

Abstract
MTH1880 is a hypothetical protein from Methanobacterium thermoautotrophicum, a target organism of structural genomics. The solution structure determined by NMR spectroscopy demonstrates a typical α + β-fold found in many proteins with different functions. The molecular surface of the protein reveals a small, highly acidic pocket comprising loop B (Asp36, Asp37, Asp38), the end of β2 (Glu39), and loop D (Ser57, Ser58, Ser61), indicating that the protein would have a possible cation binding site. The NMR resonances of several amino acids within the acidic binding pocket in MTH1880, shifted upon addition of calcium ion. This calcium binding motif and overall topology of MTH1880 differ from those of other calcium binding proteins. MTH1880 did not show a calcium-induced conformational change typical of calcium sensor proteins. Therefore, we propose that the MTH1880 protein contains a novel motif for calcium-specific binding, and may function as a calcium buffering protein.
Keywords: NMR spectroscopy, Methanobacterium thermoautotrophicum, calcium binding, structural proteomics
In the postgenomic era, it is now possible to use structural biology methods to annotate gene function before a new gene or protein is well characterized (Kim 1998). Attempts to do this on a large, even genome-wide scale have already started to yield structure-based, functional annotations for hypothetical proteins (Kennedy et al. 2002). Very recently, a number of structural genomics (or proteomics) efforts have provided correlations between biological function and fold classification for unknown gene products (Yang et al. 2003). In addition, the development of high-throughput technologies, for protein purification and structure determination, enables the determination of new protein structures in a short period of time, often revealing previously unknown biochemical functions as well as cellular mechanisms of new gene products (Zhang and Kim 2003).
As part of an on-going structural genomics effort on proteins from the thermophilic methanogen, Methanobacterium thermoautotrophicum (Christendat et al. 2000), we have determined the solution structure of MTH1880. MTH1880 is a hypothetical protein from M. thermoautotrophicum, and shares high sequence similarity to only one other protein, Y858, from Methanococcus janaschii (~80% sequence identity). However, the molecular function and structure of Y858 are still unknown. The solution structure was searched for structural homologs, which may indicate clues to its biochemical or cellular function. From its molecular surface and charge distribution, we identified a putative cation, calcium binding site.
Results
NMR resonance assignments and secondary structures
Resonance assignments of all backbone resonances were made using HNCA, HNCACB, CBCA(CO)NNH, and HN(CO)CA data. Most of the side-chain assignments were accomplished by combined use of 3D HCCH-TOCSY and 15N-edited TOCSY-HSQC experiments. Secondary structures were readily identified from the chemical shift index (CSI; Wishart and Sykes 1994; Wishart et al. 1995) and NOE information (Wagner et al. 1986). The secondary structure of MTH1880 consisted of a three-stranded antipa-rallel β-sheet (residues 6–11, 39–44, and 51–55) and three short α-helices (residues 22–30, 63–73, and 82–87). A number of hydrogen bonds are found within the main-chain atoms of the secondary structures.
Solution structure of MTH1880
Most of the residues involved in forming the hydrophobic core of MTH1880 are found between the β-sheets (β3 and β4) and Helix 2. Although Helix 1 participates in the formation of the hydrophobic core, it has a dynamic feature based on rapid amide hydrogen exchange rates. Helix 3 at the C terminus packs against the β-sheets to cover the hydrophobic core residues (Fig. 1 ▶). There are five loops, comprising residues 12–21, 31–38, 45–50, 56–64, and 74–81. Interestingly, the charged residues in these loops endow MTH1880 with a strong dipolar nature. Two loops (B [31–38] and D [56–64]) generate a highly acidic surface on the protein, whereas loop C (45–50) and loop E (74–81) create a highly basic surface. The loops B–D are rather rigid relative to those of the loops C–E, which might provide some clues to infer the molecular function of MTH1880.
Figure 1.

Solution structure of MTH1880. The backbone superposition of 20 final simulated annealing structures is displayed in stereo view.
Structure–function of MTH1880
MTH1880 is classified as typical “α and β (α + β)” fold with antiparallel β-sheets (segregated α and β regions), common to many proteins with various functions (Murzin 1995). Structural homologs of MTH1880 were searched for using the DALI and SCOP servers. No structural homologs were identified for MTH1880 from the DALI search. However, several proteins were identified from a SCOP classification. Five structural folds, SH2-like, profilin, actin depolarizing, ferredoxin-like, and crystalin-like folds, were found based on the structural diversity (Fig. 2 ▶). To gain insight about the biochemical function of MTH1880, we investigated the molecular surface for potential ligand binding pockets. The Connolly surface of MTH1880 reveals a highly acidic pocket comprised of loop B (Asp36, Asp37, Asp38), the end of Helix 2 (Glu39), and loop D (Ser57, Ser58, Ser61). Because the total surface charge calculated with the Delphi module of InsightII is -3, it could be possible that the negative charged cluster is a putative cation binding site. For further investigation, we performed 15N−1H HSQC and HCCH- TOCSY experiments in the presence of divalent cations. For the case of Mg2+ ion, no spectral change was observed during titration. For Ca2+ ion, the resonance frequencies of a number of residues (Asp37 and Asp38 in loop B, Glu39 in Helix 2, and Leu56, Ser57, Ser58, Tyr59, and Asn60 in loop D) were changed (Fig. 3 ▶), indicative of Ca2+ binding to the acidic pocket of the protein.
Figure 2.

The proteins with the α + β fold displayed in ribbon diagram together with MTH1880. All structures were based on the results of SCOP classification. (A) MTH1880 showing secondary structural topology, (B) SH2 domain of C-ABL (PDB: 1AB2), (C) uracil-DNA glycosylase (PDB: 1UDI), (D) severin domain 2 (PDB: 1SVY), (E) profilin (PDB: 1PNE), (F) nucleoside diphosphate kinase (PDB: 1EHW). The overall structures of SH2 and gelsolin are very similar to that of MTH1880; however, two proteins have different function. The structures are generated by MOLMOL (Koradi et al. 1996), MOLSCRIPT (Kraulis 1991), and Raster3D (Merritt and Bacon 1997) programs.
Figure 3.


Chemical shift differences of (A) backbone amide proton and (B) nitrogen chemical shifts between free and calcium-bound form. The chemical shift difference (ppm) was calculated from the absolute value of the change in the 1H chemical shift and 15N chemical shift. (C) A superposition of the 2D 1H−15N HSQC spectra in the presence (red) and absence (blue) of calcium ions.
Chemical shift perturbations of side-chain atoms during Ca2+ titration were more evident in HCCH-TOCSY spectra (data not shown). In the presence of Ca2+, the side-chain resonances of Asp37, Glu39, Leu56, Ser57, and Tyr59 disappeared, whereas the line width of Ser58 was broadened upon Ca2+ addition. These residues are located on the acidic pocket comprising of loops B and D. Based on these observation, we indicate that MTH1880 binds Ca2+ specifically, and the Ca2+ binding site is located on the B–D loop region.
Because the resonances of the secondary structural elements were largely unchanged, indicating little or no change in the structured part of the protein, we could readily construct a calcium-bound model. The mode of calcium binding was inferred from the model of the calcium-bound structure (Fig. 4A ▶). The carboxylic groups of Asp37 and Glu39 provide a coordination of the negative charges for the calcium cation (Fig. 4B ▶). The carbonyl or hydroxyl groups of other residues are also involved in the interaction with calcium ion. The rigid conformations of loops B and D provide a well-defined binding site for calcium. This binding mechanism is also found in those of other calcium binding proteins, especially thermitase (Fig. 4C ▶; Table 2). Ther-mitase possesses a number of binding pockets for calcium ions formed by loop residues (Teplyakov et al. 1990). This model indicates that MTH1880 has a common feature with many well-characterized calcium binding proteins, but that MTH1880 itself contains a novel motif for calcium binding.
Figure 4.


Calcium binding motif of MTH1880. (A) The structural model of MTH1880 bound to a calcium ion. Regions in red color represent the residues whose resonances changed upon calcium binding. (B) Proposed coordination of the calcium binding site based on NMR data. The residues involved in calcium binding were displayed with side-chain atoms. (C) A class of calcium binding motifs. (a) Calmodulin (PDB: 3CLN), (b) crystallin (PDB: 1HDF), (c) gelsolin (PDB: 4CPV), and (d) thermitase (PDB: 1THM). The calcium ion is shown as a black sphere and sodium ion as a white sphere.
Table 2.
Biochemical and structural data of calcium binding proteins
| Characteristic motif | Structural feature | Involved amino acids | Conformational change after Ca2+-binding | Function (with Ca2+) | |
| MTH1880 | — | Beta-sheets and loops | D, E, S, L | N | Calcium buffer |
| Gelsolin | — | Beta-sheet, loop and helix | D, G, P | Y | Actin-binding |
| Calmodulin | EF-hand | Helix-loop-helix | D, E, Q, N, T | Y | Calcium sensor |
| Thermitase | Subtilisin family | Loops | D, Q, R, S, T, A, Y, V, I | Y | Thermostability |
| Crystallin | Greek key | Beta-sheets | D, E, N, Q, K, S, Y | N | Calcium buffer |
Source: structural features and characters are reported as followed: Gelsolin (PDB: 1SVY, 1D0N), Calmodulin (PDB: 3CLN), Thermitase (PDB: 1THM, reference cited in text: 17), γ-Crystallin (PDB: 1HDF).
Discussion
MTH1880 has a common α + β type of fold similar to that found in many proteins with diverse functions. To define the biochemical function of MTH1880, we focused on a readily visible, small acidic binding pocket formed by loops B and D. Although MTH1880 has no classical cation binding motif, such as an EF-hand, a typical motif search indicates that the molecular shape and electric potential surface is typical of divalent cation binding capabilities. However, the calcium binding motif and overall topology of MTH1880 are different from those of other calcium binding proteins. Modeling of potential cations into the acidic pocket showed that the calcium ion fits well, but the magnesium ion does not, even though the magnesium ion is smaller than the calcium ion. These data may indicate that MTH1880 is a calcium specific protein. The cation specificity derived from the “size-exclusion” effect is commonly observed in calcium binding proteins with the EF-hand motif (Snyder et al. 1990). To identify the potential biological role for MTH1880, we have searched calcium binding proteins with various functions. NMR studies for both synaptotagmin and syntaxin proteins showed that Ca2+ binding effect to these proteins occurs only via the side-chain atoms, not the backbone atoms (Shao et al. 1998). This property is very similar to that of MTH1880.
SCOP results showed that the orientation of secondary structural elements and the calcium-binding motif of gelsolin are very similar to those of MTH1880. However, the calcium ion induces a functionally important conformational change in gelsolin. It has been known that calcium-buffering proteins do not undergo a conformational change in the presence of the calcium ion (Rajini et al. 2001), indicating that MTH1880 could be a Ca2+-buffering protein. Because MTH1880 has sequence homology with Y858 (~80%) from M. janashii, we propose that Y858 may also be a calcium-buffering protein and possess a similar molecular topology with MTH1880. Therefore, we propose the two hypothetical proteins, MTH1880 and Y858, could play a role in the control of archaeal calcium concentration through a calcium-buffering mechanism.
Materials and methods
Gene cloning, protein expression, and purification
The MTH1880 gene was obtained from M. thermoautotrophicum ΔH genomic DNA by PCR amplification and subcloned into pET13b plasmid (Novagen Inc.) at the NdeI and BamHI sites. This construct contains a hexahistidine tag (HisTag) with a thrombin cleavage site in the N-terminal extension.
The protein was overexpressed in the Escherichia coli strain BL21(DE3) pLysS and transformed with the pET13b/MTH1880 plasmid construct. Cells were grown on M9 minimal medium with15NH4Cl and/or [U−13C]-glucose (Cambridge Isotope Laboratories Inc.) at 37°C. For optimum growth condition, thiamine 0.1% (W/ V) and ampicillin, with a final concentration of 50 μg/mL, were added. Proteins were induced with 0.7 mM isopropyl β-D-thiogalactoside (IPTG) when A600 was reached at 0.6. Cells were harvested by centrifugation at 6700 × g for 30 min, and resuspended in 25 mL of binding buffer (5 mM imidazole, 10 mM Tris, 500 mM NaCl, 1 mM PMSF, in pH 8.0) and sonicated. The lysate was centrifuged at 17,500 × g for 30 min and the insoluble material was removed. The supernatant was applied to a His-Bind Resin (Novagen Inc.); bound protein was washed with 12 volumes of 30 mM imidazole and eluted with 300 mM imidazole. To remove His-Tag, the protein solution was incubated with bovine thrombin (Pharmacia Biotech Inc.) in the ratio of 10 Unit/mg at 25°C for 6 h. After thrombin digestion, gel filtration was performed using HiLoad 16/60 Sephadex 75 column (Pharmacia Biotech Inc.). In this procedure, the loading buffer was used with the NMR buffer (25 mM sodium phosphate, 300 mM NaCl, 0.002% NaN3). The purified protein was concentrated to approximately 2 mM with Centricon-3 concentrators (Millipore Inc.).
NMR spectroscopy
NMR spectra were recorded at 500 MHz Varian UNITY Inova or on Bruker DRX500 with a triple resonance probe. All experiments were conducted at 37°C. 2D 15N−1H HSQC (Kay et al. 1992) and 3D 15N-edited NOESY-HSQC and TOCSY-HSQC experiments were performed on [U-15N]-labeled MTH1880. 3D HNCA, 3D HNCACB, HN(CO)CA, CBCA(CO)NH (Muhandiram and Kay 1994), HCCH-TOCSY (Kay et al. 1993), and 13C-edited NOESY-HSQC experiments were conducted on [U-13C, U-15N]-labeled MTH1880. A lyophilized sample of [U-15N] MTH1880 was reconstituted in D2O solution to monitor the exchange of backbone amide protons using the 15N-1H HSQC experiment. Backbone vicinal coupling constants were measured using the 3D HNHA experiment (Kuboniwa et al. 1994). All data were processed with NMRPipe (Delaglio et al. 1995) and analyzed with the Sparky program (Goddard and Kneller 2003).
Structure calculations
Structure calculations were carried out using the hybrid distance geometry and dynamical simulated annealing protocol (Nilges et al. 1998) using the program CNS 1.0 (Brunger et al. 1998) on a Linux workstation. Cross-peaks in NOESY spectra were classified as strong, medium, and weak intensities, corresponding to upper bounds of 2.5, 3.0, 4.0, or 5.0 Å for distance constraints. In addition, the pseudo-atom corrections for methylene, methyl groups, and aromatic ring protons were made during structure calculations (Wüthich et al. 1983). For 3JHNα >8 Hz, φ was restrained to −120(±40)° and for 3JHNα <6 Hz, φ was restrained to −57(±20)°. Hydrogen bond constraints for slow exchanging amide protons (dO—HN = 1.8–2.2 Å, dO—N = 2.8–3.3 Å) were also used during structural calculations.
Initially, a set of 50 structures were generated by a simulated annealing protocol in the CNS program, and the 20 structures with the lowest energy were selected for further analysis. Structural search of MTH1880 were performed manually with SCOP (Murzin et al. 1995) and automatically with Dali (Holm and Sander 1995).
Molecular modeling for calcium binding
The average structure 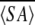 k was calculated from the geometric average of 20
k was calculated from the geometric average of 20 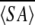 k structure coordinates and subjected to a restrained energy minimization (REM) structure, yielding an
k structure coordinates and subjected to a restrained energy minimization (REM) structure, yielding an 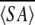 kr structure of MTH1880. The starting structure for the Ca+2 bound form was constructed with
kr structure of MTH1880. The starting structure for the Ca+2 bound form was constructed with 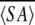 kr Using the cvff force field, a structure with the calcium ion was modeled and further optimized by the molecular dynamics simulation, using the InsightII program and DISCOVER 3 module (Accelys Inc.). Initially, the calcium ion was located adjacent to the negatively charged region of the protein, but no specific distance restraints were imposed between the calcium ion and protein; except the electrostatic interaction between the calcium ion and side-chain atoms of the protein. To obtain the lowest energy conformation with calcium ion, a restrained molecular dynamics simulation was performed for 250 psec at 298 K. During the dynamics simulation, the structural ensemble was sampled every 100 intervals (0.1 psec). Finally, the lowest energy conformation was optimized by restraint energy minimization, with the BFG method, until the deviation of its energy gradient reached 0.001 kcal mole−1.
kr Using the cvff force field, a structure with the calcium ion was modeled and further optimized by the molecular dynamics simulation, using the InsightII program and DISCOVER 3 module (Accelys Inc.). Initially, the calcium ion was located adjacent to the negatively charged region of the protein, but no specific distance restraints were imposed between the calcium ion and protein; except the electrostatic interaction between the calcium ion and side-chain atoms of the protein. To obtain the lowest energy conformation with calcium ion, a restrained molecular dynamics simulation was performed for 250 psec at 298 K. During the dynamics simulation, the structural ensemble was sampled every 100 intervals (0.1 psec). Finally, the lowest energy conformation was optimized by restraint energy minimization, with the BFG method, until the deviation of its energy gradient reached 0.001 kcal mole−1.
Accession numbers
Backbone and side-chain chemical shift assignments for MTH1880 have been deposited to BioMagResBank (accession code BMRB-5129). Coordinates for the 20 structures and ensemble structure have been deposited in the RCSB PDB with accession code 1IQO and 1IQS, respectively.
Table 1.
Structural restraints and statistics for the NMR structures
| Restraints for structure calculation | ||
| Total NOE restraints | 723 | |
| Intraresidue | 275 | |
| Sequential | 162 | |
| Medium range (1 < |i - j| ≤ 5) | 89 | |
| Long range (|i - j| > 5) | 143 | |
| Hydrogen bonds | 54 | |
| Dihedral angles (3) | 39 | |
| Statistics for structure calculations | ||
| RMSD from distance restraints | ||
| NOE restraints (Å) | 0.016 ± 0.006 | |
| Dihedral restraints (°) | 0.46 ± 0.42 | |
| RMSD from idealized geometry | ||
| Bonds (Å) | 0.016 ± 0.003 | |
| Angle (°) | 0.35 ± 0.03 | |
| Impropers (°) | 0.19 ± 0.06 | |
| Mean energies (kcal/mole) | ||
| Etotal | 75.27 ± 19.50 | |
| Ebond | 3.89 ± 1.33 | |
| Eangle | 50.67 ± 8.78 | |
| Evdw | 7.81 ± 7.61 | |
| Enoc | 11.40 ± 7.26 | |
| Edihe | 0.91 ± 0.91 | |
| RMSD of atomic coordinates (Å) | Backbone | Heavy atoms |
〈SA〉ka vs. 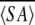 kr kr
| ||
| All residues | 1.195 | 1.907 |
| Secondary structures | 0.747 | 1.292 |
a 〈SA〉k is the ensemble of 20 final simulated annealing structures of MTH1880.
〈SA〉kr is the mean structure obtained by averaging the individual structures following a superimposition of the backbone heavy atoms.
Acknowledgments
This study was supported by the Ministry of Science and Technology of Korea/the Korea Science and Engineering Foundation through the NRL program of MOST NRDP (M1-0203-00-0020); the Ministry of Education and Human Resource through the BK21 project (W.L.); the Ontario Research and Development Challenge Fund; and Genome Canada (C.H.A.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03472104.
References
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kun-stleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Christendat, D., Yee, A., Dharamsi, A., Kluger, Y., Savchenko, A., Cort, J.R., Booth, V., Mackereth, C.D., Saridakis, V., Ekiel, I., et al. 2000. Structural proteomics of an archaeon. Nat. Struct. Biol. 7 903–909. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard, T.D and Kneller, D.G. 2003. SPARKY 3. University of California, San Francisco, CA.
- Holm, L. and Sander, C. 1995. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 20 478–480. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114 10663–10665. [Google Scholar]
- Kay, L.E., Xu, G., Singer, A.U., Muhandiram, D.R., and Forman-Kay, J.D. 1993. A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13 C correlations in H2O samples of proteins. J. Magn. Reson. Ser. B. 101 333–337. [Google Scholar]
- Kennedy, M.A., Montelione, G.T., Arrowsmith, C.H., and Markley, J.L. 2002. Role for NMR in structural genomics. J. Struct. Funct. Genomics 2 155–169. [DOI] [PubMed] [Google Scholar]
- Kim, S.H. 1998. Shining a light on structural genomics. Nat. Struct. Biol. 5 643–645. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 29–32. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kuboniwa, H., Grzesiek, S., Delaglio, F., and Bax, A. 1994. Measurement of HN-H α J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J. Biomol. NMR 4 871–878. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Muhandiram, D.R. and Kay, L.E. 1994. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. Ser. B 103 203–216. [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Clore, G.M., and Gronenborn, A.M. 1988. Determination of three-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamical simulated annealing calculations. FEBS Lett. 229 317–324. [DOI] [PubMed] [Google Scholar]
- Rajini, B., Shridas, P., Sundari, C.S., Muralidhar, D., Chandani, S., Thomas, F., and Sharma, Y. 2001. Calcium binding properties of γ-crystallin: Calcium ion binds at the Greek key β γ-crystallin fold. J. Biol. Chem. 276 38464–38471. [DOI] [PubMed] [Google Scholar]
- Shao, X., Fernandez, I., Sudhof, T.C., and Rizo, J. 1998. Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: Does Ca2+ induce a conformational change? Biochemistry 37 16106–16115. [DOI] [PubMed] [Google Scholar]
- Snyder, E.E., Buoscio, B.W., and Falke, J.J. 1990. Calcium(II) site specificity: Effect of size and charge on metal ion binding to an EF-hand-like site. Biochemistry 29 3937–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplyakov, A.V., Kuranova, I.P., Harutyunyan, E.H., Vainshtein, B.K., From-mel, C., Hohne, W.E., and Wilson, K.S. 1990. Crystal structure of thermi-tase at 1.4 Å resolution. J. Mol. Biol. 214 261–279. [DOI] [PubMed] [Google Scholar]
- Wagner, G., Neuhaus, D., Worgotter, E., Vasak, M., Kagi, J.H., and Wüthrich, K. 1986. Nuclear magnetic resonance identification of “half-turn” and 3(10)-helix secondary structure in rabbit liver metallothionein-2. J. Mol. Biol. 187 131–135. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994. Chemical shifts as a tool for structure determination. Methods Enzymol. 239 363–392. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Holm, A., Hodges, R.S., and Sykes, B.D. 1995. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5 67–81. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K., Billeter, M., and Braun, W. 1983. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton–proton distance constraints with nuclear magnetic resonance. J. Mol. Biol. 169 949–961. [DOI] [PubMed] [Google Scholar]
- Yang, Z., Savchenko, A., Yakunin, A., Zhang, R., Edwards, A., Arrowsmith, C., and Tong, L. 2003. Aspartate dehydrogenase, a novel enzyme identified from structural and functional studies of TM1643. J. Biol. Chem. 278 8804–8808. [DOI] [PubMed] [Google Scholar]
- Zhang, C. and Kim, S.H. 2003. Overview of structural genomics: From structure to function. Curr. Opin. Chem. Biol. 7 28–32. [DOI] [PubMed] [Google Scholar]