

Abstract
We have introduced the mutation glycine 29 to alanine, designed to increase the rate of protein folding, into the B-domain of protein A (BdpA). From NMR lineshape analysis, we find the G29A mutation increases the folding rate constant by threefold; the folding time is 3 μsec. Although wild-type BdpA folds extremely fast, simple-point mutations can still speed up the folding; thus, the folding rate is not evolutionarily maximized. The short folding time of G29A BdpA (the shortest time yet reported) makes it an attractive candidate for an all-atom molecular dynamics simulation that could potentially show a complete folding reaction starting from an extended chain. We also constructed a fluorescent variant of BdpA by mutating phenylalanine 13 to tryptophan, allowing fluorescence-based time-resolved temperature-jump measurements. Temperature jumps and NMR complement each other, and give a very complete picture of the folding kinetics.
Keywords: rapid protein folding, mutation, denaturant, helix propensity, dynamic NMR
Fast events in protein folding reactions have been the focus of many recent studies (Gruebele 1999; Eaton et al. 2000; Myers and Oas 2002; Ferguson and Fersht 2003). Because of the development of methods to probe faster reactions, such as laser-induced temperature jump and dynamic NMR, proteins known to fold in under a millisecond are becoming increasingly common. We have chosen a small protein with a very simple topology as a model system to study fast protein folding. The B-domain of protein A (BdpA) from Staphylococcus aureus is a 60-residue up/down/up three-helix bundle (Fig. 1 ▶). We previously showed that BdpA folds very rapidly in about 10 μsec, without detectable intermediate states (Myers and Oas 2001).
Figure 1.
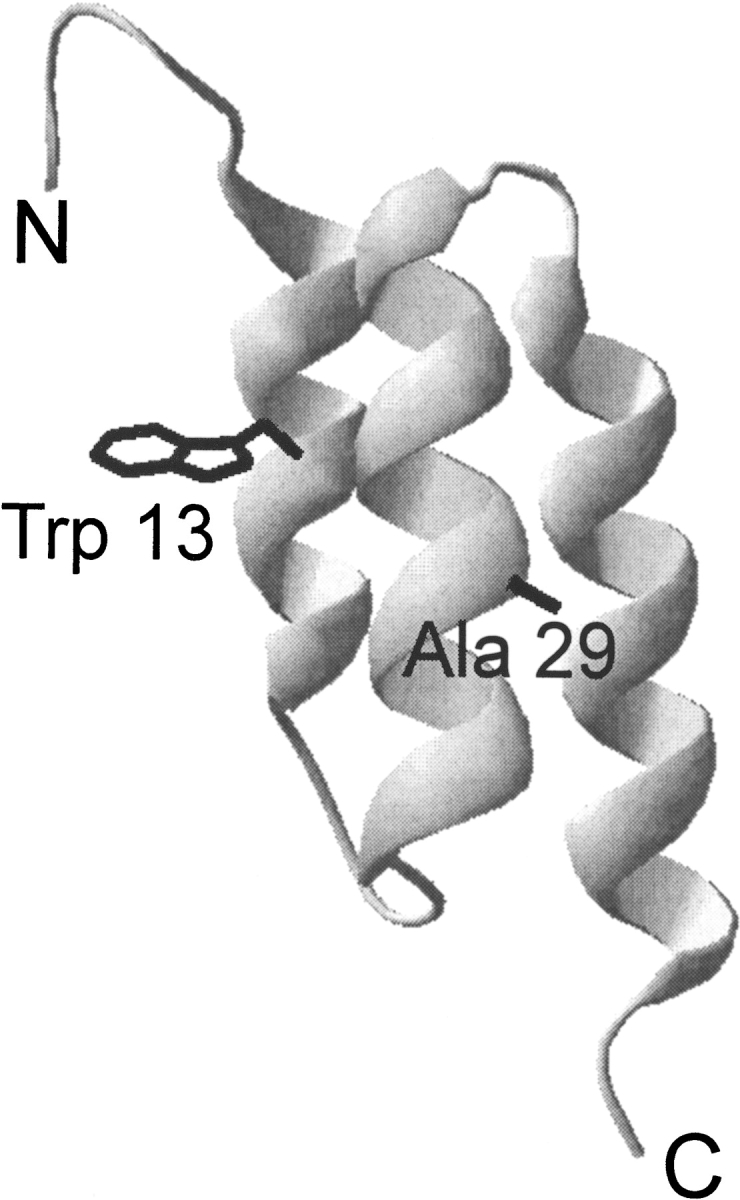
Ribbon diagram of G29A/F13W BdpA showing the helical architecture of the domain and the location of the point mutations studied in this work, based on the NMR structure of the Z-domain (PDB ID 2spz; Tashiro et al. 1997).
There are a growing number of proteins known to fold in the microsecond time regime. Wild-type BdpA (folding rate constant kf = 120,000 sec−1; Myers and Oas 2001), villin headpiece (230,000 sec−1; Kubelka et al. 2003; Wang et al. 2003), cytochrome b562 (200,000 sec−1; Wittung-Stafshede et al. 1999), N-terminal domain of lambda repressor GGA variant (88,000 sec−1; Burton et al. 1996), FBP28 WW domain W30A variant (40,000 sec−1; Ferguson et al. 2001), and engrailed homeodomain (40,000 sec−1; Mayor et al. 2003) all fold in 5–25 μsec. Along with these bona fide proteins, the designed structured peptides BBA5 and TC5B fold on a similar timescale (kf = 130,000 sec−1 and 250,000 sec−1, respectively; Qiu et al. 2002; Snow et al. 2002). It has been proposed that proteins can fold no faster than a “speed limit,” dictated by the fastest rate of chain diffusion (Hagen et al. 1996, 2001), estimated to be ~1 μsec. One interesting question is whether some proteins do fold at this speed limit, or perhaps even faster? To answer this question, we have attempted to find point mutations that increase the folding rate of BdpA.
In this paper we study a point mutation, glycine 29 to alanine, in the middle of helix 2, and on the solvent-exposed face of the helix. G29A was originally one of two mutations used to create the so-called “Z-domain” of protein A, a more stable version of BdpA (Nilsson et al. 1987). The G29A substitution should stabilize helix 2 in both the intact protein and the isolated helix (Myers et al. 1997). The diffusion-collision model of protein folding (Karplus and Weaver 1976, 1994), which predicts realistic folding times for BdpA (Myers and Oas 2001; Islam et al. 2002), suggests that stabilizing the helices of the protein should increase the folding rate.
We also report the construction of a fluorescent variant of BdpA by the introduction of a tryptophan residue, allowing fluorescence to be used as a spectroscopic probe. In collaboration with others, the fluorescent version of BdpA has been subjected to rapid temperature jumps while monitoring fluorescence (Dimitriadis et al. 2004). These results, along with temperature jumps performed on wild-type BdpA using infrared spectroscopy (Vu et al. 2004), are compared to our NMR results. The nanosecond temperature jump technique is complementary to NMR for measuring kinetics. Dynamic NMR utilizes measurements taken at equilibrium, while temperature jumps are a time-resolved method, perturbing the system and monitoring the return to equilibrium. Both equilibrium and time-resolved kinetic methods have advantages and disadvantages. Equilibrium methods do not require rapid perturbation of the system; however, they are insensitive to transiently populated intermediates. Nor do equilibrium methods give any information on the rates of individual folding steps. Transient or time-resolved methods can reveal distinct kinetic phases, and detect transient intermediates, but the rapid perturbation of conditions can cause artifacts. Together, the two methods give a more complete understanding of the folding reaction.
Results
Wild-type (WT) BdpA was modified with two goals in mind: to increase helix stability in an attempt to increase the folding rate, and to introduce a tryptophan to follow folding with fluorescence. Inspection of the sequence of BdpA reveals one glycine at position 29, in the middle of helix 2. Glycine has a low helical propensity (Pace and Scholtz 1998), and was therefore replaced with alanine. This same substitution was made in a stabilized version of BdpA called the Z-domain (Nilsson et al. 1987). Position 29 is on the solvent-exposed face of the helix, and the introduction of a methyl group at this position is not expected to affect tertiary structure (Fig. 1 ▶). The mutant is referred to as G29A. An intrinsic fluorophore was added to BdpA by substitution of one of the phenylalanine residues with tryptophan (there are no tryptophans in wild-type BdpA). Position 13 was chosen because it partially buries the tryptophan, and potential steric clashes are minimal. The F13W mutation was made in the G29A background, to produce G29A/F13W. The fluorescence of native G29A/F13W has a maximum at 339 nm; this maximum shifts to 346 nm and the signal intensity decreases twofold upon denaturation (data not shown), thus fluorescence is an excellent probe of unfolding.
The conformational stability of each mutant was measured by denaturation with guanidine hydrochloride (GuHCl), monitored by circular dichroism (CD), and in the case of G29A/F13W, fluorescence (Fig. 2 ▶). The CD spectra of WT, G29A, and G29A/F13W are identical, indicating that a major alteration of the native secondary structure has not occurred as a result of the mutations (data not shown). The stability parameters are shown in Table 1. The change in the conformational stability of the protein resulting from the G29A mutation is only 0.4 kcal/mole, a marginal increase in stability. Typical glycine-to-alanine mutations at solvent-exposed positions in helices stabilize the helix by ~1 kcal/mole (Serrano et al. 1992; Myers et al. 1997). G29A/F13W is about 0.2 kcal/mole less stable than G29A, indicating that the F13W mutation alone is slightly destabilizing. The GuHCl m values of the mutants are very close to the m value of WT, consistent with the same amount of solvent-accessible surface area being exposed upon unfolding (Myers et al. 1995).
Figure 2.

Guanidine hydrochloride (GuHCl) denaturation of WT (circles), G29A (squares), and G29A/F13W (triangles) BdpA followed by circular dichroism at 222 nm. Solid lines are fits to a two-state denaturation model (Pace and Scholtz 1997).
Table 1.
Equilibrium denaturation parameters at 310 K
| Variant | Methoda | Solventb | Denaturantc | D1/2 (M) | m (kcal/mole • M) | ΔG° (kcal/mole)d |
| WT | CD | D2O | GuHCl | 2.87e | 1.50e | 4.30c |
| G29A | CD | D2O | GuHCl | 3.17 | 1.50 | 4.76 |
| NMR Hisf | D2O | thiourea | 5.67 | 0.98 | 5.55 | |
| NMR Tyrf | D2O | thiourea | 5.43 | 1.07 | 5.81 | |
| G29A/F13W | CD | H2O | GuHCl | 2.96 | 1.42 | 4.14 |
| Fluorescence | H2O | GuHCl | 2.94 | 1.41 | 4.15 | |
| CD | D2O | GuHCl | 3.03 | 1.49 | 4.51 | |
| NMR Hisf | D2O | thiourea | 5.16 | 0.97 | 5.06 | |
| NMR Tyrf | D2O | thiourea | 5.25 | 0.84 | 4.41 |
a CD = circular dichroism at 222 nm; fluorescence = fluorescence emission at 340 nm; NMR His = lineshape fitting of His 18 proton resonance; NMR Tyr = chemical shift of Tyr 15 proton.
b Buffer conditions were 20 mM sodium acetate, pH 5.0, 100 mM NaCl.
c “Thiourea” indicates urea and thiourea in a 3.33 : 1 molar ratio.
d Unfolding free energy in the absence of denaturant = D1/2 * m. For CD measurements, error estimated to be ±0.05 in D1/2, ±0.1 in m, and ±0.3 in ΔG°.
e Data for the wild-type protein from Myers and Oas (2001).
f NMR data was corrected for the 20% denatured population that is in slow exchange due to one or more incorrect proline isomers (Myers and Oas 2001).
Equilibrium parameters from NMR experiments (see below) are also given in Table 1. For G29A/F13W, we are able to compare results from four separate denaturation probes: CD, fluorescence, His 18 NMR resonance (from the lineshape fitting; see below), and the Tyr 15 NMR resonance (by following changes in chemical shift). Comparison of the NMR experiments performed in D2O with the CD performed in D2O gives similar extrapolated unfolding free energies. CD and fluorescence denaturation experiments performed in H2O are in excellent agreement with each other. Dimitriatis et al. (2004) found that thermal denaturation curves of G29A/F13W followed by CD and fluorescence also overlay. Altogether, this evidence is consistent with a two-state denaturation model for BdpA. Our results also indicate that D2O slightly stabilizes the protein, which is a common observation (Oas and Toone 1997).
In a previous study, we found the epsilon proton (Hɛ) of histidine 15 to be a convenient probe for NMR lineshape analysis to determine the folding and unfolding kinetics. His 18 is mostly protonated at pH 5.0 (the measured pK of the side chain is 7.0; J.K. Myers and T.G. Oas, unpubl.); however, deprotonation causes a much larger change in chemical shift than does unfolding. For this reason, changes in the small population of protonated (charged) side chain can have large effects on the chemical shift. In the present study, we found a complex, nonlinear dependence of chemical shift as a function of GuHCl in the native baseline region (data not shown). This nonlinearity was particularly pronounced in G29A and G29A/F13W, although it was not obvious in our previous study of WT. Because GuHCl is a salt, there could be complex effects on the electrostatic environment of the side chain, and is therefore not the ideal denaturant when following the resonance of a polar residue like His 18. Urea is the most common nonionic denaturant; however, the urea m value for BdpA is so low that a complete unfolding transition, including good native and denatured baseline regions, is not obtainable. Thiourea is a stronger denaturant than even GuHCl (Pace and Marshall 1980), is nonionic, and can be mixed with urea up to the solubility limits of both compounds (about 3 M for thiourea and 10 M for urea at room temperature) to make a very potent denaturant. Mixtures of thiourea and urea have been used in DNA denaturing electrophoresis gels when extra denaturing power is required. A disadvantage of thiourea is that it absorbs strongly in the UV region; thus, CD and fluorescence experiments are impossible. However, it is quite suitable for NMR, and we can compare thermodynamic and kinetic parameters extrapolated to 0 M denaturant from NMR experiments in thiourea/urea to those obtained in GuHCl by T-jump fluorescence, steady-state fluorescence, or circular dichroism. The use of thiourea/urea resulted in a much better behaved native NMR baseline.
The folding and unfolding kinetics of G29A and G29A/ F13W were measured by NMR lineshape analysis using thiourea/urea as the denaturant. Representative traces of the histidine 15 Hɛ resonance as a function of denaturant are shown in Figure 3 ▶. Fitting of the lineshapes gives the apparent rate constant and the equilibrium constant, from which the folding rate constant kf and the unfolding rate constant ku can be derived. The rate constants are plotted as a function of denaturant in Figure 4 ▶. Both variants give rate constants whose logarithm is a linear function of denaturant. Extrapolation to 0 M denaturant gives the kf and ku in water, which are 370,000 sec−1 and 37 sec−1 for G29A, and 450,000 sec−1 and 100 sec−1 for G29A/F13W. For G29A, the folding rate constant is threefold higher than WT, and the unfolding rate constant is twofold lower. The G29A/ F13W variant has nearly identical folding kinetics to G29A, but unfolds threefold faster. The βT parameter, derived from the slopes of the fits shown in Figure 4 ▶, is related to the solvent accessibility of the ensemble of transition states (Fersht 1999). It is 0.75 for G29A and 0.77 for G29A/ F13W, similar to the value of 0.79 found for WT found using GuHCl as the denaturant (Myers and Oas 2001). The values close to 1 indicate a relatively compact transition state, in terms of its solvent accessibility.
Figure 3.

NMR resonance of the G29A/F13W His 18 Hɛ at different concentrations of denaturant (thiourea + urea in a 1 : 3.33 molar ratio). The shifting of the signal as the population of denatured protein increases is clearly visible, along with the broadening present when both native and denatured states are populated and undergoing rapid exchange. The number alongside each peak gives the denaturant concentration, and the data have been moved along the Y-axis for convenient viewing.
Figure 4.

Folding (filled symbols) and unfolding (open symbols) rate constants vs. denaturant for G29A (squares) and G29A/F13W (triangles), from lineshape analysis of the His 18 Hɛ. The extrapolated rate constants for WT from Myers and Oas (2001) are shown as circles for comparison.
Discussion
We have demonstrated the rapid, two-state folding of two variants of BdpA. The folding time of both the G29A and G29A/F13W variants is only 3 μsec, very close to the proposed speed limit of 1 μsec. The G29A mutation stabilizes the native state by only 0.4 kcal/mole, less than the ~1 kcal/mole expected for a glycine-to-alanine substitution in a solvent-exposed position. It has been observed previously that helix stabilizing mutations may not have their full energetic impact in the intact protein due to helix stabilization in the denatured state (Munoz et al. 1996), even though helix propensities appear to be the same in proteins and isolated helices (Myers et al. 1997). Stabilization of helical structure in the denatured state is a likely explanation for the observed increase in the folding rate, consistent with a diffusion-collision folding mechanism.
In collaboration with A. Smith, S. Radford, and coworkers, G29A/F13W has been subjected to analysis by temperature jump and fluorescence (Dimitriadis et al. 2004). T-jumps were performed at a variety of starting temperatures and GuHCl concentrations. No evidence for transiently populated intermediates was observed, and all kinetic traces fit well to a single exponential. The logarithm of the rate constants appear to be linear functions of denaturant. The rate constants found by extrapolating to 0 M denaturant and 37°C are somewhat slower than our NMR results (kf = 230,000 sec−1 versus 370,000 sec−1). The absolute rate constants do not agree presumably because the NMR was carried out in D2O and the T-jumps in H2O. Thermodynamic data indicate that D2O stabilizes BdpA (Table 1), leading to faster folding rates and slower unfolding rates. No transient or stable intermediates have been observed with either NMR or T-jump. Importantly, exponential kinetics are still observed, even for a protein folding at this speed.
Laser T-jump followed by infrared spectroscopy on the wild-type protein also shows good agreement with NMR measurements (Vu et al. 2004). Bi-exponential kinetics were observed: a fast phase consistent with the helix/coil transition, and a slower phase consistent with the folding and unfolding of the domain. The folding rate constant of the slow phase, extrapolated from high temperature to 37°C, agrees with the value found by dynamic NMR. The fast phase is probably due to rapid helix formation in the denatured state of the protein, preceding formation of helix/helix interfaces. These results directly support a diffusion-collision mechanism for the folding of BdpA. Fluorescence T-jump only found one kinetic phase; however, the tryptophan probe in G29A/F13W would not necessarily be expected to be sensitive to the helix/coil phase, because the fluorescence reports on the burial of the tryptophan side chain. Taken together, these three studies present an extremely comprehensive thermodynamic and kinetic picture of the folding reaction.
BdpA has been used in many theoretical folding studies as a model system (Garcia and Onuchic 2003; Vila et al. 2003, and references therein). The very short folding time of G29A raises the exciting possibility that a full-atom, explicit solvent molecular dynamics simulation of obtainable length will show a complete folding reaction, starting from an extended chain. A recent 400-nsec molecular dynamics simulation of G29A BdpA, using techniques similar to a 1–μsec simulation of villin headpiece (Duan and Kollman 1998), shows the folding of the chain into a very native-like structure (H. Duan, pers. comm.). In the simulation, substantial helix formation precedes the collapse of the chain. Another fast folding helical protein that has been characterized using T-jump, and simulation is engrailed homeodomain studied by Fersht and coworkers. This protein collapses to a helical intermediate state in about 1 μsec, and completes folding to its native state in around 25 μsec (Mayor et al. 2003). Simulations of the folding (by unfolding the protein at high temperature) feature a transition state with a large amount of helix, and the individual helices are found to be fairly stable even after unfolding. This is congruent with a diffusion-collision mechanism, and similar to the folding simulation of BdpA by Duan and coworkers mentioned above. The sequences of both BdpA and engrailed homeodomain have a high intrinsic tendency to be helical, leading to helical denatured states, transition states, and intermediates along the folding pathways. Slower folding helical proteins probably have a decreased intrinsic helical propensity (Laurents et al. 2000), and some may get stuck in kinetic traps involving nonnative structure (Ferguson et al. 1999). A strong tendency towards forming native local structure would help the protein avoid kinetic traps. Pockets of native secondary structure would tend to present native-like hydrophobic regions to each other, facilitation their packing into progressively larger regions of structure, leading to a hierarchic assembly process (Lesk and Rose 1981).
There is growing evidence of the importance of early secondary structure formation in the folding pathways of proteins (Myers and Oas 2002; Ferguson and Fersht 2003). One method of probing the structure of folding transition states is φ value analysis (Fersht 1999). Point mutations are introduced into the protein, and for each mutant the energetic effect on the apparent transition state and on the native state is compared as a ratio. It is interesting to note that the G29A mutation studied here, designed to stabilize secondary structure, has a relatively high φ value of 0.6, suggesting helix 2 is largely formed in the ensemble of structures representing the transition state. Contrastingly, the F13W mutation, mainly affecting tertiary packing, has a φ value close to zero. We are currently introducing a battery of single site mutations into G29A/F13W so that a more complete φ value analysis can be undertaken. Other helix stabilizing mutations are also being explored. It will be interesting to discover if BdpA’s helices can be stabilized to such a point that the protein forms full helices first, then assembles the pre-formed helices, as in the framework model of protein folding (Kim and Baldwin 1990). We previously suggested that BpdA folds by a diffusion-collision mechanism (Myers and Oas 2001). In this mechanism, collisions between partially formed (and fluctuating) helices predominate early in the folding process, and the formation of stable helices is coupled to the formation of tertiary structure. Tight coupling between the formation of secondary and tertiary structure appears to be a general feature of protein folding (Krantz et al. 2002; Uversky and Fink 2002).
The functional role of rapid cycling between native and denatured states is not well explored. Certainly unfolding may have important functional implications in vivo, for example, degradation and membrane translocation, because these processes apparently happen via denatured proteins (Matouschek 2003). In the case of protein A, the protein must be transported across the S. aureus cell membrane where it is eventually anchored to the cell wall. Presumably all five of the immunoglobulin binding domains (A through E) must unfold at some point (but not necessarily simultaneously) to make this possible. If the kinetics of the folding/unfolding equilibrium is faster than translocation, then the translocation rate is limited by the amount of unfolded protein, and the thermodynamics of the unfolding reaction determine the translocation rate. However, if the protein unfolds slowly, then the unfolding rate becomes rate limiting, and determines how fast the protein can be transported. In general, the cell may prefer to have transport or degradation under either thermodynamic or kinetic control, depending on the protein, and therefore, evolution adjusts the folding and unfolding kinetics accordingly. It is interesting to note that protein folding and unfolding rate constants vary over a much wider range than do folding equilibrium constants. Many proteins have been found that are primarily unfolded in their purified form but fold when bound to ligands (Henkels et al. 2001; Uversky 2002). Perhaps fast-folding members of this class serve in the cell as rapid-acting on/off “switches.”
Materials and methods
Mutagenesis and protein production
The G29A and F13W/G29A mutations were made by the Quick Change procedure (Stratagene) using oligonucleotides from MWG biotech. Correct gene sequence was confirmed by DNA sequencing. BdpA was expressed and purified as previously described (Myers and Oas 2001). The correct molecular weights of G29A and G29A/F13W were confirmed by electrospray mass spectrometry.
Thermodynamic measurements
All thermodynamic and kinetic measurements were performed at 310 K. Fluorescence data were collected using a SLM model 8100 fluorimeter. The excitation and emission wavelength were 280 nm and 340 nm, respectively. The protein concentration was 1 μM in a buffer of 20 mM sodium acetate pH 5.0, 100 mM NaCl. Circular dichroism experiments were performed on an Aviv model 202 spectrapolarimeter. For CD titrations, 10 μM protein samples were used in a buffer of 20 mM sodium acetate pH 5.0, 100 mM NaCl. Protein concentrations were measured in 6 M GuHCl, using extinction coefficients for tryptophan and tyrosine given by Pace et al. (1995). Deuterated denaturant was prepared with multiple rounds of dissolution in D2O followed by lyophilization under vacuum. GuHCl concentrations were determined by refractive index measurements (Pace and Scholtz 1997). To analyze denaturation curves, an equation combining a two-state denaturation model and a linear dependence of free energy on denaturant was fit to the data to determine the midpoint of the denaturation curve (D1/2) and the dependence of the unfolding free energy on denaturant (m value; Pace and Scholtz 1997).
Kinetic measurements
NMR experiments were performed on a Varian Unity 500 MHz NMR spectrometer. Samples were prepared with 1.5 mM protein, 20 mM sodium acetate pH 5.0 (uncorrected pH meter reading), 100 mM NaCl, 0.1% NaN3, 0.1% TMSP, in 99% D2O, with various concentrations of deuterated thiourea/urea in a 1 : 3.33 molar ratio. Thiourea/urea stock solutions were prepared gravimetrically. Proton NMR spectra were collected using water presaturation with a sweep width of 6500 Hz. Spectra were processed using Felix 95 software. Peaks were fit to ideal Lorentzian lines to determine the chemical shifts, and referenced to TMSP. In the baseline regions, where fully native or denatured protein predominates, the chemical shift was fit to a linear dependence on denaturant concentration. In the transition region, the resonance of the His 18 epsilon proton was fit to equations describing the effect of chemical exchange on line broadening (Huang and Oas 1995; Burton et al. 1996) to give folding and unfolding rate constants at each denaturant concentration.
Acknowledgments
We thank J.M. Word for help with fluorescent mutant design, and S. Radford, A. Smith, and members of the Radford, Smith, and Oas laboratories for helpful discussions. We also thank M. Fitzgerald for the use of his mass spectrometer. This work was supported in part by the NIH (Grant GM-45322 to T.G.O.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03541304.
References
- Burton, R.E., Huang, G.S., Daugherty, M.A., Fullbright, P.W., and Oas, T.G. 1996. Microsecond protein folding through a compact transition state. J. Mol. Biol. 263 311–322. [DOI] [PubMed] [Google Scholar]
- Dimitriadis, G., Drysdale, A., Myers, J.K., Arora, P., Radford, S.E., Oas, T.G., and Smith, D.A. 2004. Microsecond folding dynamics of the F13W/G29A Mutant of the B-domain of protein A by laser-induced temperature jump. Proc. Natl. Acad. Sci. (in press). [DOI] [PMC free article] [PubMed]
- Duan, Y. and Kollman, P.A. 1998. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science 282 740–744. [DOI] [PubMed] [Google Scholar]
- Eaton, W.A., Munoz, V., Hagen, S.J., Jas, G.S., Lapidus, L.J., Henry, E.R., and Hofrichter, J. 2000. Fast kinetics and mechanisms in protein folding. Annu. Rev. Biophys. Biomol. Struct. 29 327–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, N. and Fersht, A.R. 2003. Early events in protein folding. Curr. Opin. Struct. Biol. 13 75–81. [DOI] [PubMed] [Google Scholar]
- Ferguson, N., Capaldi, A.P., James, R., Kleanthous, C., and Radford, S.E. 1999. Rapid folding with and without populated intermediates in the homologous four-helix proteins Im7 and Im9. J. Mol. Biol. 286 1597–1608. [DOI] [PubMed] [Google Scholar]
- Ferguson, N., Johnson, C.M., Macias, M., Oschkinat, H., and Fersht, A. 2001. Ultrafast folding of WW domains without structured aromatic clusters in the denatured state. Proc. Natl. Acad. Sci. 98 13002–13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht, A.R. 1999. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. W.H. Freeman, New York.
- Garcia, A.E. and Onuchic, J.N. 2003. Folding a protein in a computer: An atomic description of the folding/unfolding of protein A. Proc. Natl. Acad. Sci. 100 13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruebele, M. 1999. The fast protein folding problem. Annu. Rev. Phys. Chem. 49 485–516. [DOI] [PubMed] [Google Scholar]
- Hagen, S.J., Hofrichter, J., Szabo, A., and Eaton, W.A. 1996. Diffusion-limited contact formation in unfolded cytochrome c: Estimating the maximum rate of protein folding. Proc. Natl. Acad. Sci. 93 11615–11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen, S.J., Carswell, C.W., and Sjolander, E.M. 2001. Rate of intrachain contact formation in an unfolded protein: Temperature and denaturant effects. J. Mol. Biol. 305 1161–1171. [DOI] [PubMed] [Google Scholar]
- Henkels, C.H., Kurz, J.C., Fierke, C.A., and Oas, T.G. 2001. Linked folding and anion binding of the Bacillus subtilis ribonuclease P protein. Biochemistry 40 2777–2789. [DOI] [PubMed] [Google Scholar]
- Huang, G.S. and Oas, T.G. 1995. Submillisecond folding of monomeric lambda repressor. Proc. Natl. Acad. Sci. 92 6878–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, S.A., Karplus, M., and Weaver, D.L. 2002. Application of the diffusion-collision model to the folding of three-helix bundle proteins. J. Mol. Biol. 318 199–215. [DOI] [PubMed] [Google Scholar]
- Karplus, M. and Weaver, D.L. 1976. Protein-folding dynamics. Nature 260 404–406. [DOI] [PubMed] [Google Scholar]
- ———. 1994. Protein folding dynamics: The diffusion-collision model and experimental data. Protein Sci. 3 650–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, P.S. and Baldwin, R.L. 1990. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 59 631–660. [DOI] [PubMed] [Google Scholar]
- Krantz, B.A., Srivastava, A.K., Nauli, S., Baker, D., Sauer, R.T., and Sosnick, T.R. 2002. Understanding protein hydrogen bond formation with kinetic H/D amide isotope effects. Nat. Struct. Biol. 9 458–463. [DOI] [PubMed] [Google Scholar]
- Kubelka, J., Eaton, W.A., and Hofrichter, J. 2003. Experimental tests of villin subdomain folding simulations. J. Mol. Biol. 329 625–630. [DOI] [PubMed] [Google Scholar]
- Laurents, D.V., Corrales, S., Elias-Arnanz, M., Sevilla, P., Rico, M., and Padmanabhan, S. 2000. Folding kinetics of phage 434 Cro protein. Biochemistry 39 13963–13973. [DOI] [PubMed] [Google Scholar]
- Lesk, A.M. and Rose, G.D. 1981. Folding units in globular proteins. Proc. Natl. Acad. Sci. 78 4304–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matouschek, A. 2003. Protein unfolding—An important process in vivo? Curr. Opin. Struct. Biol. 13 98–109. [DOI] [PubMed] [Google Scholar]
- Mayor, U., Guydosh, N.R., Johnson, C.M., Grossmann, J.G., Sato, S., Jas, G.S., Freund, S.M., Alonso, D.O., Daggett, V., and Fersht, A.R. 2003. The complete folding pathway of a protein from nanoseconds to microseconds. Nature 421 863–867. [DOI] [PubMed] [Google Scholar]
- Munoz, V., Cronet, P., Lopez-Hernandez, E., and Serrano, L. 1996. Analysis of the effect of local interactions on protein stability. Fold. Des. 1 167–178. [DOI] [PubMed] [Google Scholar]
- Myers, J.K. and Oas, T.G. 2001. Preorganized secondary structure as an important determinant of fast protein folding. Nat. Struct. Biol. 8 552–558. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Mechanisms of fast protein folding. Annu. Rev. Biochem. 71 783–815. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997. A direct comparison of helix propensity in proteins and peptides. Proc. Natl. Acad. Sci. 94 2833–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson, B., Moks, T., Jansson, B., Abrahmsen, L., Elmblad, A., Holmgren, E., Henrichson, C., Jones, T.A., and Uhlen, M. 1987. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1 107–113. [DOI] [PubMed] [Google Scholar]
- Oas, T.G. and Toone, E.J. 1997. Thermodynamic solvent isotope effects and molecular hydrophobicity. Adv. Biophys. Chem. 6 1–52. [Google Scholar]
- Pace, C.N. and Marshall, H.F. 1980. A comparison of the effectiveness of protein denaturants for β-lactoglobulin and ribonuclease. Arch. Biochem. Biophys. 199 270–276. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure: A practical approach (ed. T.E. Creighton), pp. 299–321. IRL Press, Oxford, UK.
- ———. 1998. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, L., Pabit, S.A., Roitberg, A.E., and Hagen, S.J. 2002. Smaller and faster: The 20-residue Trp-cage protein folds in 4 micros. J. Am. Chem. Soc. 124 12952–12953. [DOI] [PubMed] [Google Scholar]
- Serrano, L., Neira, J.L., Sancho, J., and Fersht, A.R. 1992. Effect of alanine versus glycine in α-helices on protein stability. Nature 356 453–455. [DOI] [PubMed] [Google Scholar]
- Snow, C.D., Nguyen, H., Pande, V.S., and Gruebele, M. 2002. Absolute comparison of simulated and experimental protein-folding dynamics. Nature 420 102–106. [DOI] [PubMed] [Google Scholar]
- Tashiro, M., Tejero, R., Zimmerman, D.E., Celda, B., Nilsson, B., and Montelione, G.T. 1997. High-resolution solution NMR structure of the Z domain of staphylococcal protein A. J. Mol. Biol. 272 573–590. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. 2002. What does it mean to be natively unfolded? Eur. J. Biochem. 269 2–12. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. and Fink, A.L. 2002. The chicken-egg scenario of protein folding revisited. FEBS Lett. 515 79–83. [DOI] [PubMed] [Google Scholar]
- Vila, J.A., Ripoll, D.R., and Scheraga, H.A. 2003. Atomically detailed folding simulation of the B domain of staphylococcal protein A from random structures. Proc. Natl. Acad. Sci. 100 14812–14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu, D.M., Myers, J.K., Oas, T.G, and Dyer, R.B. 2004. Probing the folding and unfolding dynamics of secondary and tertiary structures in a three-helix bundle protein. Biochemistry (in press). [DOI] [PubMed]
- Wang, M., Tang, Y., Sato, S., Vugmeyster, L., McKnight, C.J., and Raleigh, D.P. 2003. Dynamic NMR line-shape analysis demonstrates that the villin headpiece subdomain folds on the microsecond time scale. J. Am. Chem. Soc. 125 6032–6033. [DOI] [PubMed] [Google Scholar]
- Wittung-Stafshede, P., Lee, J.C., Winkler, J.R., and Gray, H.B. 1999. Cytochrome b562 folding triggered by electron transfer: Approaching the speed limit for formation of a four-helix-bundle protein. Proc. Natl. Acad. Sci. 96 6587–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]