer around the horizontal axis of the view shown on the left. The extreme C-terminal residues of all monomers are shown in red. On SecB the residues that interact with the C terminus of SecA (D20, E24, L75, and E77 in the E. coli protein, corresponding to D27, E31, I84, and E86 in the Hemophilus structure) are shown as CPK representations in orange. Note that the C-terminal 21 residues of SecA that interact with the ring of residues on the β-sheet of SecB are not resolved in the crystal structure but would extend from the red residues shown.
Results
Energetics of binding
We have used titration calorimetry to determine the thermodynamic binding parameters of the interactions. Titration of SecA with SecB was performed by successive injections of SecB into a solution of 7 μM SecA dimer held at 8°C in the reaction cell. Each injection resulted in an exothermic heat effect until SecA was saturated (Fig. 2A ▶). The reaction heat, obtained by integration of the deflection from baseline for each injection and corrected by subtraction of the integrated heat of dilution, was normalized to the moles of injectant and is plotted in Figure 2B ▶. The best fit of these data gave a Kd of 1.8 μM, a change in enthalpy (ΔH) of −25,000 cal/mole, and a stoichiometry for SecA dimer to SecB tetramer of 0.8. We assume the true stoichiometry is 1.0 and either 20% of the SecA is not active or there are errors in our estimates of concentration of the proteins. Table 1 shows the binding parameters for the interaction between wild-type SecA and SecB determined from nine independent titrations by using six different preparations of wild-type SecA and three of wild-type SecB. Two of the variants of SecB, SecBD20A and SecBL75Q, that were shown to be completely defective in binding membrane-associated SecA (Fekkes et al. 1998; Woodbury et al. 2000) bind SecA in solution with parameters similar to those of the wild-type SecB. The affinity for SecBL75Q was only approximately twofold lower (Kd, 4.2 μM), whereas that for SecBD20A was only slightly weaker (Kd, 2.6 μM). Even removal of the entire C-terminal 21 residues of SecA to give a truncated form, SecAN880, resulted in only a twofold lower affinity. The most extreme difference in affinity observed, from a Kd of 1.9 to 4.2 μM, represents a decrease of only 5% in energy of stabilization of the complex. These results confirm our previous conclusion that the contact between the C terminus of SecA and the ring of charges on SecB cannot be the sole site of interaction (Woodbury et al. 2000).
Figure 2.

Representative calorimetric titration of SecB into SecA. SecA at 7 μM dimer was held in the cell at 8°C. SecB, held in the syringe at 190 μM tetramer, was added in a sequence of 21 injections, each of 10 μL, spaced at 8-min intervals. (Top) Raw data. (Bottom) Integrated area of heat as a function of the mole ratio of the reactants. The points are experimental data, and the solid line is the calculated best fit using a least-squares deconvolution algorithm.
Table 1.
Parameters for interaction of SecA and SecB
| SecA | SecB | Kda (μM) | ΔG(kcal/mole)b | na,c |
| Wild type | wild type | 1.9 ± 0.3 | −7.8 | 0.88 ± 0.08 |
| SecAN880 | wild type | 3.7 ± 0.3 | −7.4 | 0.88 ± 0.08 |
| Wild type | SecBL75Q | 4.2 ± 0.6 | −7.4 | 0.9 ± 0.08 |
| Wild type | SecBD20A | 2.6 ± 0.2 | −7.6 | 0.72 ± 0.04 |
| Wild type | SecB142 | 0.70 ± 0.07 | −8.4 | 0.69 ± 0.02 |
a The errors are standard deviation for wild-type, done nine times, and for SecAN880 and SecB142, each done twice. SecBL75Q and SecBD20A were each done once, and the error of the fit is shown.
b ΔG was calculated from ΔG = −RT ln Ka and Ka = 1/Kd.
c Stoichiometry expressed as tetramer B to dimer A.
Analysis by sedimentation velocity centrifugation
As previously demonstrated using size-exclusion chromatography (Woodbury et al. 2000), the inactive complexes, which are those formed in the absence of contact with the C termini of SecA, exhibit hydrodynamic properties different from those of the wild-type complex. The hydrodynamic properties of proteins in solution are influenced by both the size and shape of the particles. To determine whether the active and inactive complexes differ in size or in conformation, we turned to sedimentation velocity ultracentrifugation. The velocity of sedimentation of a particle in a centrifugal field is a function of both the mass, which for protein can be considered equivalent to size because the density of all protein is essentially the same, and the shape of the particle as is readily seen from the relationship,
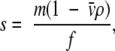 |
where s is the sedimentation coefficient in a solvent of density ρ, of a particle of mass m, having a frictional coefficient of f and a partial specific volume of v̄ In the case of the complexes between SecA and SecB, differences in size could arise either from different stoichiometries or simply from differences in the Kds of the complexes. What we assess during analysis is not an individual molecule but rather the entire population of species present. We see the average behavior; therefore, unless we work at concentrations several orders of magnitude above the Kd, we see the average sedimentation of the separate species and the associated complex.
When protein is loaded into a sample cell and subjected to centrifugation, the boundary of protein moves away from the meniscus. As seen in Figure 3A ▶, the boundary not only sediments with time but spreads because of diffusion. Analysis of SecA–SecB interaction is complicated because the system contains multiple species, many of which are involved in multiple equilibria. Therefore, it is advantageous to extract the s value independently of the diffusional spreading. We accomplish this by using the method of van Holde and Weischet (1978; for a detailed description, see Demeler et al. 1997). In this analysis the sedimenting boundaries are divided into 50 equally spaced segments along the concentration axis, and the apparent sedimentation coefficient, s*, is calculated for each segment (Fig. 3 ▶). As one moves through the sedimenting boundary from the center of rotation to the bottom of the cell, protein in the boundary fractions below the midpoint lies on the side of the boundary toward the meniscus, and protein in the upper boundary fractions lies on the side toward the bottom of the cell. The midpoint of the boundary is boundary fraction 0.5. As one can see from Figure 3A ▶, the protein in fractions below the midpoint sediments apparently more slowly and that in fractions above the midpoint apparently faster than does the protein at the midpoint. These apparent differences in s values are the effect of diffusion and do not reflect the true s values of the molecules in the population. Plots of s* as a function of the inverse of the square root of time allow extrapolation to infinite time. Because diffusion is proportional to the square root of time whereas sedimentation is proportional to the time of centrifugation, the effect of diffusion is eliminated (Fig. 3B ▶). In the case of a pure protein, the extrapolated values converge on the y-axis. The example shown in Figure 3B ▶ is analysis of sedimentation velocity data from a mixture of SecA and SecB, which are reversibly associating during centrifugation. In this case the extrapolated s values do not converge but rather show a distribution. The lowest and highest s values in the distribution depend on the s values of the individual interacting species, the magnitude of the equilibrium constant and the loading concentration used at the start of the sedimentation. The van Holde-Weischet analyses of the type shown in Figure 3B ▶ are displayed in the remainder of this study as plots of the extrapolated s values versus the boundary fraction, as shown in Figure 3C ▶. The distribution of extrapolated s values for SecB, a pure species that does not dissociate, is vertical; that is, the sedimentation coefficient of the protein in each of the 50 segments is shown to be the same, as must be the case for a population composed of molecules that are not associating. Both SecA, a pure self-associating species, and a mixture of SecA and SecB exhibit distributions in which the s values smoothly increase as a function of concentration (Fig. 3C ▶). At high concentrations of protein, nonideal behavior is exhibited by a decrease in s value. Nonideal behavior can be attributed to one of a number of causes, including asymmetric shape of the particle or high charge density.
Figure 3.
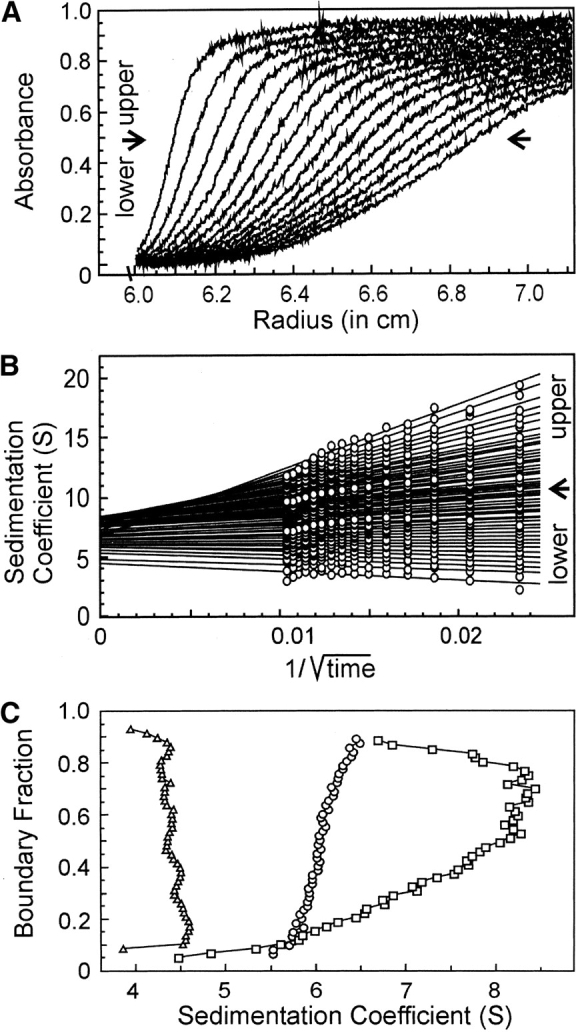
Sedimentation velocity centrifugation of SecA and SecB. (A) Raw data. The analytical ultracentrifuge cell contained SecA and SecB at 3 μM each in 10 mM Hepes-KOH, 300 mM KOAc, 5 mM Mg(OAc)2, and 1 mM TCEP (pH 7.6). Sixteen successive scans are shown to display the sedimenting boundary. Scans 2 through 16 were subjected to analysis as shown in B. (B) van Holde and Weischet analysis (1978). Each point is the apparent sedimentation coefficient (s*) of each fraction of the boundary, and each vertical array of 50 points represents one boundary. Each line is the best fit through the points for a particular boundary fraction. (C) Distribution plot of extrapolated s values. Protein samples loaded into three cells were subjected to centrifugation, and the raw data were analyzed by the method of van Holde and Weischet as described above. The intercepts of each line in the van Holde and Weischet analysis were plotted vs. the boundary fraction to which the line pertained. The samples subjected to analysis were SecB, 4 μM tetramer (triangles); SecA 5 μM dimer (circles); SecA and SecB mixed at 3 μM each (squares).
Complexes differ in hydrodynamics properties
Analysis of the SecA–SecB system is particularly challenging because both the self-association of SecA and the intermolecular interactions between SecA and SecB are characterized by equilibrium constants in the micromolar range. To distinguish effects of mass from those of shape, comparison of sedimentation of the different complexes must be made, not at the same concentration but at concentrations that give the same degree of association. Figure 4 ▶ shows analysis of SecA alone (s value 6.5 S) and mixtures of SecA and SecB. When the cell contained wild-type SecB and SecA each at 4 μM, the s value smoothly increased to a maximum of 9.0 S at the highest boundary fraction. Because the Kd for this interaction is 1.9 μM, one would expect 68% of the population to be in complex. Analysis of SecA and SecBL75Q at 20 μM each, conditions under which the complex should be 83% populated (Kd = 4.2 μM), showed a maximum s value of 7.5 S, significantly lower than that of the wild-type complex (Fig. 4 ▶). We conclude that the complexes differ in shape because the weight average mass of the protein in the wild-type mixture (68% associated) should be less than that of the mixture of SecA with the variant of SecB (83% associated), and yet the s value is higher. This conclusion depends on the assumption that all complexes have the same stoichiometry, as further elaborated in the Discussion.
Figure 4.

Comparison of sedimentation velocity of wild-type and inactive complexes. Distribution plots were generated as described in legend to Fig. 3C ▶ for van Holde and Weischet (1978) analyses of SecA, 10 μM dimer (triangles); a mixture of SecA and SecB L75Q at 20 μM each (squares); a mixture of SecA and SecB at 4 μM each (circles).
The lower s values of the inactive complexes relative to that of the wild-type complex suggest that these complexes are less compact than is the wild-type. Complexes between SecA and SecB that do not make an interaction between the C terminus of SecA and the flat β-sheet on SecB, such as the complex between SecA and SecBL75Q, might be expected to have a higher frictional ratio simply because the C-terminal portion of SecA is not tightly associated with the rest of the complex. However, this cannot be the explanation because all inactive complexes display similar hydrodynamic properties during centrifugation whether the defect has its origin in amino acyl substitutions in SecB, truncation of SecA, lack of zinc as a result of mutational alteration of the Zn2+ binding site (Fig. 5 ▶), or simply loss of zinc during purification of SecA (data not shown). Thus, it seems likely that contact between the C terminus of SecA and the ring of residues on SecB is crucial to stabilization of the conformation that results in the higher s value of the active complex. Support for this idea is provided by the demonstration that addition of a synthetic peptide having the sequence of the final 24 residues of SecA to a wild-type SecA–SecB complex converts the complex to the hydrodynamic state characteristic of the inactive forms. Wild-type SecA and SecB at 3 μM each were loaded into a sample cell and centrifuged for 3 h to acquire data showing the distribution in s values for the mixture (Fig. 6 ▶). The centrifuge was then stopped, and after addition of the peptide and Zn2+ to the sample cell at a fourfold molar excess over the SecA monomer, the mixture was again subjected to centrifugation. The presence of the peptide caused the complex to sediment with the same distribution of s values as the complexes formed by SecA and SecBL75Q, which cannot make contact with the C terminus of SecA. In the absence of Zn2+, the peptide had no effect (data not shown). As one would predict, the inclusion of the peptide and Zn2+ had no effect on the s value distribution of complexes that lack the contact between the C terminus of SecA and the β-sheet of SecB–SecA:SecBL75Q, SecA–SecBD2OA, and SecAN880–SecB (data not shown).
Figure 5.

Sedimentation velocity centrifugation of various species of SecA and SecB in complex. Distribution plots as described in legend to Fig. 3C ▶ for analyses of SecAN880 only (filled diamonds), SecA and SecB (triangles), SecA and SecBL75Q (open diamonds), SecAN880 and SecB (squares), SecAC98SC887S and SecB (circles). All proteins were present at 3 μM. The distribution of s values is not shown for SecA, but it was indistinguishable from that of SecAN880. SecAC98SC887S had a distribution centered around 5.9 S, indicating that the alterations increase the monomer–dimer equilibrium constant.
Figure 6.

Effect of C-terminal peptide of SecA on wild-type complex of SecA and SecB. Distribution plots of s values for SecA only at 3 μM (circles), a mixture of SecA and SecB at 3 μM (diamonds), SecA and SecB with addition of SecA C-terminal 21 amino acid peptide and Zn2+ (24 μM each; squares), and addition of 1 mM EDTA to SecA, SecB, peptide and Zn2+ (triangles). See text for details.
Conversion between the two states is reversible
We made use of the requirement for Zn2+ to demonstrate that the conversion of the complexes between the two hydrodynamic states is readily reversible. The C terminus of SecA contains three Cys residues (C885, C887, C896) and one His residue (H897) that provide the ligands for the Zn2+ in the intact SecA (Fekkes et al. 1999). The synthetic peptide binds Zn2+ so tightly that EDTA cannot remove it. This was demonstrated by the fact that addition of 1 mM EDTA to the centrifuge cell containing SecA, SecB, the peptide, and Zn2+ did not reverse the effect of the peptide on the complex (Fig. 6 ▶). When a modified 24-residue peptide, synthesized with alanine in place of the histidine, was added in the presence of Zn2+, this peptide affected sedimentation in the same way as did the wild-type peptide, but addition of 1 mM EDTA to the same sample restored the distribution of s values to that seen in absence of the peptide (Fig. 7 ▶). We interpret these results to mean that the Ala-containing peptide binds Zn2+ but with an affinity that is sufficiently low so that EDTA can remove the Zn2+. The simplest explanation for these observations is that the peptide competes with the C terminus of SecA for a binding site on SecB and that this contact stabilizes the active complex. However, the situation may be more complicated, and the C terminus of SecA might be involved in more than one type of interaction. SecA has been shown to undergo at least two monomer–dimer equilibrium reactions in solution (Woodbury et al. 2002) and to exist in multiple conformations (Shinkai et al. 1991; den Blaauwen et al. 1996; Ramamurthy and Oliver 1997; Chen et al. 1998; Wang et al. 2003). It is possible that the C terminus of SecA makes contacts with other regions within SecA itself and is involved in stabilizing a particular state.
Figure 7.

Reversible conversion between hydrodynamic states. Distribution plots of s values for SecA only, 4 μM (diamonds); SecA and SecB, 4 μM (circles); SecA and SecB with addition of Ala-containing peptide and Zn2+ (squares); and addition of 1 mM EDTA to SecA, SecB, peptide, and Zn2+ (triangles). See text for details.
Second site of interaction
The demonstration of a complex between SecB and the truncated species of SecA, SecAN880, clearly indicates that sites of interaction exist in addition to those involving the extreme C terminus of SecA. Although we do not yet know the part of SecA that is involved, we have defined the region on SecB as the C-terminal α-helix. SecB142, a species that lacks 13 amino acids from the C terminus of SecB, shows no interaction with SecAN880 as assessed by either size-exclusion chromatography (Fig. 8 ▶) or analytical centrifugation (Fig. 9 ▶). SecB142 does make a complex with wild-type SecA (Figs. 8, 9 ▶ ▶). This complex is stabilized solely by the interaction between the C terminus of SecA and the β-sheet of SecB. Thus, addition of the Zn2+-containing peptide mimic of the C terminus of SecA to a complex between SecA and SecB142 in the centrifuge completely disrupts the complex, resulting in an s value distribution similar to that of SecAN880 and SecB142 which can make neither contact (Fig. 9 ▶).
Figure 8.

Size-exclusion chromatography of SecAN880 and SecB142. Two-hundred μL samples were subjected to size-exclusion chromatography at 6°C. The concentration of protein was 3 μM of each, and the proteins were SecB142 only (line with short dashes), SecAN880 only (dotted line), mixture of SecB142 and SecAN880 (solid line), and mixture of SecA and SecB142 (line with short and long dashes).
Figure 9.

Sedimentation velocity centrifugation of SecAN880 and SecB142. The distribution of s values are shown for SecAN880 (circles approaching 6.5 S), SecAN880 and SecB (squares), SecA and SecB (circles approaching 9 S), SecAN880 and SecB142 (diamonds), SecA and SecB142 (open triangles), the C-terminal SecA peptide and Zn2+ added to SecA and SecB142 (filled triangles). All proteins were 4 μM each.
The complexes between SecA and SecB142 afford the opportunity to determine the contribution to the binding energy provided by the contact between β-sheet and C terminus of SecA. The Kd of the complex between the wildtype species, in which both interactions are made, was 1.9 μM, reflecting a favorable ΔG of 7.8 kcal/mole. When the only contact in the complex is between SecA and the C-terminal α-helix of SecB, the binding energy was 7.4 kcal/mole (calculated from the Kd for a complex between truncated SecA, SecAN880, and intact SecB, Table 1). If the binding energies were additive, then the energy due to the interaction with the β-sheet of SecB would contribute only 0.4 kcal/mole, which is the difference. However, calorimetric titrations of intact SecA with SecB142 show that the complex is stabilized by a ΔG of 8.4 kcal/mole (calculated from the determined Kd of 0.7 μM ± 0.07). When both contacts are available, it appears that the complex is constrained such that the C-terminal helix of SecB makes a negative contribution to the binding energy.
Discussion
During protein export in E. coli, SecA is involved in a cycle that results in translocation of precursor proteins through the membrane-associated translocase. SecA is an ATPase that is fully activated when it is associated with precursor polypeptide, the translocase, and phospholipids (Cunningham and Wickner 1989; Lill 1990; Miller et al. 1998). The binding of SecB also stimulates the ATPase activity (Miller et al. 2002). The energy of ATP binding and hydrolysis drives a cycle of insertion and deinsertion of SecA at the membrane, which in turn translocates the precursor (Economou and Wickner 1994; Economou et al. 1995). A number of exported proteins depend on the cytosolic chaperone SecB for delivery to SecA. As the precursor polypeptide is passed from SecB to SecA and finally through the translocase to the periplasm, SecA undergoes changes in conformation and oligomeric state. These changes are induced by ATP binding and hydrolysis and by interaction with protein partners as well as with lipids (Ulbrandt et al. 1992; Economou et al. 1995; den Blaauwen et al. 1996; Ding et al. 2001; Baud et al. 2002; Or et al. 2002; Wang et al. 2003). We are examining the details of the interactions between SecA and SecB in order to develop a molecular understanding of how the conformational changes are triggered. This and previous studies (Breukink et al. 1995; Fekkes et al. 1999; Woodbury et al. 2000) show there are at least two sites of contact between the two proteins. The extreme C-terminal region of each SecA monomer with Zn2+ bound interacts with a ring of negative charges on the β-sheet of each SecB dimer. A second site of interaction, identified here, is between the C-terminal α-helices of SecB (13 amino acids in length) and regions on SecA that are not yet defined.
Both contacts must be made for the complex to be active (Woodbury et al. 2000). The complexes that do not make contact between SecA and the β-sheet of SecB sediment more slowly during centrifugation than do the active wild-type complexes. The simplest interpretation of this difference in s values is that the active conformation is more compact. However, it must be remembered that this interpretation assumes the same stoichiometry for both types of complex; that is, one SecA dimer binds one SecB tetramer. The calorimetry studies indicate a stoichiometry of A : B, expressed in terms of monomers of 2 : 4 for all complexes. We cannot at this time eliminate the possibility that in the case of the inactive complexes, this reflects a complex comprising one A monomer bound to a B dimer. If this were the case, the different s values would reflect differences in mass as well as shape.
Complexes of SecA and SecB are capable of binding membrane vesicles only when the C terminus of SecA makes proper contact with the β-sheet of SecB (Woodbury et al. 2000). This is intriguing because the C terminus of SecA, which must bind SecB to render the complex active, has also been identified as the region of SecA that interacts with phospholipids (Breukink et al. 1995). In addition, this region was among those shown to be exposed to modifying reagents added to the periplasmic side of the membrane (Kim et al. 1994; van der Does et al. 1996; Ramamurthy and Oliver 1997). Obviously, after the SecA–SecB complex binds the membrane and SecB is released, conformational changes or rearrangement of subunits must occur to poise SecA for the translocation step.
We have investigated the energetics of the interactions between SecA and SecB as a start in elucidation of changes the complexes undergo during the export cycle. The binding energy provided by these two different interactions is not additive. When the only contact in a complex is that involving the C termini of the SecB tetramer, the complex has ~7.4 kcal/mole energy of stabilization, only 0.4 kcal/mole less than the energy of stabilization of the complex with both interactions. However, when this contact is absent and the complex is stabilized only by interaction of the Zn2+-containing region of SecA with the β-sheet of SecB, the binding is actually approximately twofold tighter (ΔG stabilization, 8.4 kcal/mole) than that seen in the complex with both partners intact (ΔG stabilization, 7.8 kcal/mole). This means that the C-terminal α-helices make a negative energy contribution when SecA is bound to the β-sheets of SecB. Examination of the structures shown in Figure 1 ▶ offers an explanation. The C-terminal helices are resolved in the structure of Hemophilus SecB to a different extent for each subunit. The least ordered terminus is resolved only to residue 151 (corresponding to 142 in E. coli out of a total of 155 residues). The best resolved extends to residue 163 (151 in E. coli). This disorder observed in the crystals is consistent with NMR studies carried out by Volkert et al. (1999), showing that in solution the C termini are mobile. These investigators further showed that removal of the mobile C terminus to generate SecB142, the species used in this study, resulted in a defect in export in vivo, even though the truncation caused only a twofold decrease in affinity for precursor (Diamond and Randall 1997). Because the export defect was overcome by increased production of SecA in vivo, the investigators concluded that the defect was likely to be in the interaction between SecA and SecB. They pointed out that to perform its physiological function efficiently, SecB must dissociate from SecA, and they proposed that the mobile C termini might decrease the affinity to ensure that SecA does not bind SecB too tightly.
An extension of this idea is that unoccupied SecB should have a lower affinity for SecA than when it has a precursor ligand bound so it would be released upon transfer of the ligand to SecA. The C-terminal helices of SecB are known to be involved in precursor binding, and we proposed earlier (Randall and Hardy 2000) that the helices might fold over the precursor ligand along its pathway from the binding groove on one side of the SecB tetramer to the other side (Fig. 1 ▶). Such a cap would not only stabilize ligand binding but also mask differences on the surface created by the various precursors that SecB might have in its binding site. In this way, all SecB-precursor complexes could present a common face to SecA for binding. This capping would also immobilize the helices and might well eliminate the negative contribution to the energy of binding between SecB and SecA. One can envision that the C-terminal α-helices of SecB undergo a dynamic cycle of order and disorder coupled to the binding and release of a precursor polypeptide ligand, which in turn would affect the affinity of SecB for SecA. The net result might be transfer of the precursor from SecB to SecA with release of SecB. SecA is likely to be an active participant in this process because the binding of ATP to SecA triggers the transfer of precursor to SecA and the release of SecB (Fekkes et al. 1998). There is a myriad of interactions that could have mutual affects on the interacting partners. We are currently exploring the possibilities.
Materials and methods
Bacterial strains and plasmids
Wild-type SecA was purified from several strains: RR1/pMAN400 (Kawasaki et al. 1989), Cy15077/pMAN400 (Ross et al. 1997), and BL21.9(λDE3)-harboring plasmid pT7SecA2 (Dapic and Oliver 2000). SecAN880, a truncated SecA consisting of the first 880 aminoacyl residues, was purified from E. coli strain HB1732 (Woodbury et al. 2000). Wild-type SecB was purified from HB1042, which is BL21(λDE3)-harboring plasmid pJW25 (Weiss et al. 1988); SecB142 from HB1596, which is CK2212-harboring plasmid pTV6 (Volkert et al. 1999); and SecBL75Q from HB1579 and SecBD20A from HB1561, which are CK2212-harboring plasmids as described (Kimsey et al. 1995).
Protein purification
SecA was purified as previously described (Woodbury et al. 2000) with the following changes. The sonication step was omitted because it resulted in oxidation of cysteines and a decrease in zinc retention, centrifugation of the cell suspension was increased to 362,000g for 3 h (60Ti rotor, Beckman), and the Macroprep DEAE column was replaced by a Q-Sepharose Fast Flow column (Amersham). All species of SecB were purified as described previously (Randall et al. 1998), with the following modifications. The sonication step was eliminated from preparation of the cellular lysate, and the two low speed spins were replaced with a high-speed spin (362,000g, 3 h, 60Ti rotor, Beckman). In the first step of column chromatography, the Q-Sepharose Fast Flow ion-exchange column (Amersham) was equilibrated with 20 mm Tris-Cl (pH 7.6), and the gradient was developed over 600 mL. This was followed by an HPLC size-exclusion column, BioSep-SEC-S3000 (60 × 2.1 cm, Phenomenex), equilibrated with 10 mm Hepes-KOH, 300 mM KOAc, and 2 mM DTT (pH 7.6). The second ion-exchange column was eliminated. Purified SecA and SecB were dialyzed against 10 mM Hepes-KOH, 300 mM KOAc, and 2 mM TCEP (pH 7.6) and stored at −70°C. The protein concentrations were determined spectrophotometrically at 280 nm by using coefficients of extinction as follows: 47,600 M−1 cm−1 for SecB tetramer and 157, 800 M−1 cm−1 for SecA dimer. All concentrations are expressed as tetrameric SecB and dimeric SecA. The zinc content of purified SecA was determined by using a spectroscopic assay as described (Zhou et al. 1999). We routinely find 0.7 to 0.9 mole Zn2+ per mole of SecA, but extreme care must be taken to avoid oxidation of cysteines during purification.
Titration calorimetry
All calorimetric titrations were carried out by using the VP-ITC titration calorimeter from MicroCal, Inc. The system has been described in detail (Wiseman et al. 1989). Titrations of SecA with SecB were all carried out following essentially the same procedure. SecA was held in the cell at 7 μM dimer in 10 mM Hepes-KOH, 300 mM KOAc, 5 mM Mg(OAc)2, and 1 mM TCEP (pH 7.6) at 8°C (cell volume, 1.44 mL). SecB (held in the syringe at between 170 and 200 μM tetramer depending on the species of SecB) in this same buffer was added in a sequence of between 17 and 21 injections spaced at 8-min intervals. The integrated area of heat of each injection was corrected for the heat of dilution and plotted as a function of the molar ratio of the reactants. The best fit of the data was calculated by using the Origin software supplied with the instrument. The model used was the “one set of sites” curve fitting model. The errors shown in Table 1 are standard deviations with the exception of the titrations for SecBL75Q and SecBD20A. These were each done once, and the error given is the error of the fit. The fitting error for all fits of the data where standard deviations are shown was ≤±0.1 μM.
Analytical centrifugation
Solutions containing mixtures of proteins at the concentrations indicated in 10 mM Hepes-KOH, 300 mM KOAc, 5 mM Mg(OAc)2, and 1 mM TCEP (pH 7.6) were subjected to centrifugation by using the XL-I ultracentrifuge (Beckman Instruments). Samples (415 μL) were loaded into cells with two-sector centerpieces in either the An-60 Ti or An-50 Ti rotor, and after equilibration to 6°C, were centrifuged at 50,000 rpm (An-60 Ti) or 45,000 rpm (An-50 Ti) for up to 4 h. Radial scans to measure the absorbance profile at 280 nm of the column of liquid in each of the cells in the rotor were taken at ~4-min intervals for the An-60 Ti and ~9-min intervals for An-50 Ti. The data were analyzed by the method of van Holde and Weischet (1978), using the Ultrascan Data Analysis Program version 6.0 from Borries Demeler (University of Texas Science Center, San Antonio, Texas). The values used for the density and viscosity of the buffer relative to water were 1.014 and 1.063, respectively. The s values reported are all corrected to water at 20°C.
Size-exclusion chromatography
Chromatography was carried out by using a TSK G3000SW (To-soHaas) column (7.5 mm inner diameter × 60 cm) equilibrated in 10 mM Hepes-KOH, 300 mM KOAc, 5 mM Mg(OAc)2, and 1 mM TCEP (pH 7.6). Separation was carried out at 8°C at 0.7 mL/min, and absorbance was monitored at 280 nm.
Acknowledgments
We thank Virginia F. Smith and Chunfeng Mao for critically reading the manuscript. We are grateful to Gerhard Munske for synthesis of the peptides used in this work and Traci B. Topping for several of the SecA purifications. This work was supported in part by NIH research grant GM29798 to L.L.R. and an endowment from the Hugo Wurdack Trust at the University of Missouri.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03410104.
References
- Akita, M., Shinkai, A., Matsuyama, S., and Mizushima, S. 1991. SecA, an essential component of the secretory machinery of Escherichia coli, exists as homodimer. Biochem. Biophys. Res. Commun. 174 211–216. [DOI] [PubMed] [Google Scholar]
- Baud, C., Karamanou, S., Sianidis, G., Vrontou, E., Politou, A.S., and Economou, A. 2002. Allosteric communication between signal peptides and the SecA protein DEAD motor ATPase domain. J. Biol. Chem. 277 13724–13731. [DOI] [PubMed] [Google Scholar]
- Breukink, E., Nouwen, N., van Raalte, A., Mizushima, S., Tommassen, J., and de Kruijff, B. 1995. The C terminus of SecA is involved in both lipid binding and SecB binding. J. Biol. Chem. 270 7902–7907. [DOI] [PubMed] [Google Scholar]
- Chen, X., Brown, T., and Tai, P.C. 1998. Identification and characterization of protease-resistant SecA fragments: SecA has two membrane-integral forms. J. Bacteriol. 180 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, K. and Wickner, W. 1989. Specific recognition of the leader region of precursor proteins is required for the activation of translocation ATPase of Escherichia coli. Proc. Natl. Acad. Sci. 86 8630–8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapic, V. and Oliver, D. 2000. Distinct membrane binding properties of N- and C-terminal domains of Escherichia coli SecA ATPase. J. Biol. Chem. 275 25000–25007. [DOI] [PubMed] [Google Scholar]
- Demeler, B., Saber, H., and Hansen, J.C. 1997. Identification and interpretation of complexity in sedimentation velocity boundaries. Biophys. J. 72 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Blaauwen, T., Fekkes, P., de Wit, J.G., Kuiper, W., and Driessen, A.J. 1996. Domain interactions of the peripheral preprotein translocase subunit SecA. Biochemistry 35 11994–12004. [DOI] [PubMed] [Google Scholar]
- den Blaauwen, T., Terpetschnig, E., Lakowicz, J.R., and Driessen, A.J. 1997. Interaction of SecB with soluble SecA. FEBS Lett. 416 35–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, D.L. and Randall, L.L. 1997. Kinetic partitioning: Poising SecB to favor association with a rapidly folding ligand. J. Biol. Chem. 272 28994–28998. [DOI] [PubMed] [Google Scholar]
- Ding, H., Mukerji, I., and Oliver, D. 2001. Lipid and signal peptide-induced conformational changes within the C-domain of Escherichia coli SecA protein. Biochemistry 40 1835–1843. [DOI] [PubMed] [Google Scholar]
- Driessen, A.J. 1993. SecA, the peripheral subunit of the Escherichia coli precursor protein translocase, is functional as a dimer. Biochemistry 32 13190–13197. [DOI] [PubMed] [Google Scholar]
- Economou, A. 2002. Bacterial secretome: The assembly manual and operating instructions. Mol. Membr. Biol. 19 159–169. [DOI] [PubMed] [Google Scholar]
- Economou, A. and Wickner, W. 1994. SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78 835–843. [DOI] [PubMed] [Google Scholar]
- Economou, A., Pogliano, J.A., Beckwith, J., Oliver, D.B., and Wickner, W. 1995. SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell 83 1171–1181. [DOI] [PubMed] [Google Scholar]
- Fekkes, P., van der Does, C., and Driessen, A.J. 1997. The molecular chaperone SecB is released from the carboxy-terminus of SecA during initiation of precursor protein translocation. EMBO J. 16 6105–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekkes, P., de Wit, J.G., van der Wolk, J.P., Kimsey, H.H., Kumamoto, C.A., and Driessen, A.J. 1998. Preprotein transfer to the Escherichia coli translocase requires the co-operative binding of SecB and the signal sequence to SecA. Mol. Microbiol. 29 1179–1190. [DOI] [PubMed] [Google Scholar]
- Fekkes, P., de Wit, J.G., Boorsma, A., Friesen, R.H., and Driessen, A.J. 1999. Zinc stabilizes the SecB binding site of SecA. Biochemistry 38 5111–5116. [DOI] [PubMed] [Google Scholar]
- Gannon, P.M. and Kumamoto, C.A. 1993. Mutations of the molecular chaperone protein SecB which alter the interaction between SecB and maltose-binding protein. J. Biol. Chem. 268 1590–1595. [PubMed] [Google Scholar]
- Hartl, F.U., Lecker, S., Schiebel, E., Hendrick, J.P., and Wickner, W. 1990. The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E. coli plasma membrane. Cell 63 269–279. [DOI] [PubMed] [Google Scholar]
- Hunt, J.F., Weinkauf, S., Henry, L., Fak, J.J., McNichols, P., Oliver, D.B., and Deisenhofer, J. 2002. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science. 297 2018–2026. [DOI] [PubMed] [Google Scholar]
- Kawasaki, H., Matsuyama, S., Sasaki, S., Akita, M., and Mizushima, S. 1989. SecA protein is directly involved in protein secretion in Escherichia coli. FEBS Lett. 242 431–434. [DOI] [PubMed] [Google Scholar]
- Kim, Y.J., Rajapandi, T., and Oliver, D. 1994. SecA protein is exposed to the periplasmic surface of the E. coli inner membrane in its active state. Cell 78 845–853. [DOI] [PubMed] [Google Scholar]
- Kimsey, H.H., Dagarag, M.D., and Kumamoto, C.A. 1995. Diverse effects of mutation on the activity of the Escherichia coli export chaperone SecB. J. Biol. Chem. 270 22831–22835. [DOI] [PubMed] [Google Scholar]
- Lill, R., Dowhan, W., and Wickner, W. 1990. The ATPase activity of SecA is regulated by acidic phospholipids, SecY, and the leader and mature domains of precursor proteins. Cell 60 271–280. [DOI] [PubMed] [Google Scholar]
- Miller, A., Wang, L., and Kendall, D.A. 1998. Synthetic signal peptides specifically recognize SecA and stimulate ATPase activity in the absence of preprotein. J. Biol. Chem. 273 11409–11412. [DOI] [PubMed] [Google Scholar]
- ———. 2002. SecB modulates the nucleotide-bound state of SecA and stimulates ATPase activity. Biochemistry 41 5325–5332. [DOI] [PubMed] [Google Scholar]
- Or, E., Navon, A., and Rapoport, T. 2002. Dissociation of the dimeric SecA ATPase during protein translocation across the bacterial membrane. EMBO J. 21 4470–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy, V. and Oliver, D. 1997. Topology of the integral membrane form of Escherichia coli SecA protein reveals multiple periplasmically exposed regions and modulation by ATP binding. J. Biol. Chem. 272 23239–23246. [DOI] [PubMed] [Google Scholar]
- Randall, L.L. and Hardy, S.J. 2000. The promiscuous and specific sides of SecB. Nat. Struct. Biol. 7 1077–1079. [DOI] [PubMed] [Google Scholar]
- ———. 2002. SecB: One small chaperone in the complex milieu of the cell. Cell. Mol. Life Sci. 59 1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall, L.L., Topping, T.B., Smith, V.F., Diamond, D.L., and Hardy, S.J. 1998. SecB: A chaperone from Escherichia coli. Methods Enzymol. 290 444–459. [DOI] [PubMed] [Google Scholar]
- Ross, J.B., Szabo, A.G., and Hogue, C.W. 1997. Enhancement of protein spectra with tryptophan analogs: Fluorescence spectroscopy of protein–protein and protein–nucleic acid interactions. Methods Enzymol. 278 151–190. [DOI] [PubMed] [Google Scholar]
- Schmidt, M.G., Rollo, E.E., Grodberg, J., and Oliver, D.B. 1988. Nucleotide sequence of the secA gene and secA(Ts) mutations preventing protein export in Escherichia coli. J. Bacteriol. 170 3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai, A., Mei, L.H., Tokuda, H., and Mizushima, S. 1991. The conformation of SecA, as revealed by its protease sensitivity, is altered upon interaction with ATP, presecretory proteins, everted membrane vesicles, and phospholipids. J. Biol. Chem. 266 5827–5833. [PubMed] [Google Scholar]
- Ulbrandt, N.D., London, E., and Oliver, D.B. 1992. Deep penetration of a portion of Escherichia coli SecA protein into model membranes is promoted by anionic phospholipids and by partial unfolding. J. Biol. Chem. 267 15184–15192. [PubMed] [Google Scholar]
- van der Does, C., den Blaauwen, T., de Wit, J.G., Manting, E.H., Groot, N.A., Fekkes, P., and Driessen, A.J. 1996. SecA is an intrinsic subunit of the Escherichia coli preprotein translocase and exposes its carboxyl terminus to the periplasm. Mol. Microbiol. 22 619–629. [DOI] [PubMed] [Google Scholar]
- van Holde, K.E. and Weischet, W.O. 1978. Boundary analysis of sedimentation velocity experiments with monodisperse and paucidisperse solutes. Biopolymers 17 1387–1403. [Google Scholar]
- Volkert, T.L., Baleja, J.D., and Kumamoto, C.A. 1999. A highly mobile C-terminal tail of the Escherichia coli protein export chaperone SecB. Biochem. Biophys. Res. Commun. 264 949–954. [DOI] [PubMed] [Google Scholar]
- Wang, H.W., Chen, Y., Yang, H., Chen, X., Duan, M.X., Tai, P.C., and Sui, S.F. 2003. Ring-like pore structures of SecA: Implication for bacterial protein-conducting channels. Proc. Natl. Acad. Sci. 100 4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, J.B., Ray, P.H., and Bassford Jr., P.J. 1988. Purified secB protein of Escherichia coli retards folding and promotes membrane translocation of the maltose-binding protein in vitro. Proc. Natl. Acad. Sci. 85 8978–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman, T., Williston, S., Brandts, J.F., and Lin, L.N. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179 131–137. [DOI] [PubMed] [Google Scholar]
- Woodbury, R.L., Topping, T.B., Diamond, D.L., Suciu, D., Kumamoto, C.A., Hardy, S.J., and Randall, L.L. 2000. Complexes between protein export chaperone SecB and SecA: Evidence for separate sites on SecA providing binding energy and regulatory interactions. J. Biol. Chem. 275 24191–24198. [DOI] [PubMed] [Google Scholar]
- Woodbury, R.L., Hardy, S.J., and Randall, L.L. 2002. Complex behavior in solution of homodimeric SecA. Protein Sci. 11 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z., Knafels, J.D., and Yoshino, K. 2000. Crystal structure of the bacterial protein export chaperone secB. Nat. Struct. Biol. 7 1172–1177. [DOI] [PubMed] [Google Scholar]
- Zhou, Z.S., Peariso, K., Penner-Hahn, J.E., and Matthews, R.G. 1999. Identification of the zinc ligands in cobalamin-independent methionine synthase (MetE) from Escherichia coli. Biochemistry 38 15915–15926. [DOI] [PubMed] [Google Scholar]