

Abstract
The accuracy of an alignment between two protein sequences can be improved by including other detectably related sequences in the comparison. We optimize and benchmark such an approach that relies on aligning two multiple sequence alignments, each one including one of the two protein sequences. Thirteen different protocols for creating and comparing profiles corresponding to the multiple sequence alignments are implemented in the SALIGN command of MODELLER. A test set of 200 pairwise, structure-based alignments with sequence identities below 40% is used to benchmark the 13 protocols as well as a number of previously described sequence alignment methods, including heuristic pairwise sequence alignment by BLAST, pairwise sequence alignment by global dynamic programming with an affine gap penalty function by the ALIGN command of MODELLER, sequence-profile alignment by PSI-BLAST, Hidden Markov Model methods implemented in SAM and LOBSTER, pairwise sequence alignment relying on predicted local structure by SEA, and multiple sequence alignment by CLUSTALW and COMPASS. The alignment accuracies of the best new protocols were significantly better than those of the other tested methods. For example, the fraction of the correctly aligned residues relative to the structure-based alignment by the best protocol is 56%, which can be compared with the accuracies of 26%, 42%, 43%, 48%, 50%, 49%, 43%, and 43% for the other methods, respectively. The new method is currently applied to large-scale comparative protein structure modeling of all known sequences.
Keywords: protein sequence alignment, sequence profiles, comparative protein structure modeling
Nucleic acid and protein sequence alignments are central to many problems in biology, including gene assignment, phylogeny construction, protein structure modeling, protein design, and functional annotation of proteins (Barton 1996, 1998; Gotoh 1999). An alignment between two sequences of residues is usually calculated by optimizing an alignment scoring function. The two common ingredients of the scoring function are a gap penalty function and a matrix of substitution scores for matching every residue in one sequence to every residue in the other sequence. The alignment score is usually a sum of the gap penalties that depend linearly on the gap lengths, and the pairwise substitution scores that depend on the matched residue types. The original and still widely used optimization method for sequence alignment is based on dynamic programming (Needleman and Wunsch 1970; Sellers 1974). Since its inception, the scoring function and its optimization by dynamic programming have been improved for alignment accuracy and speed, and applied to a variety of alignment problems.
One of the most significant improvements in alignment accuracy was achieved through the use of multiple sequence alignments and the corresponding sequence profiles (Gribskov et al. 1987, 1990; Gribskov 1994). For proteins, a sequence profile lists a preference for the 20 standard amino acid residue types at each position in a given multiple sequence alignment. The PSI-BLAST program relies on the BLAST algorithm (Altschul et al. 1990) to collect homologs of a query sequence and construct its profile by iteratively scanning a sequence database (Altschul et al. 1997; Park et al. 1998); a basic step in this calculation is a comparison of the query sequence profile with each sequence in the database. A multiple sequence alignment can also be transformed into a Hidden Markov Model (HMM), a class of probabilistic models that are generally applicable to a time series or linear sequences (Eddy 1998). A particularly successful method in this class is implemented in the SAM package (Hughey and Krogh 1996) that outperforms other sequence-based methods for fold recognition (Karplus et al. 1998; Park et al. 1998; Madera and Gough 2002). The SATCHMO algorithm in the LOBSTER package simultaneously constructs a similarity tree and compares multiple sequence alignments of each internal node of the tree using HMMs (Edgar and Sjolander 2003a,b). The CLUSTALW program compares two multiple sequence alignments by scoring an alignment of two positions, one from each profile, as the average of all pairwise substitution scores for the amino acid residues in the two profiles (Higgins and Sharp 1988; Thompson et al. 1994). The LAMA program aligns two multiple sequence alignments by first transforming them into profiles and then comparing the two to each other by the Pearson correlation coefficient (Pietrokovski 1996). Similarly, the FFAS program was developed to align two sequence profiles with each other (Jaroszewski et al. 2000; Rychlewski et al. 2000). A related approach was also used by Yona and Levitt (2000) Yona and Levitt (2002) to construct the ProtoMap database of protein sequence families (Yona et al. 1999, 2000; Yona and Levitt 2002). Al-Lazikani and co-workers (2001) combined multiple structure and sequence comparisons to improve the accuracy of alignments of SH2 domains. Most recently, the COMPASS program was developed to locally align two multiple sequence alignments with assessment of statistical significance (Sadreyev and Grishin 2003). These methods compare two profiles by constructing a matrix of scores for matching every position in one profile to each position in the other profile, followed by either local or global dynamic programming to calculate the optimal alignment. It was noted previously that profile–profile alignment methods are capable of detecting more remote relationships compared to the sequence-profile methods, such as PSI-BLAST (Rychlewski et al. 2000; Panchenko 2003; Sadreyev and Grishin 2003).
Another significant improvement of the alignment accuracy in the low similarity range was achieved by considering protein structure information for one of the sequences in a pairwise comparison. The methods in this class include threading and 3D template matching (Bowie et al. 1991; Godzik and Skolnick 1992; Jones et al. 1992; Kelley et al. 2000; Shi et al. 2001; Fischer 2003. For review, see Jones 1997; Levitt 1997; Smith et al. 1997; Torda 1997; David et al. 2000).
Yet another approach is implemented in the SEA program, which aligns a pair of remotely related sequences by optimizing a match between the predicted conformations of their short segments (Ye et al. 2003). The resulting alignments were more accurate than the pairwise sequence alignments by BLAST (Altschul et al. 1990) and ALIGN (Myers and Miller 1988), as well as the profile–profile alignments by FFAS (Jaroszewski et al. 2000; Rychlewski et al. 2000).
For closely related protein sequence pairs, with sequence identity over 40%, an accurate alignment is almost always trivial to obtain. In contrast, despite the methodological advances listed above, alignments in the so-called “twilight zone” of less than 30% sequence identity still contain many errors (Rost 1999). Unfortunately, most sequences share less than 30% sequence identity to a known structure (Sanchez and Sali 1998; Pieper et al. 2002). On average, pairwise alignments between sequences at 30% sequence identity have ~20% of the residues aligned incorrectly (Johnson et al. 1993). Some pairs of related proteins have almost no correctly aligned positions when aligned by sequence-based alignments methods (Venclovas et al. 2001).
Alignment accuracy in the twilight zone is crucial for several applications, including comparative protein structure prediction (Blundell et al. 1987; Marti-Renom et al. 2000; Baker and Sali 2001). To calculate an accurate comparative model, it is necessary to identify and correctly align at least one template structure to the target sequence. An incorrect alignment invariably leads to an inaccurate model, because none of the existing comparative model building methods can generally recover from an incorrect alignment (Marti-Renom et al. 2000). A number of studies assessed both the sensitivity of alignment methods in the detection of remote homologs (Park et al. 1997; Muller et al. 1999; Kelley et al. 2000; Rychlewski et al. 2000; Yona and Levitt 2000; Panchenko 2003; Sadreyev and Grishin 2003) and alignment accuracy (Jaroszewski et al. 2000; Sauder et al. 2000; Blake and Cohen 2001; Panchenko 2003; Sadreyev and Grishin 2003), as well as optimized the alignment accuracy for protein structure prediction (Jaroszewski et al. 2000; John and Sali 2003).
In this study, we optimized alignments specifically for comparative protein structure prediction. We begin by describing 13 profile–profile alignment protocols, the training and testing alignment sets, and measures of alignment accuracy (Materials and Methods). Next, we benchmark the alignment accuracy of our profile–profile alignment protocols relative to representative sequence-sequence, profile-sequence, and other profile–profile alignment methods (Results). Finally, we discuss our improvements in sequence alignment from the point of view of comparative protein structure modeling (Discussion).
Materials and methods
We first describe the source of multiple sequence alignments used for calculating the profiles. We proceed by defining the 13 profile–profile alignment protocols in terms of four alternative schemes for transforming a multiple sequence alignment into a profile or a matrix and six alternative measures for comparison of two profiles. We also describe the training and testing alignment sets and measures of alignment accuracy.
Multiple sequence alignment
For each sequence in a pair of sequences to be aligned, a multiple sequence alignment with its homologs was prepared by scanning the nonredundant protein sequence database at NCBI (June 2002) with the program PSI-BLAST, version 2.11 (Altschul et al. 1997). The scanning was performed without filtering out compositionally biased segments, was run for up to 20 iterations, and included all matches with an e-value smaller than 0.0005. Up to 1000 sequences with the most significant e-values were retained in the multiple sequence alignment. The default values were used for all other parameters. The multiple sequence alignment and the profile were saved after each iteration. The PSI-BLAST multiple sequence alignment of a sequence was defined to be the sequence-profile alignment with the most significant e-value from any of the iterations.
Sequence weighting
Sequence weighting is part of the calculation of a sequence profile from a multiple sequence alignment, and is used to compensate for nonuniform distribution of the homologs in the alignment. We applied two different weighting schemes.
First, we tested the often used position-based sequence weighting (Henikoff and Henikoff 1994) that assigns low weights to overrepresented sequences and high weights to unique sequences:
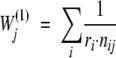 |
(1) |
where ri is the number of different residue types at position i and ni,j is the frequency of the residue type in sequence j at position i.
Second, we also tested our variation of the position-based sequence weighting that increases the weights of those sequences that are more similar to the query sequence:
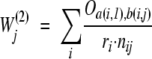 |
(2) |
where Oa(i,1),b(i,j) are the Blosum62 odds ratios for matching the residue type a in the query sequence with the residue type b in sequence j, defined as “qij/eij” in the original paper (Henikoff and Henikoff 1992).
Sequence profile
A sequence profile of a given set of similar sequences specifies a preference for each of the 20 standard amino acid residue types at each of the residue positions in the set. A number of different estimation schemes have been suggested, because a multiple alignment may not contain a sufficiently large number of homologs to calculate a statistically robust profile solely from the occurrence of each residue type in the multiple alignment. They generally depend on prior or expected probabilities of residue occurrences and/or residue-residue substitutions (Henikoff and Henikoff 1996). We tested three different profile-building methods.
First, profiles generated by pseudo-counting (Henikoff and Henikoff 1996) as implemented in the PSI-BLAST program (Altschul et al. 1997): the use of pseudo-counting for profile generation was chosen for its simplicity of implementation and comparable performance to other tested approaches (Henikoff and Henikoff 1996).
Second, profiles generated by pseudo-counting (Henikoff and Henikoff 1996) as implemented by us in the MODELLER-7 program: the probability of a residue type a to occur at position i in a multiple alignment is estimated by:
 |
(3) |
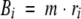 |
(4) |
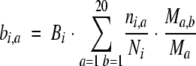 |
(5) |
Ni is the sum of the weights Wj(1) (eq. 1) for the sequences that do not have a gap at position i. ni,a is the sum of the weights Wj(1) for the sequences with residue type a at position i. Bi is the total number of pseudo-counts at position i and depends on the parameter m that is set to the optimal value of 5 (Henikoff and Henikoff 1996). bi,a is the number of pseudo-counts for residue type a at position i. Ma is the probability of residue type a in the background distribution that is obtained from the Blosum62 matrix. Ma,b are the Blosum62 probabilities (Henikoff and Henikoff 1992) for matching the residue type a in the query sequence with the residue type b in sequence j. Both ni,a/Ni and bi,a/Bi are estimates of Pi,a, based on the observed and pseudo-counts, respectively. Correspondingly, Pi,a is a weighted sum of the two estimates, with the contributions determined by Ni and Bi. If Ni is larger than Bi, Pi,a is dominated by the observed counts, whereas if Bi is larger than Ni, Pi,a is dominated by pseudo-counts.
Third, our variation of the Henikoff and Henikoff schema with sequences weighted proportionally to their similarity to the query sequence, using Wj(2) (eq. 2) instead of Wj(1) (eq. 1).
Profile–profile substitution scores
Ideally, an optimal alignment of two profiles P and Q would be obtained by relying on a matrix of probabilities Si,j that any pair of profile positions Pi and Qj are “equivalent.” It is not clear what the best definition of “equivalent” is and how to calculate such a probability of equivalence, given two profile distributions Pi and Qj. As a result, we are forced into a “parametric” approach, whereby we calculate a “substitution score” that approximates the probability of equivalence. Such substitution scores, together with a gap penalty function, can then be used to obtain an optimal alignment of two profiles by dynamic programming. Six recipes for calculating profile–profile substitution scores Si,j for each pair of profile positions i and j were tested.
First, the dot product between two distributions Pi and Qi at profile positions i and j, respectively:
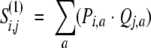 |
(6) |
Second, the correlation coefficient between two distributions Pi and Qj:
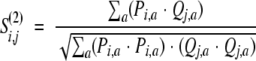 |
(7) |
Third, the Euclidean distance between two distributions Pi and Qi:
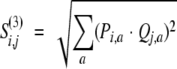 |
(8) |
Fourth, a substitution score based on the Jensen-Shannon divergence measure DJS for two distributions (Lin 1991; Yona and Levitt 2000):
 |
(9) |
 |
(10) |
 |
(11) |
The R vector can be seen as the most likely parent distribution of Pi and Qj. DKL is the Kullback-Leibler distance, also called the “cross-entropy measure” in information theory. λ is a parameter between 0 and 1, set to 0.5 in this study. λ and its complement (1−λ) are the weights given to the Pi and Qj distributions, respectively. The Jensen-Shannon divergence, though not being a true metric, is bound by 0 and 1. It is 0 when the two compared distributions are identical and 1 when they are not related at all.
Fifth, for each position in a multiple sequence alignment, a pairwise residue substitution probability matrix was calculated as a weighted sum of the Blosum62 substitution probability matrix and the matrix of relative residue substitution frequencies observed at the given position in the multiple sequence alignment. Next, the substitution score for two multiple alignment positions i and j was calculated by averaging over these residue substitution probabilities for all pairs of residues containing a residue from each of the two compared positions:
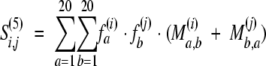 |
(12) |
 |
(13) |
 |
(14) |
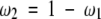 |
(15) |
where fa(i) is the observed frequency of residue type a at position i in the first multiple alignment corrected for sequence weights as defined above (using equation 1), Ma,b(i) is the substitution probability matrix for residue types a and b at position i in the first multiple alignment, Ma,b is the Blosum62 substitution probability matrix for residue types a and b, and ω1 and ω2 are scalar weights. Variable n is the number of the pairwise residue-residue substitutions within the multiple alignment at position i, and σ is a smoothing parameter (set to 0.1 by optimization of the alignment accuracy on a learning set of alignments).
Sixth, the score Si,j(6) was defined as the correlation coefficient between the corresponding values in two posterior substitution matrices Ma,b (i) and Mb,a (j) for positions i and j in the first and second multiple alignments, respectively.
After the substitution scores were computed according to one of the six recipes above, they were scaled to fit the range from 0 to 1000.
Alignment methods
The testing pairs of sequences were aligned by (1) heuristic pairwise sequence alignment as implemented in BLAST 2.1.2 (Altschul et al. 1990), (2) pairwise sequence alignment by global dynamic programming with an affine gap penalty function as implemented in the ALIGN command of MODELLER-7 (Sali et al. 2001), (3) sequence-profile alignment as implemented by PSI-BLAST 2.1.2 (Altschul et al. 1997), (4) Hidden Markov Model (HMM) as implemented in SAM 3.3.1 (Hughey and Krogh 1996) and LOBSTER (Edgar and Sjolander 2003a), (5) pairwise sequence alignment based on matching predicted local structure as implemented in the SEA Web server (Ye et al. 2003), (6) multiple sequence alignment by CLUSTALW 1.81 (Thompson et al. 1994), (7) profile–profile alignments as implemented by COMPASS 1.24 (Sadreyev and Grishin 2003), and (8) the 13 schemes of profile–profile alignment by global dynamic programming with an affine gap penalty function as implemented by the SALIGN command of MODELLER-7.
For BLAST, a high e-value threshold of 100 was used for accepting an alignment between two sequences. Otherwise, the pair of sequences was ignored. Here, we focus on the alignment accuracy rather the accuracy of the methods to detect relationships. Therefore, we increased the e-value threshold relative to the commonly used value of ~10−4 to produce the maximum number of pairwise alignments obtained from the BLAST program. All other parameters were kept at their default values. Only four pairs of sequences did not have any fragments that could be aligned by this method.
For ALIGN, the default parameters were used. They include the AS1 residue type similarity matrix calculated from the reference structure alignments (file “as1.mat” in the MODELLER distribution; Overington et al. 1992), the initiation gap penalty u of −450, and the extension gap penalty v of −50; the penalty for a gap of n residue positions is u + v n.
For PSI-BLAST, multiple sequence alignments of each one of the two sequences were calculated as described above. The sequence-profile alignment with the most significant e-value from any of the iterations with either of the two sequences as queries was used as the PSI-BLAST alignment. Only two pairs of sequences did not have any fragments that could be aligned by this method.
For SAM, the following protocol was used (R. Karchin, pers. comm.). The w0.5 script with default parameters was applied to build HMMs for the target and template sequences, using their PSI-BLAST multiple sequence alignments. Next, the program hmmscore in the SAM package (sw = 0; select_align = 8; adpstyle = 5) was employed to align the HMM of the target and the template with the template and the target sequences, respectively, resulting in two generally different template-target alignments. The alignment with the most significant e-value as reported by the hmmscore program was selected.
For LOBSTER, the COACH algorithm was used through the -coach option to align a multiple sequence alignment against a Hidden Markov Model. First, the program was used to build HMMs for the target and the template sequences, using their PSI-BLAST multiple sequence alignments. Next, we aligned the HMMs of the target and the template to the template and target sequences, respectively, resulting in two generally different template-target alignments. The alignment with the higher bit score as reported by LOBSTER was selected.
For SEA, the Web server at http://ffas.ljcrf.edu/sea/ was used with the default parameters: the FRAGlib library (http://ffas.ljcrf.edu/frag/) for extracting structural fragments with a cutoff of −1.5, local alignment with the initiation gap penalty u of −5 and the extension gap penalty v of −1, and the weight for local similarity of 0.5.
For CLUSTALW, the profile alignment option (i.e., number 3 in the main CLUSTALW menu) with the default parameters was used (Thompson et al. 1994). We used this option over the multiple sequence alignment option (i.e., number 2 in the main CLUSTALW menu) to benchmark CLUSTALW using the same profiles as for the other tested programs.
For COMPASS, the default parameters were used to align the target and template multiple sequence alignments (Sadreyev and Grishin 2003).
For SALIGN, the 13 different protocols were tested, combining three different ways to construct a profile with four different ways to score a match between two profile positions, as well as two protocols based on posterior substitution probability matrices, as described above (Table 1). The PSI-BLAST profiles cannot be used with the Jensen-Shannon scheme for calculating the profile–profile substitution scores because this scheme relies on probabilities Pi and Qj that are not reported in the PSI-BLAST output.
Table 1.
Thirteen protocols implemented in the SALIGN command in MODELLER-7
| Protocol name | Profile scheme | Profile–profile comparison scheme | Initiation gap penalty | Extension gap penalty | σ smoothing |
| CCPBP | PSI-BLAST | correlation coefficient7 | −300 | 0 | n/a |
| CCHH | Henikoff-Henikoff1 | correlation coefficient7 | −300 | 0 | n/a |
| CCHS | Henikoff-Henikoff with similarity bias2 | correlation coefficient7 | −150 | 0 | n/a |
| CCMAT | Henikoff-Henikoff matrix13 | correlation coefficient7 | −100 | 0 | 0.1 |
| EDPBP | PSI-BLAST | Euclidean distance8 | −450 | −30 | n/a |
| EDHH | Henikoff-Henikoff1 | Euclidean distance8 | −550 | 0 | n/a |
| EDHS | Henikoff-Henikoff with similarity bias2 | Euclidean distance8 | −450 | −10 | n/a |
| DPPBP | PSI-BLAST | dot product6 | −250 | −30 | n/a |
| DPHH | Henikoff-Henikoff1 | dot product6 | −550 | 0 | n/a |
| DPHS | Henikoff-Henikoff with similarity bias2 | dot product6 | −100 | −30 | n/a |
| JSHH | Henikoff-Henikoff1 | Jensen-Shannon distance9 | −150 | 0 | n/a |
| JSHS | Henikoff-Henikoff with similarity bias2 | Jensen-Shannon distance9 | −250 | 0 | n/a |
| AveMAT | Henikoff-Henikoff matrix13 | Average value12 | −100 | −50 | 0.1 |
The protocols are defined by the schemes used to calculate the profiles and the profile–profile comparison scores. In addition, the table lists the optimal gap initiation and extension penalties, as well as the smoothing parameter σ, obtained by optimizing the accuracy of the protocols on the training set of alignments. Equation numbers (text) corresponding to the schemes are shown in superscript.
The alignment of two multiple sequence alignments by SALIGN requires approximately 40 sec for ~250 sequences with about ~250 residues in each of the two profiles on a typical Pentium 4 computer. The total CPU time is dominated by the computing of the scoring matrix, rather than the dynamic programming step. This CPU time is approximately proportional to the product of the numbers of sequences in the two profiles and the profile lengths.
Training and testing alignment sets
Because our aim is to improve the accuracy of comparative protein structure modeling, the reference alignments were pairwise, structure-based alignments. They were extracted from our comprehensive database of pairwise structure-based alignments, DBAli (Marti-Renom et al. 2001). The alignments in DBAli were calculated by superposing all pairs of proteins of known structure in the Protein Data Bank (PDB, Feb. 1999; Berman et al. 2002) that are classified into the same H class in the CATH database (Orengo et al. 1999), using the program CE (Shindyalov and Bourne 1998). There are 33,920 such alignments with a Z-score higher than 3.8 (Shindyalov and Bourne 1998). They cover the entire spectrum of sequence and structure similarities.
First, 387 alignments were extracted from DBAli by requiring up to 40% sequence identity, at least 100 aligned residues, at least 50% of the residues aligned, and that at least 90% of the residues of one chain are covered in the alignment. Second, structure pairs that did not have at least 50% of the residues in the shorter chain aligned by MAMMOTH (Ortiz et al. 2002) were also eliminated, resulting in the final set of 300 reference alignments. These 300 alignments were randomly divided into the training and testing sets of 100 and 200 alignments, respectively. The training set of alignments was used to optimize the gap initiation and gap extension penalties for all of our alignment protocols and the parameter σ for the two posterior substitution probability matrix protocols, and the testing set was used to assess the performance of all examined alignment methods. The PDB chain identifiers, chain lengths, percentage sequence identities, root-mean-square deviations (RMSDs) for the aligned Cα atoms, average percentages of the aligned Cα atoms, and percentages of structurally equivalent residues (below) are listed separately for the training and testing alignments in Supplementary Table 1 (http://salilab.org/suppmat/suppmat.shtml). Distributions of these features for all 300 alignments are shown in Figure 1 ▶, indicating that the selected structure pairs indeed represent difficult alignment cases, with an average pair sharing only 20% sequence identity and 65% of structurally equivalent Cα atoms superposed with an RMSD of 3.5 Å.
Figure 1.

Composition of the 300 reference alignments that constitute the training and testing sets. (A) Distributions corresponding to the 100 alignments in the training set. (B) Distributions corresponding to the 200 alignments in the testing set. The percentage sequence identity is defined by the ratio of the alignment positions with the same residue types and the number of aligned positions. The RMSD is calculated over the aligned Cα atoms. The percentage of structurally equivalent residues was calculated as the percentage of residues within 3.5 Å after rigid superimposition.
Measures of alignment accuracy
The accuracy of an alignment was measured by relying on the aligned native structures extracted from the PDB (Berman et al. 2002). First, the RMSD between the corresponding Cα atoms in the two structures was calculated upon rigid-body least-squares superposition of all the Cα atoms, as implemented in the SUPERPOSE command of MODELLER (Sali et al. 2001).
Second, the percentage of structurally equivalent positions was defined as the percentage of the Cα atoms in the shorter of the sequences that are within a certain cutoff (e.g., 1, 2, 3, 4, and 5 Å, and their average) of the corresponding atoms in the superposed structure (“structure overlap”). Unless indicated otherwise, the structure overlap quoted is the average over all cutoffs.
Additionally, the alignment methods were assessed by the percentage of alignments with the structure overlap higher than 30% (“success rate”); structure pairs with at least as much overlap have the same fold (Abagyan and Batalov 1997).
In addition to the assessment of structure similarity implied by an alignment, we evaluated the accuracy of the alignment through a comparison with the CE structure-based alignment. First, the fraction of correctly aligned positions was defined as the percentage of positions in the tested alignment that were identical to those in the CE structure-based alignment (“CE overlap”); the residue-gap matches are ignored in this calculation.
Secondly, the shift score, which ranges from −e for two completely different alignments to 1 for identical alignments, was also calculated (Cline and Karplus 1998). We used e = 0.2, as suggested (Cline and Karplus 1998). The shift score incorporates both coverage and error.
Optimization of gap penalties and σ
The optimal gap initiation and extension penalties for the 11 profile–profile alignment protocols were identified by maximizing the average percentage of correctly aligned positions for the training set of sequence pairs. The maximization scanned all combinations of the initiation penalties from −1000 to 0 in steps of 50 and the extension penalties from −200 to 0 in steps of 10. The gap initiation, gap extension, and the σ parameters for the two posterior substitution probability matrix protocols were optimized on a 3D grid, with σ ranging from 0.001 to 10 (0.001, 0.01, 0.1, 1, and 10). The optimal parameters for each alignment protocol are listed in Table 1.
Significance of an observed difference in the alignment accuracy
A statistical analysis of the differences between alignment accuracies of various methods was performed. For this analysis, the alignment accuracy of a method was measured independently by the average shift score and CE overlap, both calculated for the 200 testing pairs of sequences. The significance of the differences was computed using Student’s t-test statistics (Marti-Renom et al. 2002).
Results
As described in Materials and Methods, we devised and implemented a profile–profile alignment method in the SALIGN command of MODELLER-7 (available at http://salilab.org/modeller/). There are 13 variations in the calculation of the profiles and the profile–profile substitution scores. The opening and extension gap penalties as well as the σ parameter were optimized separately for each one of the 13 protocols, by relying on the 100 training alignments. To assess SALIGN and a variety of other alignment methods, we used the 200 reference structure-based alignments. First, we assessed the differences in accuracy between the 13 different SALIGN protocols. Next, we compared two of the SALIGN protocols for profile–profile alignment by global dynamic programming to a heuristic pairwise sequence alignment (BLAST), a pairwise sequence alignment by global dynamic programming (ALIGN), a heuristic sequence-profile alignment (PSI-BLAST), two HMM methods (as implemented in SAM and LOBSTER), a pairwise sequence alignment by matching predicted local structures (SEA), and two profile–profile alignment methods (CLUSTALW and COMPASS). Finally, to illustrate the utility of our method, we describe two examples of comparative protein structure modeling that benefit from profile–profile alignment.
SALIGN protocols
The SALIGN protocols that on average aligned most positions correctly were DPHH, CCHH, and CCHS, (c.f. Table 1) with the average CE overlaps of 56.4%, 56.3%, and 55.5%, respectively (Table 2). These three protocols are marginally superior to the other 10 SALIGN protocols, but the differences are significant only for CCHH, and DPHH, (Fig. 2A ▶). When the shift score is used to assess the alignment accuracy, we cannot differentiate in accuracy between CCHS, CCHH, CCPBP, DPHS, and DPPBP at the confidence level of 95% (Fig. 2B ▶). For assessment of SALIGN relative to other alignment methods, we have chosen the protocols CCHH and CCHS based on their marginal superiority over the other protocols according to both accuracy measures as well as previous studies that reported good performance of the correlation coefficient (Pietrokovski 1996; Rychlewski et al. 2000; Edgar and Sjolander 2003a; Panchenko 2003; Sadreyev and Grishin 2003).
Table 2.
Accuracy of the SALIGN protocols
| SALIGN protocol | CE overlap [%] | Shift score |
| CCPBP | 55 ± 23 | 0.61 ± 0.24 |
| CCHH | 56 ± 23 | 0.61 ± 0.24 |
| CCHS | 56 ± 24 | 0.62 ± 0.23 |
| CCMAT | 51 ± 25 | 0.55 ± 0.27 |
| EDPBP | 54 ± 24 | 0.60 ± 0.25 |
| EDHH | 54 ± 24 | 0.59 ± 0.26 |
| EDHS | 55 ± 24 | 0.59 ± 0.26 |
| DPPBP | 55 ± 23 | 0.61 ± 0.24 |
| DPHH | 56 ± 23 | 0.60 ± 0.25 |
| DPHS | 55 ± 24 | 0.61 ± 0.24 |
| JSHH | 53 ± 24 | 0.60 ± 0.24 |
| JSHS | 54 ± 24 | 0.60 ± 0.24 |
| AveMAT | 49 ± 26 | 0.52 ± 0.29 |
| TOP | 62 ± 20 | 0.67 ± 0.20 |
The average accuracies and standard deviations of the protocols are obtained from the runs on the testing set of 200 alignments. The last row (TOP) corresponds to the average of the scores for the best of the 13 alignments, chosen independently for each of the 200 test pairs.
Figure 2.

Statistical significance of the differences in the accuracies of the tested SALIGN protocols. (Upper diagonal) Gray and white squares indicate pairs of methods whose performance is and is not significantly different at a confidence level of 95%, respectively. (Lower diagonal) The intensity of gray indicates the degree of the average difference between the corresponding methods. (A) The accuracy of a method measured by the average CE overlap. (B) The accuracy of a method measured by the average shift score.
We also assessed the average best accuracy obtained by any of the 13 protocols for each one of the 200 alignments in the test set (Table 2). The average best CE overlap is 6% higher than that of CCHH and CCHS, and the average best shift score is 0.06 points better than that of CCHH and CCHS.
The accuracy of a profile–profile alignment method must depend on the accuracy of the input multiple sequence alignments. In general, a multiple sequence alignment prepared by PSI-BLAST depends significantly on the number of iterations and the e-value cutoff for inclusion of a sequence in the alignment. As described in Materials and Methods, our protocol for constructing a multiple sequence alignment already selects automatically the iteration that results into the highest statistical significance of the alignment between the profile and the other sequence. This protocol is based on the empirical observation that accuracy of the alignments correlates with their statistical significance, albeit weakly in the low-similarity range (Altschul et al. 1997; Brenner et al. 1998; Sauder et al. 2000). In addition, we tested the impact of the e-value cutoff used for the construction of PSI-BLAST multiple sequence alignments on the accuracy of the resulting SALIGN alignments. There is no dependence of the SALIGN alignment accuracy on this e-value cutoff, in the range from 10−20 to 10−4 (e.g., the average structure overlap varies from 35% to 36%).
Assessment of SALIGN relative to other alignment programs
For SAM, LOBSTER, ALIGN, CLUSTALW, and SALIGN, the alignment covers 100% of the residues in the input sequences. The average coverage by the SEA server is 97.2%; only five alignments (2.5% of all alignments in the test set) have less than 90% coverage. Therefore, the coverage of SEA is comparable to that of SAM, LOBSTER, ALIGN, CLUSTALW, and SALIGN. However, the coverage of BLAST, PSI-BLAST, and COMPASS alignments is smaller than any of the other methods (Fig. 3 ▶). For BLAST, only 24 alignments (12% of the testing set) cover over 75% of both chains, 70 for PSI-BLAST (35%), and 108 for COMPASS (54%). BLAST could not find any significant hits for four alignments, whereas PSI-BLAST could not find a hit for two of the 200 test alignments.
Figure 3.

The extent of the protein sequence that is aligned by the assessed alignment methods. (A) BLAST, (B) PSI-BLAST, (C) SEA, and (D) COMPASS. In contrast, ALIGN, SAM, CLUSTALW, and our profile–profile alignment protocols generally align the whole of the input protein sequences, either because they rely on global dynamic programming or because the aligned sequences are of similar lengths.
The SALIGN method (protocols CCHH and CCHS) has higher accuracy according to the CE overlap and shift score measures than BLAST, ALIGN, PSI-BLAST, CLUSTALW, COMPASS, SEA, SAM, and LOBSTER (Table 3, Fig. 4 ▶). BLAST has the lowest accuracy by CE overlap (26.1%). SEA, SAM, and LOBSTER align correctly about 50% of the residues (49.2%, 48.4%, and 49.9% CE overlap, respectively); COMPASS aligns 43.2%, whereas SALIGN (CCHH) correctly aligns about 56.4% of the residues (Table 3). There are no statistically significant differences in the average accuracy between LOBSTER, SAM, and SEA, nor between ALIGN, PSI-BLAST, and CLUSTALW (Fig. 4A ▶). Individual SALIGN protocols differ from LOBSTER, SEA, SAM, COMPASS, PSI-BLAST, CLUSTALW, ALIGN, and BLAST by 5.5%–31.0% in CE overlap (Fig. 4A ▶). On average, SALIGN (CCHH protocol) outperforms LOBSTER, SEA, SAM, COMPASS, PSI-BLAST, CLUSTALW, ALIGN, and BLAST by 6.3%, 7.0%, 7.8%, 13.1%, 13.9%, 13.7%, 14.8%, and 31.0% in CE overlap, respectively. LOBSTER, SEA, and SAM are statistically better than COMPASS, PSI-BLAST, CLUSTALW, ALIGN, and BLAST by 5.1%–24.3%. However, LOBSTER, SEA, and SAM are statistically worse than the SALIGN protocols, correctly aligning on the average 5.5%–7.8% less residues (Fig. 4A ▶). Although these differences may not seem large, the statistical analysis demonstrates that SALIGN is significantly more accurate than all other benchmarked methods at the 95% confidence level (Fig. 4 ▶).
Table 3.
Comparison of the accuracies of the SALIGN protocols CCHHand CCHSwith those of BLAST, ALIGN, SAM, SEA, CLUSTALW, and PSI-BLAST
| Method | CE overlap [%] | Shift score | RMSD [A] | Structure overlap [%] |
| CE | 100 ± 0 | 1.00 ± 0.00 | 2.7 ± 0.6 | 59.8 ± 12.9 |
| BLAST | 26 ± 29 | 0.32 ± 0.33 | 5.6 ± 3.7 | 20.6 ± 23.7 |
| PSI-BLAST | 43 ± 31 | 0.48 ± 0.35 | 6.5 ± 3.9 | 30.3 ± 24.9 |
| SAM | 48 ± 26 | 0.50 ± 0.34 | 9.2 ± 4.7 | 28.9 ± 24.8 |
| LOBSTER | 50 ± 27 | 0.51 ± 0.32 | 9.1 ± 4.9 | 31.1 ± 25.2 |
| SEA | 49 ± 27 | 0.53 ± 0.29 | 8.4 ± 4.4 | 33.4 ± 24.3 |
| ALIGN | 42 ± 25 | 0.44 ± 0.28 | 10.6 ± 5.0 | 25.7 ± 24.1 |
| CLUSTALW | 43 ± 27 | 0.44 ± 0.31 | 10.2 ± 4.9 | 26.4 ± 24.3 |
| COMPASS | 43 ± 32 | 0.49 ± 0.35 | 4.8 ± 3.2 | 32.3 ± 24.7 |
| CCHH | 56 ± 23 | 0.61 ± 0.24 | 7.8 ± 4.2 | 36.7 ± 22.9 |
| CCHS | 56 ± 24 | 0.62 ± 0.24 | 7.8 ± 4.2 | 36.5 ± 23.2 |
See Materials and Methods for program versions. The average and standard deviation of the alignment-based and structure-based accuracy criteria are shown for the benchmarking runs on the testing set of 200 alignments. For comparison, the values are also shown for the reference alignments obtained by structure superposition with CE. On average, BLAST, PSI-BLAST, and SEA cover 42%, 70%, and 97% of the aligned sequences, respectively, while the other methods align entire sequences.
Figure 4.

Statistical significance of the differences in the accuracies of the tested alignment methods. (Upper diagonal) Gray and white squares indicate pairs of methods whose performance is and is not significantly different at a confidence level of 95%, respectively. (Lower diagonal) The intensity of gray indicates the magnitude of the average difference between the corresponding methods: white indicates no difference in accuracy; black indicates maximum difference. (A) The accuracy of a method measured by the average CE overlap. (B) The accuracy of a method measured by the average shift score.
The alignments produced by the SALIGN protocols CCHH and CCHS have an average Cα RMSD of 7.8 Å (Table 3). SEA and SAM have a marginally higher average RMSD (8.4 and 9.2 Å, respectively). The average Cα RMSD upon superimposition by PSI-BLAST, BLAST, and COMPASS alignments of 6.5, 5.6, and 4.8 Å, respectively, is lower than that of SALIGN. However, this difference is a consequence of a much smaller number of aligned residues (Fig. 3 ▶). A different trend is observed for “structure overlap” (Table 3). The two SALIGN protocols have higher average structure overlap than any of the other compared methods (36.7% and 36.5%, respectively). The SEA method has a lower average structure overlap of 33.4%. In summary, although the average Cα RMSD of SALIGN, PSI-BLAST, SEA, and SAM are comparable, SALIGN has much higher coverage. Therefore, it is more useful for comparative protein structure modeling because it allows modeling of a larger fraction of a target sequence without sacrificing the RMSD accuracy of a model relative to the other tested alignment methods.
For the sequence-sequence alignment methods (ALIGN and BLAST), the alignment success rate (i.e., the fraction of the test alignments with at least 30% structure overlap) averaged over all superposition cutoffs is ~30%; for sequence-profile methods (PSI-BLAST, SAM, and LOBSTER), it is ~40%; for the sequence-structure method (SEA), it is ~45%; for the COMPASS profile–profile method, it is 49%; for SALIGN, it is ~53% (Table 4). CLUSTALW, a profile–profile method that aligns two consensus sequences based on multiple sequence alignment, has the alignment success rate of ~36%. For the 5 Å cutoff alone, the SALIGN CCHH and CCHS protocols also have a higher alignment success rate than any other tested method, aligning more than 80% of the test alignments with at least 30% structure overlap (Table 4). This performance is 13% higher than that of SEA and ~20% higher than that of SAM, LOBSTER, COMPASS, and PSI-BLAST. Only COMPASS’s success rate for the higher-resolution cutoffs (i.e., 1, 2, and 3 Å) was similar to that of SALIGN, indicating that the local alignments by COMPASS are more accurate than those by PSI-BLAST and BLAST. The two SALIGN protocols have a higher alignment success rate than any of the other tested methods over the whole range of structural overlap cutoffs (Fig. 5 ▶). Even pairs of sequences with as little as 4% sequence identity can sometimes be aligned reasonably well by the CCHH and CCHS protocols.
Table 4.
The alignment success rate of the different methods
| Alignment success rate | ||||||
| Method | 1Å | 2Å | 3Å | 4Å | 5Å | Average |
| CE | 20.5 | 82.5 | 100.0 | 100.0 | 100.0 | 100.0 |
| BLAST | 8.0 | 21.5 | 30.0 | 35.0 | 37.5 | 28.5 |
| PSI-BLAST | 8.0 | 31.0 | 45.5 | 55.0 | 60.0 | 43.0 |
| SAM | 7.5 | 28.5 | 42.0 | 52.0 | 63.0 | 39.5 |
| LOBSTER | 8.5 | 30.5 | 46.0 | 58.5 | 64.5 | 44.0 |
| SEA | 10.5 | 35.0 | 47.5 | 60.5 | 70.0 | 45.5 |
| ALIGN | 8.5 | 23.0 | 35.0 | 45.5 | 55.5 | 32.5 |
| CLUSTALW | 7.5 | 27.5 | 38.5 | 45.0 | 55.0 | 36.5 |
| COMPASS | 10.5 | 35.0 | 52.0 | 58.5 | 61.0 | 49.0 |
| CCHH | 10.0 | 35.5 | 58.0 | 71.5 | 84.0 | 53.5 |
| CCHS | 10.0 | 36.0 | 54.0 | 71.0 | 83.0 | 53.0 |
An alignment is “successful” when the structure overlap is at least 30%. The success rates are listed for the 1, 2, 3, 4, and 5 Å cutoffs used in the calculation of the structure overlap, as well as their average.
Figure 5.

Percentage of the 200 testing alignments as a function of the minimal fraction of structurally equivalent positions at the 5 Å cutoff. The vertical line indicates the threshold of alignments that have structural overlap of at least 30%.
The results presented above indicate that SALIGN on average outperforms all other methods tested in this study. We now ask to what extent this is true for the individual test alignments. To answer the question, we tabulated the percentages of the 200 test alignments obtained by one method that were of higher accuracy than those obtained by the other methods (Table 5). The CCHH protocol has a lower RMSD for 83.0%, 37.5%, 50.0%, 70.0%, 68.0%, 59.0%, 82.5%, and 28.5% of the alignments with respect to ALIGN, BLAST, PSI-BLAST, SAM, LOBSTER, SEA, CLUSTALW, and COMPASS, respectively (Table 5, top).
Table 5.
Comparison of the tested methods by the individual pairwise alignments
| ALIGN | BLAST | PSI-BLAST | SAM | LOBSTER | SEA | CLUSTALW | COMPASS | CCHH | CCHS | |
| ALIGN | 17.5 | 15.5 | 34.5 | 32.5 | 22.5 | 44.0 | 8.0 | 16.0 | 14.5 | |
| BLAST | 80.5 | 55.5 | 68.0 | 66.0 | 60.5 | 76.5 | 38.5 | 60.0 | 59.5 | |
| PSI-BLAST | 83.5 | 41.0 | 62.0 | 63.5 | 56.5 | 77.5 | 27.0 | 48.0 | 51.0 | |
| SAM | 65.5 | 30.0 | 37.0 | 48.0 | 41.5 | 64.5 | 20.0 | 29.5 | 27.5 | |
| LOBSTER | 67.0 | 32.0 | 35.5 | 52.0 | 43.5 | 69.0 | 18.5 | 29.5 | 29.5 | |
| SEA | 76.5 | 36.5 | 41.5 | 57.5 | 55.5 | 75.0 | 27.0 | 39.0 | 39.5 | |
| CLUSTALW | 55.5 | 21.5 | 21.5 | 35.0 | 29.5 | 24.0 | 8.5 | 16.0 | 16.0 | |
| COMPASS | 92.0 | 59.0 | 71.0 | 80.0 | 81.5 | 72.0 | 91.5 | 71.0 | 72.0 | |
| CCHH | 83.0 | 37.5 | 50.0 | 70.0 | 68.0 | 59.0 | 82.5 | 28.5 | 53.0 | |
| CCHS | 85.0 | 38.0 | 46.5 | 72.0 | 69.0 | 59.0 | 82.5 | 27.5 | 43.5 |
| ALIGN | BLAST | PSI-BLAST | SAM | LOBSTER | SEA | CLUSTALW | COMPASS | CCHH | CCHS | |
| Each cell lists the percentage of alignments for which the method listed in the row header is more accurate than the method listed in the column header. The accuracy is measured by the RMSD between the compared structures given the alignment (top table) and the percentage of structurally equivalent positions (bottom table). Diagonally related percentages may not sum to 100% due to identical alignments from the two compared methods. | ||||||||||
| ALIGN | 73.0 | 33.5 | 39.0 | 37.0 | 24.5 | 50.5 | 35.0 | 16.5 | 14.0 | |
| BLAST | 23.0 | 14.5 | 23.5 | 15.5 | 11.5 | 26.5 | 14.5 | 5.0 | 6.0 | |
| PSI-BLAST | 65.0 | 82.0 | 49.5 | 42.0 | 35.0 | 61.5 | 36.5 | 20.5 | 21.5 | |
| SAM | 60.5 | 74.5 | 47.5 | 44.0 | 36.5 | 61.0 | 40.0 | 25.5 | 27.5 | |
| LOBSTER | 62.5 | 82.0 | 54.0 | 55.5 | 41.0 | 69.0 | 46.0 | 22.0 | 23.0 | |
| SEA | 75.0 | 85.0 | 62.5 | 62.5 | 56.5 | 75.0 | 52.0 | 34.0 | 34.0 | |
| CLUSTALW | 48.5 | 71.5 | 37.5 | 37.5 | 28.0 | 23.5 | 29.5 | 15.5 | 14.5 | |
| COMPASS | 64.5 | 83.0 | 59.5 | 59.5 | 53.0 | 85.0 | 69.5 | 26.0 | 29.0 | |
| CCHH | 83.0 | 91.5 | 77.5 | 73.5 | 74.5 | 63.5 | 85.0 | 72.5 | 50.0 | |
| CCHS | 85.0 | 91.0 | 76.5 | 70.5 | 73.5 | 63.0 | 84.0 | 70.5 | 43.5 | |
Similar results apply to CCHS. Approximately half of the PSI-BLAST alignments have a lower RMSD than those of SALIGN, but only 20% of the PSI-BLAST alignments have higher structure overlap that those of SALIGN (Table 5, bottom). Only ~30% of the SAM and SEA alignments have higher structure overlap than those of SALIGN (Table 5, bottom).
Examples
To illustrate SALIGN, we aligned and modeled two target sequences from the fourth Critical Assessment of Techniques for Protein Structure Prediction (CASP) meeting (Moult et al. 2001): an enolase enzyme from E. coli (target T0111) and a hypothetical protein from H. influenzae (target T0092). T0111 shares 45% sequence identity to the template structure with the PDB code of 5enl, and T0092 res only 8% sequence identity with the most similar template 1d2c:A.
The model for the easy target T0111, based on the SALIGN alignment and the default MODELLER model building routine ‘model’ (Sali and Blundell 1993), is similar in accuracy to the best model presented at the CASP4 meeting (Fig. 6A ▶). For this example, PSI-BLAST generated an alignment that led to a slightly better model in terms of RMSD, but with a smaller number of correctly modeled residue positions (i.e., Cα atoms within 3 Å of their correct positions).
Figure 6.

Comparative protein structure modeling with SALIGN and PSI-BLAST alignments. The comparative protein structure models were built by satisfaction of spatial restraints, as implemented in MODELLER-7 (Sali and Blundell 1993). The default model building routine ‘model’ was used. (A) CASP4 target T0011. The RMSD errors (percentage of structurally equivalent Cα positions within the 3 Å cutoff) for the SALIGN, PSI-BLAST, and the best CASP4 model (not shown; http://predictioncenter.llnl.gov) are 1.8 Å (96.1%), 1.0 Å (95.8%), and 1.8 Å (96.7%), respectively. (B) CASP4 target T0092. The RMSD errors (percentage of structurally equivalent Cα positions within the 3 Å cutoff) for the SALIGN, PSI-BLAST, and the best CASP4 model (not shown; http://predictioncenter.llnl.gov) are 5.9 Å (67.8%), 4.0 Å (31.7%), and 6.0 Å (65.2%), respectively.
The model for the difficult target T0092, based on the SALIGN alignment and the default MODELLER model building routine ‘model’ (Sali and Blundell 1993), is better than the best model presented at the CASP4 meeting, both in terms of accuracy and the percentage of correctly modeled residues (Fig. 6B ▶). The PSI-BLAST alignment included only the Rossman fold domain. SALIGN is a com- pletely automatic method without user intervention at any stage, which is not always the case with predictions presented at CASP.
Discussion
Several methods that align protein sequences by comparing their profiles have been described (Pietrokovski 1996; Jaroszewski et al. 2000; Yona and Levitt 2002; Edgar and Sjolander 2003a; Panchenko 2003; Sadreyev and Grishin 2003). Relative to individual sequences, additional information in the profiles has been employed to increase the accuracies of both fold assignment and sequence alignment (Ortiz et al. 1998; Park et al. 1998; Muller et al. 1999; Cuff and Barton 2000; Jaroszewski et al. 2000; Bonneau et al. 2001).
We focused on the utility of sequence profiles to enhance the coverage and accuracy of sequence alignment for comparative protein structure modeling (Sanchez and Sali 1997; Marti-Renom et al. 2000; Pieper et al. 2002). We expected that the conservation and variation of residue types at a given position in a family alignment would allow us to score more accurately whether or not two given positions should be aligned than by comparing only two residue types (i.e., sequence-sequence alignment), or even a residue type and a distribution of residue types (i.e., sequence-profile alignment). We combined several schemes to generate profiles with several recipes to compare the profile positions with each other, resulting in 13 different protocols. The corresponding substitution scoring matrices were used in a global dynamic programming procedure with an affine gap penalty function to create optimal alignments. The protocols were evaluated with the aid of a testing set of alignments and subsequently compared to other existing and widely used alignment programs. This comparison was performed with a view of using the alignments for comparative protein structure modeling.
Comparative modeling is limited by the accuracy and extent of the alignment between the modeled sequence and the template structure(s) (Marti-Renom et al. 2000). Two fundamentally distinct features that cannot trivially be combined describe the quality of a model: (1) the fraction of the protein sequence that is modeled (i.e., coverage) and (2) the accuracy of the modeled region. Generally, the smaller the fraction of the target modeled, the more accurate the model. For example, the accuracy of a model can be increased at the expense of coverage by retaining only the core of the fold and eliminating loops and termini from the model.
A case in point are the local alignment methods, such as BLAST (Altschul et al. 1990), PSI-BLAST (Altschul et al. 1997), and COMPASS (Sadreyev and Grishin 2003). These algorithms generally do not align whole sequences, but only regions that are quite similar to each other. In contrast, global dynamic programming implemented in SALIGN ensures an optimal alignment that is forced to cover whole sequences. In the testing set of 200 pairwise alignments, PSI-BLAST covered less than 75% of the chain residues for 70 alignments and had 60 alignments under 50% coverage (Fig. 3 ▶). In contrast, CLUSTALW (Thompson et al. 1994), ALIGN (Sali and Blundell 1993), SAM (Hughey and Krogh 1996), LOBSTER (Edgar and Sjolander 2003a), and SALIGN always covered 100% of the sequences, whereas SEA (Ye et al. 2003) and COMPASS covered on the average 97% and 64% of the sequences, respectively. Despite larger coverage, SALIGN still outperformed COMPASS, PSI-BLAST, and BLAST in the accuracy of what was covered. The Student’s t-test statistics show that the differences observed between SALIGN and the other tested alignment methods are significant at the confidence level of 95%.
We assessed the 13 different protocols of SALIGN. The two marginally best protocols used the correlation coefficient to compare two profile positions described by (1) the Henikoff-Henikoff scheme (Henikoff and Henikoff 1992, 1994) and (2) its variation that weighs sequences proportionally to their similarity with the target sequence. Most of the differences between the 13 SALIGN protocols were not significant at the 95% confidence level (Fig. 2 ▶). However, there were significant differences between the two protocols of SALIGN that have the best average accuracy and the protocol that always picks the best of the 13 alignments. This fact encourages further development of the profile–profile alignment method.
In summary, the alignment success rate (i.e., the fraction of alignments with more than 30% structure overlap within 5 Å) of the SALIGN method is ~13% higher than that for SEA, ~20% higher than that for COMPASS, SAM, LOBSTER, and PSI-BLAST, and 25%–45% higher than those of CLUSTALW, ALIGN, and BLAST (Table 4). Moreover, the best SALIGN protocols increased the structure overlap (5 Å cutoff) by 6%–28% relative to the other benchmarked methods without sacrificing the coverage of the aligned sequences. The fraction of the correctly aligned residues relative to the structure-based alignment by our top protocol is 56%, which can be compared with the accuracies of 26%, 42%, 43%, 43%, 43%, 48%, 49%, and 50% for BLAST, ALIGN, CLUSTALW, PSI-BLAST, COMPASS, SAM, SEA, and LOBSTER, respectively.
The present results quantify the significant improvement in the accuracy of sequence alignment that is achieved by the use of multiple sequences, in agreement with previous studies (Pietrokovski 1996; Rychlewski et al. 2000; Edgar and Sjolander 2003a; Panchenko 2003; Sadreyev and Grishin 2003). Here, we emphasize an implementation in our publicly available program MODELLER (http://salilab.org/modeller) as well as an increase in the coverage and accuracy of the method, not its novelty. The tests described in the Results section indicate that the SALIGN protocol aligns pairs of sequences with significantly higher coverage and accuracy than the other benchmarked methods used with recommended settings. However, the gap penalties for SALIGN were optimized for a training set of alignments of similar difficulty as the testing set of alignments. In contrast, although we did use the recommended settings for the other benchmarked programs, these settings may not be optimal for this particular benchmark. It is difficult to estimate how much better the other benchmarked methods would perform if their options and parameters were optimized based on the current training set of alignments.
Other existing methods for profile–profile alignment were not compared in this analysis, for several reasons. The LAMA program was developed to detect sequence relationships between local conserved blocks (Pietrokovski 1996) and not to align two sequences using global dynamic programming that allows for gap insertions. The FFAS program (Rychlewski et al. 2000) was developed for fold assignment and not optimized for sequence alignment. Moreover, the SEA program, which we did test here, was shown by the authors to be superior in alignment accuracy to the FFAS program (Ye et al. 2003). Finally, we could not find programs by Yona and Levitt (2002) and Panchenko (2003). In addition, the COMPASS program, which we did benchmark, compares favorably against the program developed by Yona and Levitt (Sadreyev and Grishin 2003).
None of the methods benchmarked in this paper, including SALIGN, rely on structural information to align two multiple sequence alignments. However, there are methods that do use structure information, including threading and consensus methods, and they can produce accurate sequence-structure alignments (e.g., Kelley et al. 2000; Shi et al. 2001; Fischer 2003; John et al. 2003). We did not benchmark SALIGN against any structure-dependent methods, for three reasons. First, there already is a published benchmark on the EVA Web site (http://cubic.bioc.columbia.edu/eva; Eyrich et al. 2001; Koh et al. 2003) that compares several sequence-structure, SAM, and PSI-BLAST methods. Therefore, the EVA Web site provides an indirect estimate of SALIGN relative to the sequence-structure methods. For example, the FUGUE program (Shi et al. 2001) aligns correctly ~6% more Cα atoms within 3.5 Å than the PSI-BLAST program, and is approximately comparable to SAM (http://cubic.bioc.columbia.edu/eva/fr/Pairwise_bestof5.html, December 15, 2003). Similar results are reported by EVA for the 3D-PSSM server (Kelley et al. 2000). Second, many of the threading programs are implemented as Web servers or are not generally available from the authors. Their exact input and output are difficult to control, and consequently informative comparisons are difficult. And finally, although we were motivated by comparative modeling, even application of profile–profile alignment to pairs of sequences, none of which has a known structure, is an important problem.
SALIGN is a starting point for incorporating additional information into the alignment process, to further increase the accuracy of the resulting alignments. For example, information derived from the 3D structure of one of the aligned sequences, such as the environment-dependent substitution matrices (Overington et al. 1992) and variable structure-dependent gap penalties (Zhu et al. 1992; Koretke et al. 1996; Yang 2002), is likely to further improve the utility of sequence-structure alignment in comparative modeling applications.
Currently, SALIGN is used as a module in ModPipe, our software pipeline for large-scale modeling of all available protein sequences (Bairoch and Apweiler 2000) that are detectably related to at least one known protein structure (Eswar et al. 2003). An extrapolation of the present results indicates that SALIGN applied to large-scale modeling will result in an additional ~100,000 sequences that have more than 30% of residues aligned correctly to the closest structure, in comparison to the current models that were calculated based on the PSI-BLAST alignments.
Acknowledgments
We thank our group members for many discussions about the alignment problem. M.A.M.-R. was the Presidential Postdoctoral Fellow from the Rockefeller University, and M.S.M. was a C.H. Revson fellow. This work has been supported in part by the San-dler Family Supporting Foundation, Sun Academic Equipment Grant EDUD-7824-020257-US, and NIH grants R01 GM54762 and P50 GM62529.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03379804
References
- Abagyan, R.A. and Batalov, S. 1997. Do aligned sequences share the same fold? J. Mol. Biol. 273 355–368. [DOI] [PubMed] [Google Scholar]
- Al Lazikani, B., Sheinerman, F.B., and Honig, B. 2001. Combining multiple structure and sequence alignments to improve sequence detection and alignment: Application to the SH2 domains of Janus kinases. Proc. Natl. Acad. Sci. 98 14796–14801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D. and Sali, A. 2001. Protein structure prediction and structural genomics. Science 294 93–96. [DOI] [PubMed] [Google Scholar]
- Barton, G.J. 1996. Protein sequence alignment and database scanning. In Protein structure prediction: A practical approach. (ed. M.J.E. Sternberg). IRL Press at Oxford University Press, Oxford.
- ———. 1998. Protein sequence alignment techniques. Acta Crystallogr. D Biol. Crystallogr. 54 1139–1146. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain, S., et al. 2002. The Protein Data Bank. Acta Crystallogr. D. Biol. Crystallogr. 58 899–907. [DOI] [PubMed] [Google Scholar]
- Blake, J.D. and Cohen, F.E. 2001. Pairwise sequence alignment below the twilight zone. J. Mol. Biol. 307 721–735. [DOI] [PubMed] [Google Scholar]
- Blundell, T.L., Sibanda, B.L., Sternberg, M.J., and Thornton, J.M. 1987. Knowledge-based prediction of protein structures and the design of novel molecules. Nature 326 347–352. [DOI] [PubMed] [Google Scholar]
- Bonneau, R., Strauss, C.E., and Baker, D. 2001. Improving the performance of rosetta using multiple sequence alignment information and global measures of hydrophobic core formation. Proteins 43 1–11. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U., Luthy, R., and Eisenberg, D. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253 164–170. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Chothia, C., and Hubbard, T.J. 1998. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc. Natl. Acad. Sci. 95 6073–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline, M. and Karplus, K. 1998. On the alignment shift and its measures. UCSC-CRL-97-27 27.
- Cuff, J.A. and Barton, G.J. 2000. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40 502–511. [DOI] [PubMed] [Google Scholar]
- David, R., Korenberg, M.J., and Hunter, I.W. 2000. 3D-1D threading methods for protein fold recognition. Pharmacogenomics 1 445–455. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1998. Profile hidden Markov models. Bioinformatics 14 755–763. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. and Sjolander, K. 2003a. SATCHMO: Sequence alignment and tree construction using hidden Markov models. Bioinformatics 19 1404–1411. [DOI] [PubMed] [Google Scholar]
- ———. 2003b. Simultaneous sequence alignment and tree construction using hidden Markov models. Pac. Symp. Biocomput.: 180–191. [PubMed]
- Eswar, N., John, B., Mirkovic, N., Fiser, A., Ilyin, V.A., Pieper, U., Stuart, A.C., Marti-Renom, M.A., Madhusudhan, M.S., Yerkovich, B., et al. 2003. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31 3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyrich, V.A., Marti-Renom, M.A., Przybylski, D., Madhusudhan, M.S., Fiser, A., Pazos, F., Valencia, A., Sali, A., and Rost, B. 2001. EVA: Continuous automatic evaluation of protein structure prediction servers. Bioinformatics 17 1242–1243. [DOI] [PubMed] [Google Scholar]
- Fischer, D. 2003. 3D-SHOTGUN: A novel, cooperative, fold-recognition meta-predictor. Proteins 51 434–441. [DOI] [PubMed] [Google Scholar]
- Godzik, A. and Skolnick, J. 1992. Sequence-structure matching in globular proteins: Application to supersecondary and tertiary structure determination. Proc. Natl. Acad. Sci. 89 12098–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh, O. 1999. Multiple sequence alignment: Algorithms and applications. Adv. Biophys. 36 159–206. [DOI] [PubMed] [Google Scholar]
- Gribskov, M. 1994. Profile analysis. Methods Mol. Biol. 25 247–266. [DOI] [PubMed] [Google Scholar]
- Gribskov, M., McLachlan, A.D., and Eisenberg, D. 1987. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. 84 4355–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribskov, M., Luthy, R., and Eisenberg, D. 1990. Profile analysis. Methods Enzymol. 183 146–159. [DOI] [PubMed] [Google Scholar]
- Henikoff, J.G. and Henikoff, S. 1996. Using substitution probabilities to improve position-specific scoring matirices. Comput. Appl. Biosci. 12 135–143. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1994. Position-based sequence weights. J. Mol. Biol. 243 574–578. [DOI] [PubMed] [Google Scholar]
- Higgins, D.G. and Sharp, P.M. 1988. CLUSTAL: A package for performing multiple sequence alignment on a microcomputer. Gene 73 237–244. [DOI] [PubMed] [Google Scholar]
- Hughey, R. and Krogh, A. 1996. Hidden Markov models for sequence analysis: Extension and analysis of the basic method. Comput. Appl. Biosci. 12 95–107. [DOI] [PubMed] [Google Scholar]
- Jaroszewski, L., Rychlewski, L., and Godzik, A. 2000. Improving the quality of twilight-zone alignments. Protein Sci. 9 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, B. and Sali, A. 2003. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 31 3982–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M.S., Overington, J.P., and Blundell, T.L. 1993. Alignment and searching for common protein folds using a data bank of structural templates. J. Mol. Biol. 231 735–752. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1997. Successful ab initio prediction of the tertiary structure of NK-lysin using multiple sequences and recognized supersecondary structural motifs. Proteins 1 185–191. [DOI] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. A new approach to protein fold recognition. Nature 358 86–89. [DOI] [PubMed] [Google Scholar]
- Karplus, K., Barrett, C., and Hughey, R. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics 14 846–856. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Koh, I.Y., Eyrich, V.A., Marti-Renom, M.A., Przybylski, D., Madhusudhan, M.S., Eswar, N., Grana, O., Pazos, F., Valencia, A., Sali, A., et al. 2003. EVA: Evaluation of protein structure prediction servers. Nucleic Acids Res. 31 3311–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koretke, K.K., Luthey-Schulten, Z., and Wolynes, P.G. 1996. Self-consistently optimized statistical mechanical energy functions for sequence structure alignment. Protein Sci. 5 1043–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt, M. 1997. Competitive assessment of protein fold recognition and alignment accuracy. Proteins 1 92–104. [DOI] [PubMed] [Google Scholar]
- Lin, J. 1991. Divergence measures based on the Shannon entropy. IEEE Trans. Info. Theor. 37 145–151. [Google Scholar]
- Madera, M. and Gough, J. 2002. A comparison of profile hidden Markov model procedures for remote homology detection. Nucleic Acids Res. 30 4321–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom, M.A., Stuart, A., Fiser, A., Sanchez, R., Melo, F., and Sali, A. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29 291–325. [DOI] [PubMed] [Google Scholar]
- Marti-Renom, M.A., Ilyin, V.A., and Sali, A. 2001. DBAli: A database of protein structure alignments. Bioinformatics 17 746–747. [DOI] [PubMed] [Google Scholar]
- Marti-Renom, M.A., Madhusudhan, M.S., Fiser, A., Rost, B., and Sali, A. 2002. Reliability of assessment of protein structure prediction methods. Structure 10 435–440. [DOI] [PubMed] [Google Scholar]
- Moult, J., Fidelis, K., Zemla, A., and Hubbard, T. 2001. Critical assessment of methods of protein structure prediction (CASP): Round IV. Proteins 45 2–7. [PubMed] [Google Scholar]
- Muller, A., MacCallum, R.M., and Sternberg, M.J. 1999. Benchmarking PSI-BLAST in genome annotation. J. Mol. Biol. 293 1257–1271. [DOI] [PubMed] [Google Scholar]
- Myers, E.W. and Miller, W. 1988. Optimal alignments in linear space. Comput. Appl. Biosci. 4 11–17. [DOI] [PubMed] [Google Scholar]
- Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48 443–453. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Pearl, F.M., Bray, J.E., Todd, A.E., Martin, A.C., Lo Conte, L., and Thornton, J.M. 1999. The CATH Database provides insights into protein structure/function relationships. Nucleic Acids Res. 27 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz, A.R., Kolinski, A., and Skolnick, J. 1998. Combined multiple sequence reduced protein model approach to predict the tertiary structure of small proteins. Pac. Symp. Biocomput.: 377–388. [PubMed]
- Ortiz, A.R., Strauss, C.E., and Olmea, O. 2002. MAMMOTH (matching molecular models obtained from theory): An automated method for model comparison. Protein Sci. 11 2606–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington, J., Donnelly, D., Johnson, M.S., Sali, A., and Blundell, T.L. 1992. Environment-specific amino acid substitution tables: Tertiary templates and prediction of protein folds. Protein Sci. 1 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko, A.R. 2003. Finding weak similarities between proteins by sequence profile comparison. Nucleic Acids Res. 31 683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J., Teichmann, S.A., Hubbard, T., and Chothia, C. 1997. Intermediate sequences increase the detection of homology between sequences. J. Mol. Biol. 273 349–354. [DOI] [PubMed] [Google Scholar]
- Park, J., Karplus, K., Barrett, C., Hughey, R., Haussler, D., Hubbard, T., and Chothia, C. 1998. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 284 1201–1210. [DOI] [PubMed] [Google Scholar]
- Pieper, U., Eswar, N., Ilyin, V.A., Stuart, A., and Sali, A. 2002. ModBase, a database of annotated comparative protein structure models. Nucleic Acids Res. 30 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrokovski, S. 1996. Searching databases of conserved sequence regions by aligning protein multiple-alignments. Nucleic Acids Res. 24 3836–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12 85–94. [DOI] [PubMed] [Google Scholar]
- Rychlewski, L., Jaroszewski, L., Li, W., and Godzik, A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 9 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadreyev, R. and Grishin, N. 2003. COMPASS: A tool for comparison of multiple protein alignments with assessment of statistical significance. J. Mol. Biol. 326 317–336. [DOI] [PubMed] [Google Scholar]
- Sali, A. and Blundell, T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Sali, A., Fiser, A., Sanchez, R., Marti-Renom, M.A., Jerkovic, B., Badretdinov, A., Melo, F., Overington, J., and Feyfant, E. 2001. MODELLER, A protein structure modeling program, release 6v0, http://www.salilab.org/modeller/.
- Sanchez, R. and Sali, A. 1997. Advances in comparative protein-structure modelling. Curr. Opin. Struct. Biol. 7 206–214. [DOI] [PubMed] [Google Scholar]
- ———. 1998. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. 95 13597–13602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauder, J.M., Arthur, J.W., and Dunbrack, R.L. 2000. Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins 40 6–22. [DOI] [PubMed] [Google Scholar]
- Sellers, P.H. 1974. Theory and computation of evolutionary distances. Siam J. Appl. Math. 26 787–793. [Google Scholar]
- Shi, J., Blundell, T.L., and Mizuguchi, K. 2001. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310 243–257. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Smith, T.F., Lo Conte, L., Bienkowska, J., Gaitatzes, C., Rogers Jr., R.G., and Lathrop, R. 1997. Current limitations to protein threading approaches. J. Comput. Biol. 4 217–225. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torda, A.E. 1997. Perspectives in protein-fold recognition. Curr. Opin. Struct. Biol. 7 200–205. [DOI] [PubMed] [Google Scholar]
- Venclovas, C., Zemla, A., Fidelis, K., and Moult, J. 2001. Comparison of performance in successive CASP experiments. Proteins 45 163–170. [DOI] [PubMed] [Google Scholar]
- Yang, A.S. 2002. Structure-dependent sequence alignment for remotely related proteins. Bioinformatics 18 1658–1665. [DOI] [PubMed] [Google Scholar]
- Ye, Y., Jaroszewski, L., Li, W., and Godzik, A. 2003. A segment alignment approach to protein comparison. Bioinformatics 19 742. [DOI] [PubMed] [Google Scholar]
- Yona, G. and Levitt, M. 2000. Towards a complete map of the protein space based on a unified sequence and structure analysis of all known proteins. ISMB 8 395–406. [PubMed] [Google Scholar]
- ———. 2002. Within the twilight zone: A sensitive profile–profile comparison tool based on information theory. J. Mol. Biol. 315 1257–1275. [DOI] [PubMed] [Google Scholar]
- Yona, G., Linial, N., and Linial, M. 1999. ProtoMap: Automatic classification of protein sequences, a hierarchy of protein families, and local maps of the protein space. Proteins 37 360–378. [PubMed] [Google Scholar]
- ———. 2000. ProtoMap: Automatic classification of protein sequences and hierarchy of protein families. Nucleic Acids Res. 28 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Z.Y., Sali, A., and Blundell, T.L. 1992. A variable gap penalty function and feature weights for protein 3-D structure comparisons. Protein Eng. 5 43–51. [DOI] [PubMed] [Google Scholar]