

Abstract
The N-terminal domain of HIV-1 glycoprotein 41,000 (gp41) participates in viral fusion processes. Here, we use physical and computational methodologies to examine the secondary structure of a peptide based on the N terminus (FP; residues 1–23) in aqueous and detergent environments. 12C-Fourier transform infrared (FTIR) spectroscopy indicated greater α-helix for FP in lipid-detergent sodium dodecyl sulfate (SDS) and aqueous phosphate-buffered saline (PBS) than in only PBS. 12C-FTIR spectra also showed disordered FP conformations in these two environments, along with substantial β-structure for FP alone in PBS. In experiments that map conformations to specific residues, isotope-enhanced FTIR spectroscopy was performed using FP peptides labeled with 13C-carbonyl. 13C-FTIR results on FP in SDS at low peptide loading indicated α-helix (residues 5 to 16) and disordered conformations (residues 1–4). Because earlier 13C-FTIR analysis of FP in lipid bilayers demonstrated α-helix for residues 1–16 at low peptide loading, the FP structure in SDS micelles only approximates that found for FP with membranes. Molecular dynamics simulations of FP in an explicit SDS micelle indicate that the fraying of the first three to four residues may be due to the FP helix moving to one end of the micelle. In PBS alone, however, electron microscopy of FP showed large fibrils, while 13C-FTIR spectra demonstrated antiparallel β-sheet for FP (residues 1–12), analogous to that reported for amyloid peptides. Because FP and amyloid peptides each exhibit plaque formation, α-helix to β-sheet interconversion, and membrane fusion activity, amyloid and N-terminal gp41 peptides may belong to the same superfamily of proteins.
Keywords: CD spectroscopy, isotope-enhanced FTIR spectroscopy, computer simulations, electron microscopy, secondary structure, amyloid, prion, fusion
Earlier findings support the hypothesis that the amino-terminal peptide (FP; 23 amino acid residues; Fig. 1 ▶) of gly-coprotein 41,000 (gp41) participates in the fusion processes underlying human immunodeficiency virus (HIV-1) infection of host cells (McCune et al. 1988). Gallaher (1987) and Gonzalez-Scarano et al. (1987) each noted extensive homologies between other viral fusion peptides and those of the N terminus of gp41, and proposed that the N-terminal gp41 peptide is involved in the fusion of the HIV-1 envelope with host cells. When the HIV-1 glycoprotein 120,000 (gp120) binds to the lymphocyte CD4 receptor and one of several coreceptors of the chemokine family, the N-terminal gp41 domain is activated, which in turn, may attack the target cell surface (Jiang et al. 2002). Recent X-ray studies indicated that HIV-1 gp41 has a multimeric protein core that presents the N-terminal domain as a trimer (Lu et al. 1995; Weissenhorn et al. 1997), similar to the low-pH-induced conformation of influenza virus hemagglutinin (HA2; Bullough et al. 1994). This suggests that gp41 shares aspects of the low pH-induced “spring-loaded” mechanism for HA2, in which the N-terminal fusion peptide undergoes a major translocation (Bullough et al. 1994). According to one model, the N terminus of HIV-1 gp41 inserts deeply into the target-cell surface membrane (Gordon et al. 1992), and the viral envelope gp41 acts to bridge the host cell surface to the HIV-1 lipid bilayer (Aloia et al. 1988).
Figure 1.

Amino acid sequences of the native N-terminal peptide (FP) of HIV-1 gp41, and seven FP variants labeled with 13C at distinct positions. The N-terminal gp41 peptide is from the HIV-1 strain LAV1a with the sequence 1–23, corresponding to residues 519–541 using an earlier numbering system (Myers et al. 1991). Amino acids are represented by one-letter codes, and those residues labeled with 13C-carbonyls are indicated with bold letters and asterisks.
There is now considerable experimental evidence confirming the role of the N-terminal gp41 domain in HIV-1–mediated cytolytic and fusogenic processes. For example, site-directed mutagenesis studies indicated defective gp41 fusion activity for various modifications in the N-terminal domain, including replacement of hydrophobic amino acids with polar residues (Freed et al. 1990; Bergeron et al. 1992), deletion of short amino acid sequences (Schaal et al. 1995), or substitution of Gly or Phe residues with Val (Delahunty et al. 1996). An alternative experimental approach has been to synthesize peptides based on the known N-terminal gp41 sequence, and then to assess whether the biological activity attributed to the peptide domain in the virus is also observed with the isolated peptide. For example, synthetic peptides based on the N-terminal domain of gp41 promoted leakage of lipid vesicles (Rafalski et al. 1990; Slepushkin et al. 1992; Martin et al. 1993; Nieva et al. 1994), supporting the hypothesis that the N terminus of gp41 is partly responsible for the cytolytic actions of HIV-1 virions. Consistent with this proposal is the finding that N-terminal gp41 peptides lysed both cultured cells (Mobley et al. 1992; Dimitrov et al. 2001) and human erythrocytes (Mobley et al. 1992). Introduction of N-terminal gp41 peptides to model liposomes also induced lipid mixing (Rafalski et al. 1990; Slepushkin et al. 1990; Martin et al. 1993; Nieva et al. 1994; Kliger et al. 1997). With human erythrocytes, FP triggers not only rapid lipid mixing between cell membranes, but also the formation of multicell aggregates (Mobley et al. 1995, 1999, 2001). Of interest in this regard are the observations that selective modifications in synthetic N-terminal gp41 peptides reduced lytic and fusogenic activities, coincident with the lowered syncytia-forming properties of the corresponding mutated full-length gp41 (Mobley et al. 1995, 1999; Pereira et al. 1995; Martin et al. 1996; Kliger et al. 1997). Taken together, these results suggest that critical fusogenic actions of HIV-1 gp41 are captured by synthetic N-terminal peptides.
It is therefore important to elucidate the structure of the N-terminal gp41 peptide in aqueous and membrane environments, in view of the likely participation of this domain in HIV-1 fusion. One classical approach would be to determine its three-dimensional structure using X-ray crystallography. However, previous X-ray analyses have been performed only on gp41 proteins lacking the N-terminal region, because full-length gp41 proteins do not form crystals. Instead, circular dichroism (CD) and Fourier transform infrared (FTIR) spectroscopy have been used to investigate the structure of synthetic N-terminal peptides. These experiments indicated that N-terminal gp41 peptides exhibit variable proportions of secondary conformations (e.g., α-helix, β-sheet, β-turn, random) when in aqueous, membrane-mimics, and membrane lipids, depending on peptide length and concentration, solvent polarity, lipid charge, and cation concentrations (Rafalski et al. 1990; Slepushkin et al. 1990; Gordon et al. 1992; Martin et al. 1993, 1996; Nieva et al. 1994; Chang et al. 1997a; Kliger et al. 1997; Mobley et al. 1999). Moreover, oriented FTIR spectra of the N-terminal gp41 peptide indicated an oblique insertion of the α-helix region into lipid bilayers (Martin et al. 1993, 1996), consistent with recent theoretical predictions based on molecular dynamics simulations indicating penetration of the FP α-helix into the hydrophobic membrane interior (Kamath and Wong 2002; Maddox and Longo 2002). Nevertheless, a significant limitation of earlier CD and conventional FTIR spectral work is that these are global methodologies that cannot assign conformations or orientations to individual amino acid residues within the peptide.
Two-dimensional nuclear magnetic resonance (2D-NMR) and solid-state, 13C-enhanced nuclear magnetic resonance (SS 13C-NMR) studies each offer the potential of defining the “residue-specific” conformations of the N-terminal gp41 peptide in aqueous, membrane-mimic, and/or lipid environments. Using CD and 2D-NMR spectroscopy for FP in an acidic aqueous medium at 5°C, residues Phe-8 to Phe-11 exhibited a type-1 β-turn (Chang et al. 1997b). In a 2D-NMR, CD and molecular modeling study of FP suspended in the membrane-mimics trifluoroethanol (TFE) solvent or sodium dodecyl sulfate (SDS) detergent, however, Chang et al. (1997a,b) noted high levels of α-helix. Specifically, FP in 50% aqueous TFE assumed an α-helix conformation for residues Ile-4 to Ala-15, whereas the corresponding α-helix included Gly-5 to Gly-16 for FP bound to SDS micelles. With a subsequent 2D-NMR investigation of a FP analog in SDS, the hydrophobic core (~residues Gly-5 to Gly-16) was similarly reported to fold in an α-helical conformation (Vidal et al. 1998). Contrarily, a recent SS 13C-NMR analysis of FP bound to mixed liposomes at −50°C indicated that residues Ala-1 to Ala-15 are extended β-strands (Yang et al. 2001). Although the sharply divergent conformations reported in these NMR studies for FP in membrane mimics (TFE, SDS micelles; Chang et al. 1997a,b; Vidal et al. 1998) or mixed lipid bilayers (Yang et al. 2001) may simply reflect the intrinsic polymorphism of this peptide, questions may be raised about extrapolating FP structures elucidated with either 2D-NMR or SS 13C-NMR spectroscopy to those in biological membranes. On the one hand, the local FP conformations determined in 2D-NMR studies of membrane-mimics (Chang et al. 1997a,b; Vidal et al. 1998) may not faithfully reflect the corresponding structure in biological membranes, while on the other it is unclear whether the peptide structure assessed by SS 13C-NMR of FP in liposomes at −50°C (Yang et al. 2001) will be maintained at physiologic temperatures.
Accordingly, it is worthwhile to assess the residue-specific conformations of FP in various membrane-mimics and, if possible, biological membranes with alternative experimental methodologies. One such approach employs 13C-enhanced FTIR spectroscopy, which has previously identified specific random, β-strand and β-turn structural domains in a soluble peptide (Tadesse et al. 1991), α-helical structures in the transmembrane domain of phospholamban (Ludlam et al. 1996), and discrete antiparallel β-sheet regions in amyloid peptides (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999). In a recent 13C-FTIR structural study (Gordon et al. 2002), a suite of 13C-labeled peptides was used to determine the residue-specific conformations of FP in the membrane-mimic hexafluoroisopropanol (HFIP), the lipid 1-palmitoyl-2-oleoyl phosphatidylglycerol (POPG), and erythrocyte ghosts and ghost lipid extracts. Combining these 13C-enhanced FTIR results with molecular simulations indicated the following model for FP in HFIP: α-helix (residues 3–16) and random and β-structures (residues 1–2 and residues 17–23). Additional 13C-FTIR analysis indicated a similar conformation for FP in POPG at low peptide loading, except that the α-helix extends over residues 1–16 (Gordon et al. 2002). It is of considerable interest that the α-helical conformation (residues 5–15) detected for FP in SDS with 2D-NMR (Chang et al. 1997a) is also found in 13C-FTIR spectra of membrane lipids and erythrocyte ghosts at low peptide loading (Gordon et al. 2002). Similarly, the β-structure (residues 5–15) originally observed for FP in mixed liposomes with SS 13C-NMR (Yang et al. 2001) is also reported with 13C-FTIR spectra of FPG5-A15 added to erythrocyte ghosts or lipids at high peptide/lipid (P/L) ratios (Gordon et al. 2002).
Given its ability to complement and extend the conformational results on HIV-1 FP acquired with NMR techniques, 13C-FTIR spectroscopy is applied in this study to assess the residue-specific structures of FP in detergent and aqueous environments. As noted above, one important unresolved question is how closely the structure of FP in detergent micelles reproduces that of FP in membrane bilay-ers. Here, 13C-FTIR results on FP in SDS at low peptide loading indicated α-helix (residues 5 to 16) and disordered conformations (residues 1–4), in agreement with a prior FP structural model obtained using 2D-NMR spectroscopy (Chang et al. 1997a). However, because earlier 13C-FTIR analysis of FP in POPG lipid bilayers demonstrated α-helix for residues 1–16 at similarly low peptide loading (Gordon et al. 2002), the FP structure in SDS micelles only approximates that found with membranes. The limitations of FP-SDS micelles in mimicking the structural properties of FP–membrane systems are highlighted here using molecular dynamics simulations of FP in an explicit SDS micelle. Such simulations indicated that the fraying of the first three to four residues may be due to the FP helix moving to one end of the SDS micelle, with the peptide exposing its N-terminal residues to aqueous solvent. In further experiments, 13C-FTIR conformational mapping of FP in only PBS indicated extensive β-sheet for FP (residues 1–12). Of particular interest is our finding that the 13C-FTIR spectral signature for β-sheet residues of FP in PBS is identical to that observed for antiparallel β-sheet residues in amyloid peptides (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999). Because we also report here the presence of characteristic “amyloid-like” fibrils for FP in PBS using transmission electron microscopy (TEM), consistent with earlier studies on this peptide (Slepushkin et al. 1992; Pereira et al. 1997), our results support an earlier hypothesis that amyloid and N-terminal gp41 peptides belong to the same class of proteins (Callebaut et al. 1994; Nieva et al. 2000; Tamm and Han 2000).
Results
Conventional 12C-FTIR spectroscopy of FP in SDS micellar or PBS environments
Secondary structures for FP in the SDS micellar and aqueous environments were examined using conventional 12C-FTIR spectroscopy. Representative FTIR spectra of the amide I band for FP in the SDS and PBS systems are shown in Figure 2 ▶. A principal band occurs at 1657 cm−1 for the SDS spectrum (Fig. 2C ▶), and very similar spectra with major peaks centered at 1657 cm−1 were also observed for FP suspended in the membrane-mimic HFIP solvent (Fig. 2B ▶) or POPG liposomes (Gordon et al. 2002). Because previous FTIR studies of deuterated proteins have assigned bands in the range of 1650–1659 cm−1 as α-helical (Byler and Susi 1986; Surewicz and Mantsch 1988; Haris and Chapman 1995), FP in SDS likely assumes high α-helical content. On the other hand, the control FTIR spectrum of FP alone in the PBS buffer (Fig. 2A ▶) showed a dominant peak at 1628 cm−1, a high-field shoulder of 1643 cm−1 and a minor high-field peak at 1696 cm−1 (Gordon et al. 2002). In agreement with the FTIR spectrum of FP in PBS (Fig. 2A ▶), an earlier FTIR spectroscopic study of a pure FP monolayer in D2O buffer similarly indicated a principal peak at 1620 cm−1, with a shoulder at 1640–1650 cm−1, and a less intense component at ~1690 cm−1 (Agirre et al. 2000). The major peak at 1628 cm−1 and minor peak at 1696 cm−1 in Figure 2A ▶ probably reflect extensive antiparallel β-sheets for FP in PBS, created by a strong interstrand and, to a lesser degree, intrastrand TDC interactions (Moore and Krimm 1976a,b; Krimm and Bandekar 1986); the high field-shoulder at approx. 1643 cm−1 further denotes some random structure for FP in PBS. FTIR spectra comparable to that observed for FP in PBS (Fig. 2A ▶) were reported for several aqueous amyloid peptides, and were also attributed to high proportions of antiparallel β-sheet structures (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999).
Figure 2.

Fourier transform (FTIR) spectra of the amide I band of FP in phosphate-buffered saline (PBS), hexafluoroisopropanol (HFIP), and sodium dodecyl sulfate (SDS) at 25°C, as described in Materials and Methods. (A) FP concentration was 470 1μM in deuterated PBS (pH 7.4). The arrows at 1628 and 1696 cm−1 denote a β-sheet component, while the shoulder at 1643 cm−1 also indicates random structure (Gordon et al. 2002). (B) FP concentration was 470 μM in deuterated hexafluoroisopropanol (HFIP)/water/formic acid (70:30:0.1, v/v; Gordon et al. 2002). (C) FP concentration was 470 μM in 94 mM SDS and deuterated PBS (pH 7.4) at a peptide/lipid (P/L) ratio of 1 : 200. The arrows at 1657 cm−1 in B–C indicate a dominant α-helix component for FP in these membrane-mimic environments. Spectra have been normalized for comparison. The abscissa for each spectrum (left to right) is 1730 to 1580 cm−1, while the ordinate represents absorption (in arbitrary units).
The relative proportions of secondary structure were analyzed with subsequent curve fitting using earlier criteria (Byler and Susi 1986), as described in Materials and Methods (Gordon et al. 1996, 2000, 2002). These Fourier self-deconvolutions indicated the following α-helical levels for FP in the various environments: POPG ~ HFIP ~ SDS >> PBS (Table 1). Although the α-helix proportions for FP in SDS and POPG in Table 1 lie within experimental error (i.e., 43.9% versus 52.1%), slightly fewer residues may participate in the α-helix for peptide in the SDS medium; that is, 10 and 12 residues are α-helical for FP in SDS and POPG, respectively. The FTIR analysis further indicates that significant β- and random structures are present for peptide in the POPG, HFIP, SDS, and PBS environments (Table 1). Last, the FTIR findings suggest that moving FP from a lipid (or membrane-mimic) milieu to a more aqueous environment transforms certain α-helical residues into β-sheet, β-turn, and random conformations. Table 1 shows that β-sheet, β-turn, and random conformations are greater than 80% of the total secondary structure for FP in PBS, while the corresponding α-helical level is less than 20%.
Table 1.
Proportions of secondary structureafor FP in PBS, HFIP solvent, SDS detergent, and POPG liposomes as estimated from Fourier self-deconvolution of the FTIR spectra of the peptide amide I band
| % Conformation | ||||
| System | 3-Helix | 3-sheet | 3-Turn | Disordered |
| FTIR Spectrab | ||||
| HFIPc | 52.0 | 11.5 | 22.5 | 14.1 |
| PBSd | 19.6 | 32.3 | 23.0 | 25.1 |
| SDSe | 43.9 | 20.9 | 22.9 | 12.3 |
| POPGf | 52.1 | 6.5 | 24.9 | 16.7 |
a Data are the means of four separate determinations and have an SE 5% or better.
b FTIR spectra were deconvoluted as described in Materials and Methods.
c FP (470 μM) dried on to an ATR plate from 100% HFIP, and resolvated with deuterated HFIP/water/formic acid (70:30:0.1, v/v) (Fig. 2B ▶) (Gordon et al. 2002).
d FP (470 μM) dried on to the ATR plate from 100% HFIP, and resolvated with deuterated PBS, pH 7.4 (Fig. 2A ▶) (Gordon et al. 2002).
e FP was suspended at 470 μM in 94 mM SDS and deuterated PBS at a peptide/lipid (P/L) ratio of 1/200, dried on the ATR from the SDS suspension, and then resolvated with deuterated PBS, pH 7.4 (Fig. 2C ▶).
f FP incorporated into POPG at an initial P/L ratio of 1/70 for LUV lipo-somes suspended in PBS, pH 7.4. Peptide/liposomes were chromatographed to remove non-lipid-associated FP, and then dried onto the ATR plate, and resolvated with D2O (Gordon et al. 2002).
Isotopically enhanced 13C-FTIR spectroscopy of FP in the SDS micellar environment
Despite the above 12C-FTIR spectroscopic results indicating that FP assumes very different conformations in the SDS micellar and PBS systems (Fig. 2 ▶; Table 1), it is still not possible to assign secondary structure to specific residues. To further probe for local conformations, therefore, isotopically enhanced FTIR spectroscopy was next conducted with FP labeled with 13C-carbonyls at multiple sites, staggered to sequentially cover the peptide (Fig. 1 ▶).
With peptides in the SDS micellar and PBS (pH 7.4), environment (Fig. 2 ▶; Table 1), Figure 3 ▶ shows the natural abundance, 12C-FTIR spectrum of FP and the isotopically enhanced 13C-FTIR spectra of FP labeled with 13C-carbonyls at Ala-1 and Gly-3 (FPA1/G3), Ala-1, Gly-3, and Gly-5 and Leu-7 (FPA1/G3/G5/L7), Gly-5 to Ala-15 (FPG5-A15), or Gly-20 and Ala-21 (FPG20/A21; Fig. 1 ▶). There are major differences between the native and cassette spectra, which are attributed to 13C-carbonyl groups. In the FPG5-A15 spectrum (Fig. 3C ▶), there is a decrease in the area 1677–1630 cm−1 corresponding to an α-helical component, and a concurrent increase in the area 1630–1586 cm−1 with a major new peak centered at 1621 cm−1, indicating an isotopic shift of ~36 cm−1. This isotopic shift may also be visualized in a difference FTIR spectrum, obtained by subtracting the native FTIR spectrum (dashed line in Fig. 3C ▶) from that of the FPG5-A15 spectrum (solid line in Fig. 3C ▶); the resulting difference FTIR spectrum (Fig. S-1C) confirms the presence of negative and positive bands centered at 1657 cm-1 and 1618 cm−1, respectively. The simplest interpretation of these results is that the isotope-induced shift of ~36–39 cm−1 in Figures 3C ▶and S-1C is due to residues Gly-5 to Ala-15 participating in α-helix (Fig. 4 ▶), a peptide conformation in which TDC interactions will probably be negligible (see below; Moore and Krimm 1976a,b; Krimm and Bandekar 1986; Halverson et al. 1991). These findings suggest that the ~10 α-helical residues for FP in SDS, identified above from Fourier self-deconvolution of the 12C-FTIR spectrum (Fig. 2C ▶; Table 1), are approximately localized to the Gly-5 to Ala-15 region. Furthermore, the high α-helical content in the Gly-5 to Ala-15 region determined here with 13C-FTIR spectroscopy is in good agreement with the 2D-NMR spectroscopic study of FP bound to SDS micelles, which demonstrated α-helix for residues Gly-5 to Gly-16 (Chang et al. 1997a). Interestingly, earlier 13C-FTIR spectra of FPG5-A15 in the HFIP solvent or POPG liposomes similarly show new, predominant peaks at ~1620–1621 cm−1, representing an isotopic shift of ~37 cm−1 for the major α-helical peak at 1657 cm−1 in the respective 12C-FTIR spectra of native FP in these milieu (Gordon et al. 2002). This confirms that a high proportion of amino acid residues between FP residues Gly-5 and Ala-15 assumes an α-helical conformation, for FP in the SDS, HFIP, or POPG liposome environments (Fig. 4 ▶; Gordon et al. 2002).
Figure 3.

FTIR spectra of the amide I band for the 12C-carbonyl (i.e., “native”) FP peptide and a suite of multiply 13C-carbonyl enhanced FP peptides in sodium dodecyl sulfate (SDS; Fig. 1 ▶). Peptides were suspended at 470 μM in 94 mM SDS and deuterated PBS at a peptide/lipid (P/L) ratio of 1 : 200. Spectra were recorded at 25°C on the peptide, which was first dried on the ATR from the SDS suspension, and then resolvated with deuterated PBS: (A) FPA1/G3 is the solid line and native FP is the dashed line. The amide I band is shown for the native FP spectrum, with a dominant α-helical component centered at 1657 cm−1. The minor peak at 1608 cm−1 in the FPA1/G3 spectrum indicates random structure. (B) FPA1/G3/G5/L7 (solid line), FP (dashed line). The minor peak at 1613 cm−1 in the FPA1/G3/G5/L7 spectrum indicates a mix of α-helical and random components. (C) FPG5-A15 (solid line), FP (dashed line). The major peak at 1621 cm−1 in the FPG5-A15 spectrum indicates a strong α-helical component. (D) FPG20/A21 (solid line), FP (dashed line). The broad, minor shoulder centered at 1608 cm−1 and extending to ~1590 cm−1 in the FPG20/A21 spectrum indicates random structure and minor β-sheet.
Figure 4.

Conformational map of the N-terminal peptide (FP) of HIV-1 gp41 in HFIP, SDS micellar, and PBS environments, as estimated from the FTIR spectra of 13C-labeled peptides. Amino acids are represented by three-letter codes. Codes (in parentheses) for peptide conformations are: β-sheet (β),β-turn (βT), α-helix (α) and random (R). For FP (470 μM) in HFIP:water:formic acid (70:30:0.1. v/v), conformations were earlier determined from native and 13C-enhanced FTIR spectra (Gordon et al. 2002). For FP in SDS micelles (P/L = 1 : 200) in PBS (pH 7.4), conformations were determined from the FTIR spectra of Figures 3 ▶, S-1, and 5D–F. For FP (470 μM) in PBS (pH 7.4), conformations were determined from the FTIR spectra of Figures 5A ▶–C and 6.
It is important to determine whether intermolecular TDC interactions between 13C-carbonyls contribute either to the isotopic-shifted peak at ~1620–1621 cm−1 in the SDS FPG5-A15 spectrum (Fig. 3C ▶; Moore and Krimm 1976a,b; Krimm and Bandekar 1986) or to the corresponding isotope-shifted peaks in the HFIP and POPG FPG5-A15 spectra (Gordon et al. 2002). Following earlier protocols (Halverson et al. 1991; Ashburn et al. 1992), control experiments were performed by diluting 13C-labeled FPG5-A15 with unlabeled FP (i.e., 1 part 13C-peptide and 1 part native 12C-peptide) in the HFIP solvent. The FTIR spectrum of the “isotopically diluted” peptide mixture in the HFIP solvent (Fig. S-2A) indicates a reduced 13C-signal at ~1621 cm−1, but no frequency shift, as would be expected if intermolecular TDC interactions were diminished (Halverson et al. 1991; Ashburn et al. 1992). Also, it should be noted that the mixed experimental FTIR spectrum (solid line in Fig. S-2A) agrees well with a predicted FTIR spectrum (dashed line in Fig. S-2A), obtained by simply averaging the FTIR spectrum of native FP in HFIP with that of the 13C-labeled FPG5-A15 peptide in HFIP. Thus, these FTIR results for FPG5-A15 in HFIP are consistent with an isotopic-shifted peak at ~1620 cm−1 (Gordon et al. 2002) representing a localized amide I absorption due to α-helix (residues Gly-5 to Ala-15), with no contributions from intermolecular TDC interactions (Fig. 4 ▶). Given the close resemblances between the 13C-FTIR spectra for FPG5-A15 in the SDS (Fig. 3C ▶), HFIP and POPG (Gordon et al. 2002) environments, comparable α-helical domains (residues Gly-5 to Ala-15) probably also occur for FP in SDS micelles, HFIP (Fig. 4 ▶) and POPG liposomes (Gordon et al. 2002).
The conformational fine structure within the central core of FP in the SDS micelles was further investigated using fusion peptides labeled with 13C-carbonyls at Gly-5 and Ala-6 (FPG5/A6), Leu-7, Leu-9, and Leu-12 (FPL7/L9/L12), and Gly-13, Ala-14, Ala-15, and Gly-16 (FPG13-G16; Figs. 1 ▶,5,S-1). The new peaks at 1619 and 1617 cm-1 for the FPG5/A6 and FPL7/L9/L12 spectra (Fig. 5, D and E ▶), respectively, confirm that Gly-5, Ala-6, Leu-7, Leu-9, and Leu-12 assume α-helical conformations (Fig. 4 ▶). On the other hand, the FPG13-G16 spectrum for peptide in SDS micelles (Fig. 5F ▶), as well as the corresponding difference FPG13-G16 spectrum (Fig. S-1D), show a new positive band centered at 1608 cm−1 and extending to ~1590 cm−1, indicating that Gly-13, Ala-14, Ala-15, and Gly-16 adopt a random conformation with minor β-sheet contributions (Fig. 4 ▶). In summary, these findings suggest that this GAAG sequence represents the C-terminal cap of the α-helix, with the α-helical region spanning Gly-5 to Gly-16 for FP in SDS micelles. Interestingly, the spectra of FPG5/A6, FPL7/L9/L12, FPG5-A15, and FPG13-G16 in either HFIP solvent (Fig. 4 ▶) or POPG liposomes (Gordon et al. 2002) indicate that FP similarly assumes an α-helix between Gly-5 to Gly-16, with the only difference being that the carbonyl groups of Gly-13 to Gly-16 adopt a β-turn for FP in HFIP (Fig. 4 ▶) and POPG liposomes (Gordon et al. 2002). Accordingly, these 13C-FTIR results confirm that SDS micelles provide an environment for the FP core (i.e., Gly-5 to Gly-16), which accurately mimics that afforded by membrane lipid bilayers.
Figure 5.

FTIR spectra of the amide I band for the 12C-carbonyl (i.e., “native”) FP and a suite of FP labeled with multiple 13C-carbonyls in the central region, for peptides in the PBS or SDS micellar solutions. Peptides were suspended at 470 μM in deuterated PBS (pH 7.4), (A–C) or SDS micelles suspended in deuterated PBS (pH 7.4) (D–F) at 25°C; PBS spectra were recorded on peptide dried from 100% HFIP and resolvated on the ATR from deuterated PBS (pH 7.4), while SDS spectra were recorded on dried peptide-SDS micelles and resolvated with deuterated PBS (pH 7.4). (A) FPG5/A6 (solid line), FP (dashed line) in PBS. The FPA1/G3 spectrum indicates a residual 12C amide peak at 1636 cm−1 and a 13C amide peak at 1609 cm−1, denoting antiparallel β-sheet. (B) FPL7/L9/L12 (solid line), FP (dashed line) in PBS. The FPL7/L9/L12 spectrum indicates a residual 12C amide peak at 1643 cm−1and a 13C amide peak at 1607 cm−1 denoting antiparallel 3-sheet. (C) FPG13-G16 (solid line), FP (dashed line) in PBS. The shoulder encompassing components at approx. 1605 and 1593 cm−1, indicates random and β-sheet elements. (D) FPG5/A6 (solid line), FP (dashed line) in SDS micelles. The minor shoulder centered at 1619 cm−1 in the FPG5/A6 spectrum indicates an α-helix. (E) FPL7/L9/L12 (solid line), FP (dashed line) in SDS micelles. The minor peak at 1617 cm−1 indicates α-helical components. (F) FPG13-G16 (solid line), FP (dashed line) in SDS micelles. The broad, low-field shoulder centered at 1605 cm−1 represents β-turn, random and β-sheet structures.
Secondary conformations at the N- and C-terminal regions of FP in SDS micelles were next analyzed using peptide analogs with 13C-carbonyls at Ala-1 and Gly-3 (FPA1/G3), Gly-20, and Ala-21 (FPG20/A21) and Ala-1, Gly-3, Gly-5, and Leu-7 (FPA1/G3/G5/L7; Fig. 1 ▶). The FTIR spectrum of FPG20/A21 in SDS micelles (Fig. 3D ▶), and also the corresponding difference FTIR spectrum for FPG20/A21 (Fig. S-1E), shows a new broad shoulder centered at 1608 cm−1 and extending to ~1590 cm−1, indicating that Gly-20 and Ala-21 probably exhibit a mix of random and minor β-sheet conformations (Fig. 4 ▶). Earlier FTIR spectra of FPG20/A21 in HFIP solvent or POPG liposomes demonstrated similar random and β-sheet components (Fig. 4 ▶; Gordon et al. 2002), confirming that Gly-20 and Ala-21 share similar conformations for FP in the SDS, HFIP, and POPG environments. At the N-terminal region, the FTIR spectra of FPA1/G3 in SDS (Figs. 3A ▶,S-1A) showed a minor positive band at 1608 cm−1, consistent with Ala-1 and Gly-3, adopting random conformations (Fig. 4 ▶). In contrast, the corresponding FTIR spectra of FPA1/G3 in HFIP solvent or POPG liposomes, respectively, indicated a mix of α-helix and random conformations (positive new band at 1614 cm−1) or α-helix (positive new band at 1617 cm−1; Gordon et al. 2002). The FTIR spectra of FPA1/G3/G5/L7, another N-terminal 13C-labeled fusion peptide, in SDS micelles showed positive bands at 1613 cm−1 (Figs. 3B ▶,S-1B), suggesting a mix of α-helix and random structures (Fig. 4 ▶). On the other hand, earlier FTIR spectra of FPA1/G3/G5/L7 in HFIP solvent or POPG liposomes indicated α-helix with positive new bands centered at 1619 and 1618 cm−1, respectively (Fig. 4 ▶; Gordon et al. 2002).
With the above 13C-enhanced FTIR results, it is now possible to develop the following conformational map for FP in SDS micelles: α-helix between residues 5 and 16, with random (or disordered) structures for residues 1–4, and random and minor β-sheet for residues 20 and 21 (Fig. 4 ▶).
Electron microscopy of FP in PBS buffer
The peptide complexes formed by FP in the aqueous phosphate-buffered saline (PBS; pH 7.4) were studied with transmission electron microscopy (TEM). Figure S-3B shows that peptide complexes form after incubation of FP on the formvar-coated grid in PBS at room temperature. After 1-h incubation, these peptide aggregates consist of a branching fibrous network, with individual fibers having a diameter of approx. 75 nm and variable lengths (up to approx. 1 μm); the control grid incubated without FP indicated only a blank field (Fig. S-3A). Fiber formation appeared to be complete at 1 h, as no further differences were observed in fiber appearance in samples incubated for 5 and 24 h (not shown). Another image of FP incubated with PBS for 1 h (Fig. S-4) shows an approximately spherical aggregate (~200 nm in diameter), with numerous branched fibrils (25–75 nm in diameter) extending from the core. It is of interest to compare the present results with earlier electron microscopic investigations on N-terminal gp41 peptides. Using the related 22-amino-acid fusion peptide (i.e., FP sequence in Figure 1 ▶, but omitting the C-terminal serine), Slepushkin et al. (1992) reported large spherical aggregates (500–700 nm in diameter), with a “clew-like” appearance consisting of long filaments approximately 5 nm in diameter. Also of note is the electron microscopy study of the fractions obtained by incubating FP with liposomes, after ultracentrifugation of peptide–vesicles mixtures in an aqueous buffer (Pereira et al. 1997). Using TEM, Pereira et al. (1997) showed that the unbound peptide in the pellet fraction consisted of fibrillar bundles ~500 nm in diameter. Independent ultracentrifugation experiments confirmed that FP produces insoluble aggregates when incubated in neutral buffers (Yang et al. 2001). The formation of insoluble FP aggregates may be responsible for the peptide inactivation often seen in lytic and lipid-mixing assays using liposome suspensions (Pereira et al. 1997), as well as the relatively high FP/lipid ratios required to demonstrate lytic and fusogenic actions with cells (Mobley et al. 1992, 1995, 2001).
Isotopically enhanced 13C-FTIR spectroscopy of FP in the PBS (aqueous) environment
The technique of 13C-enhanced FTIR spectroscopy was next used to map the secondary conformations of FP in aqueous media. With peptides in PBS (pH 7.4), Figure 6 ▶ shows the native 12C-FTIR spectrum of FP, and also the 13C-FTIR spectra of FP labeled with 13C-carbonyls at Ala-1 and Gly-3 (FPA1/G3), Ala-1, Gly-3, and Gly-5 and Leu-7 (FPA1/G3/G5/L7), Gly-5 to Ala-15 (FPG5-A15), or Gly-20 and Ala-21 (FPG20/A21). In each instance, there are major differences between the native and cassette spectra, due to the presence of 13C-carbonyl groups. The amide I band is shown for the native FP spectrum for peptide in PBS, with a dominant peak at 1628 cm−1 and a minor peak at 1696 cm−1 denoting an antiparallel β-sheet, and a high-field shoulder at 1643 cm−1, indicating random structure (Fig. 6A ▶). On the other hand, the FPA1/G3 spectrum indicates the 1628 cm−1 peak has split into two; one peak at 1634 cm−1, and the other at 1616 cm−1 (Fig. 6A ▶). The high-field peak at 1634 cm−1 is attributed to the normal 12C = O peak being shifted from 1628 cm−1, while the anomalously intense peak at 1616 cm−1 is due to 13C = O vibrations. Analogous “splitting” of the dominant β-sheet peak has been earlier observed in FTIR spectra of amyloid peptides enhanced with 13C = O at specific amino acids, and has been assigned to those residues participating in antiparallel β-sheet (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999). Therefore, Figure 6A ▶ shows that Ala-1 and Gly-3 fold as antiparallel β-sheet, for FP in PBS buffer (Fig. 4 ▶). The anomalously high intensity for the 13C = O peak at 1616 cm−1 (Fig. 6A ▶) may be accounted for with an antiparallel β-sheet model that incorporates transition dipole coupling (TDC) and through-bond interactions within the Wilson GF matrix method (Brauner et al. 2000). Similar to the FPA1/G3 spectrum, the FPA1/G3/G5/L7 spectrum in Figure 6B ▶ showed a splitting of the native β-sheet peak at 1628 cm−1 into a high-field 12C = O peak and a low-field 13C = O peak. Here, however, the residual 12C = O peak position for the FPA1/G3/G5/L7 spectrum is higher than the corresponding peak position in the FPA1/G3 spectrum (i.e., 1644 versus 1634 cm−1), while the 13C = O peak frequency for the FPA1/G3/G5/L7 spectrum is lower than the corresponding peak frequency in the FPA1/G3 spectrum (i.e., 1616 versus 1610 cm−1; Fig. 6AB). These results are readily explained by further reductions in the interstrand TDC interactions in the FPA1/G3/G5/L7 spectrum, due to Gly-5 and Leu-7 participating with Ala-1 and Gly-3 in antiparallel β-sheet (Fig. 4 ▶). The more extensive the interactions between 13C = O groups in adjacent strands, the lower will be the frequency of the 13C = O peak (Baldwin 1999). Contrarily, the FPG20/A21 spectrum in Figure 6D ▶ demonstrated a 1628cm−1 band with reduced intensity, as well as a broad new shoulder extending from 1607 to 1593 cm−1, consistent with Gly-20 and Ala-21 exhibiting random and extended β-conformations (Fig. 4 ▶). In this instance, however, the β-structures probably lack extensive interchain interactions, due to the absence of a “split” 1628 cm−1 peak (Fig. 6D ▶).
Figure 6.

FTIR spectra for the 12C-carbonyl (i.e., “native”) FP peptide and a suite of multiply 13C-carbonyl enhanced FP peptides in PBS solution. Peptides were suspended at 470 μM in deuterated PBS (pH 7.4), as described in Materials and Methods. (A) FPA1/G3 is the solid line and native FP is the dashed line. The amide I band is shown for the native FP spectrum, with a dominant peak at 1628 cm−1 and a minor peak at 1696 cm−1 denoting antiparallel β-sheet, and a high-field shoulder at 1643 cm−1 indicating random structure. The FPA1/G3 spectrum indicates a residual 12C amide peak at 1634 cm−1 and a 13C amide peak at 1616 cm−1, denoting antiparallel β-sheet. (B) FPA1/G3/G5/L7 (solid line), native FP (dashed line). The FPA1/G3/G5/L7 spectrum indicates a residual 12C amide peak at 1644 cm−1 and a 13C amide peak at 1610 cm−1, denoting antiparallel β-sheet. (C) FPG5-A15 (solid line), native FP (dashed line). The FPG5-A15 spectrum indicates a minimal residual 12C amide peak at and a pronounced 13C amide peak at 1590 cm-1, denoting antiparallel β-sheet. (D) FPG20/A21 (solid line), FP (dashed line). The low-field shoulder at ~1607 and tail at ~1593 cm−1in the FPG20/A21 spectrum indicates random (disordered) and β-sheet structure.
The central region of FP in aqueous environment was also investigated with the 13C-isotopically enhanced FPG5-A15 peptide. Interestingly, the FTIR spectrum of FPG5-A15 in PBS (pH 7.4), demonstrated a dramatic shift of the 1628 cm−1 band to 1590 cm−1, suggesting that the β-sheet region in FP largely overlaps the Gly-5 through Ala-15 domain (Fig. 6C ▶). Consistent with this assignment is the prediction by Halverson et al. (1991) that, if amide carbonyl 12C were to be replaced with 13C for every residue in an antiparallel β-sheet, then the amide I band frequency should also be reduced from 1628 to ~1591 cm−1. It should be additionally noted that the 1590 cm-1 peak for the FPG5-A15 spectrum is somewhat asymmetric with a high-field tail (1600–1615 cm−1), indicating minor disordered component mixed with the predominant β-sheet for Gly-5 to Ala-15 (Fig. 4 ▶). Our hypothesis that the 1590 cm−1 peak (Fig. 6C ▶) is principally due to intermolecular 13C = O interactions between adjacent antiparallel β-strands was further tested in isotope-dilution experiments. When 13C-labeled FPG5-A15 was mixed with unlabeled FP (i.e., 1 part 13C-peptide and 1 part native 12C-peptide) in PBS, the dilution in the label in the intermolecular β-sheet caused a shift in the low frequency 13C = O vibration from 1590 to 1600 cm−1 (solid line in Fig. S-2B). Furthermore, a new peak appeared at 1631 cm−1 for the mixed spectrum in Figure S-2B, which is attributed to TDC interactions between the 12C = O groups of adjacent β-sheet strands. Because averaging the FTIR spectrum of native FP in PBS with that of the 13C-labeled FPG5-A15 peptide in PBS cannot reproduce these spectral features (see dashed line in Fig. S-2B), the simplest interpretation of the 1590 cm−1 peak in Figure 6C ▶ is that it is primarily due to 13C-13C TDC interactions between adjacent strands in antiparallel β-sheets. Prior isotopic-dilution experiments conducted on various amyloid peptides have produced similar FTIR spectral changes for 13C-enhanced amino acids participating in antiparallel β-sheets (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999).
The β-sheet structure within the central core of FP in PBS was further explored using fusion peptides labeled with 13C-carbonyls at Gly-5 and Ala-6 (FPG5/A6), Leu-7, Leu-9, and Leu-12 (FPL7/L9/L12) and Gly-13, Ala-14, Ala-15, and Gly-16 (FPG13-G16; Fig. 5A–C ▶). The FPG5/A6 and FPL7/L9/L12 spectra (Fig. 5AB ▶) each share the characteristic “split” peak from the native 1628 cm−1 absorption band (i.e., 12C = O and 13C = O peaks of 1636 and 1609 cm−1 for FPG5/A6 and 1643 and 1607 cm−1 for FPL7/L9/L12, respectively), confirming that residues Gly-5, Ala-6, Leu-7, Leu-9, and Leu-12 each adopt antiparallel β-sheet conformations in PBS (Fig. 4 ▶). Contrarily, the FPG13-G16 spectrum for peptide in PBS (Fig. 5C ▶) shows a marked decline in intensity for the 1628 cm−1 band, and a new positive shoulder from ~1605 to 1590 cm−1, indicating that Gly-13, Ala-14, Ala-15, and Gly-16 adopt both random and extended β-conformations (Fig. 4 ▶).
Molecular modeling of FP in PBS
With the above 13C-enhanced FTIR results, it is now possible to develop the following conformational map for FP in aqueous PBS buffer: antiparallel β-sheet for residues 1–12, with extended β-structures and random (or disordered) structures for residues 13–23 (Fig. 4 ▶). Figure 7A ▶ shows a ribbon model for monomeric FP with residues Ala-1 through Leu-12 assuming a β-conformation, while Figure 7B ▶ shows an idealized trimeric FP subunit in PBS as an antiparallel β-sheet. Although FP is here modeled to maximize H-bonding between opposing peptides (residues 1–12) in the antiparallel β-sheet, it should be emphasized that the precise vertical register between the fusion peptides has not been experimentally determined. Further addition of FP peptides to either side of the trimeric FP subunit may create an “infinitely long” antiparallel β-sheet with a fibril axis perpendicular to the extended polypeptide chain.
Figure 7.
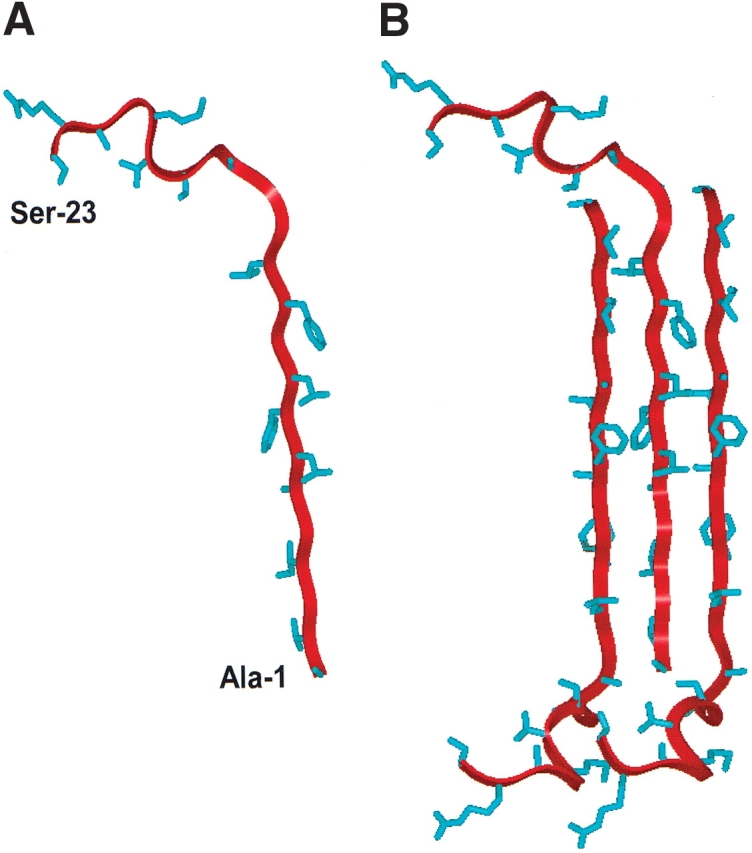
Ribbon representations of the N-terminal (FP) of HIV-1 gp41 in PBS solution as a monomer (A) and as a trimeric (B) antiparallel β-sheet. Residue-specific information on backbone conformations was derived from 13C-labeled FP peptides in the PBS solution (Figs. 4 ▶,5A–C ▶,6 ▶). Amino acid residues Ala−1to Leu-12 were determined as antiparallel β-sheet, while residues Gly-13 to Ser-23 were here assigned random and extended β-conformations (Fig. 4 ▶). (A) Monomer FP in PBS, with the conformer backbone modeled as a red ribbon and side chains as blue sticks; the N-terminal residue (Ala-1) and C-terminal residue (Ser-23) are indicated. (B) Trimeric FP in PBS with residues Ala-1 to Leu-12 as an antiparallel β-sheet, with the middle monomer assuming the same orientation as that of FP in Figure 7A. The precise vertical alignment for the monomers in this 3-sheet is undetermined.
Molecular dynamics simulations of FP in an explicit SDS micelle
FP interactions with a sodium dodecyl sulfate (SDS) micelle were explicitly simulated using CHARMM version c28b2 (Brooks et al. 1983; Kaznessis et al. 2002). The system of more than 16,000 atoms was simulated for 2.5 nsec at a constant pressure P = 1 atm and constant temperature T = 303.15 K. Initially, the SDS micelle consisted of 60 detergent molecules, built in an all-trans configuration, largely following an earlier established procedure (MacKerell 1995). Briefly, the carbon atom of the methyl group at the end of the acyl chain was positioned on the surface of a sphere with a radius of 3.5 Å, with the remainder of the molecule extending outward. Keeping the position of the last methyl group fixed, systematic body rotations of SDS molecules were used to perform a global search and reduce the number of unrealistic hardcore overlaps. Bad contacts are defined as a distance less than 2.3 Å between any two nonhydrogen atoms. Energy minimization was then conducted, allowing the internal degrees of freedom of the molecules to relax. This led to a spherical micelle of radius 23.3 Å, calculated as the average distance between the sulfur atoms of the headgroup and the center of mmass of the micelle.
The initial conformation of the FP peptide used in the FP–SDS micelle simulations was obtained from the Protein Data Bank (PDB accession code: 1ERF; http://www.rcsb.org), based on 13C-enhanced FTIR spectroscopy of FP in the HFIP solvent and molecular dynamics simulations (Gordon et al. 2002). In the membrane-mimicking HFIP solvent, FP assumes α-helix (residues 3–16) and random and β-structures for residues 1–2 and residues 17–23. The peptide with this conformation was then inserted into the above SDS micelle with its α-helical axis (Ala-15 to Ile-4) overlapping with the end-to-end distance axis of one of the SDS molecules. The N terminus of the peptide was incorporated into the detergent micelle such that the C- α atom of Ala-15 overlapped with the sulfur atom of the SDS molecule. This left the hydrophilic C terminus of the peptide effectively outside the micelle wrapped around the sulfate headgroup of the SDS molecules (Fig. 8 ▶). Of course, this insertion led to a very large number of unrealistic overlaps. With the peptide kept stationary, a global search was performed with systematic rigid rotations and translations of the SDS molecules to reduce the number of overlaps. Once again, energy minimization eliminated all the bad contacts between non-hydrogen atoms. The peptide–SDS micelle complex was subsequently solvated in a cube with 4375 TIP3P water molecules (Jorgensen et al. 1983). Finally, 0.15 M NaCl was added in the water region, positioning the ions randomly in space. Periodic boundary conditions were applied in all dimensions.
Figure 8.
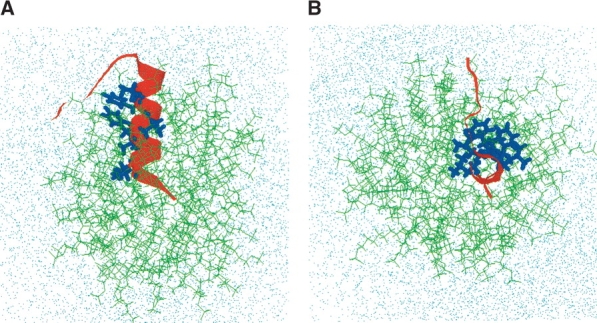
Initial conformation of hydrated FP-SDS micellar system before molecular dynamics simulations, viewed from the side (A) and down (B) the peptide α-helix (FP residues 3–16). (A) Side view of the initial configuration of the hydrated FP-SDS micelle, with the peptide’s α-helix (residues 3–16) penetrating the micelle, whereas the hydrophilic C-terminal region (residues 17–23) wraps around the micellar surface (i.e., micelle–water interface). The SDS detergent lipids are represented by green stick models; the FP backbone, as a red ribbon with the side chains for Ile-4, Leu-7, Phe-8, Leu-9, Phe-11, and Leu-12 as blue stick models; and the water molecules and sodium and chloride ions by dots. (B) Top view of the same initial configuration for the FP-SDS micellar system. The overall initial configuration of the FP-SDS micelle is spherical.
The entire system was minimized with 2000 steps of steepest descent and heated up to 303.15 K in the course of 500 psec. Two different views are shown for the system before (Fig. 8 ▶) and after minimization and heating (Fig. 9 ▶). Another 500 psec of equilibration at constant temperature and pressure were then conducted. The constant pressure-temperature module of CHARMM is used for the simulations with a leap-frog integrator (2-fsec time step). The temperature was set at 303.15 K using the Hoover temperature control with a mass of 1500 kcal psec2 for the thermal piston (Hoover 1985). For the extended system pressure algorithm employed, all the components of the piston mass array were set to 500 amu (Andersen 1980). The nonbonded van der Waals interactions were smoothly switched off over a distance of 3.0 Å, between 9 Å and 12 Å. The electrostatic interactions were simulated using the particle mesh Ewald (PME) summation with no truncation (Essman et al. 1995). A real space Gaussian width of 0.42 Å−1, a B-spline order of 6, and a FFT grid of about one point per Å (64 × 64 × 64) were used. The SHAKE algorithm was used to hold the hydrogen bonds fixed (Ryckaert et al. 1977). Throughout the simulation the backbone atoms of residues 3–16 were not constrained as α-helix.
Figure 9.
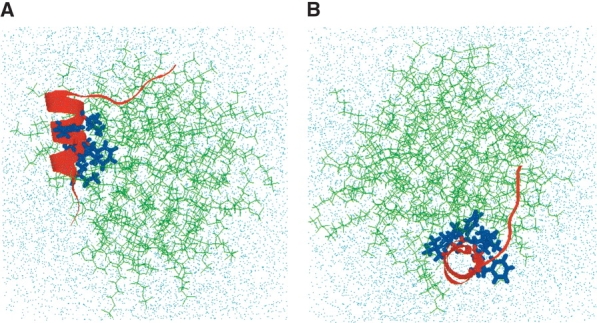
Final conformation of hydrated FP-SDS micellar system after molecular dynamics simulations, viewed from the side (A) and down (B) the peptide helix (FP residues 4–15). (A) Side view of the final configuration of the hydrated FP-SDS micelle, with the FP moving to one end of the micelle. The central core of FP remains buried in SDS, with the primary hydrophobic interactions with the micelle coming from the side chains of Ile-4, Leu-7, Phe-8, and Leu-12. However, the opposing residues on the amphipathic helix (i.e., Gly-5, Ala-6, Gly-10, Gly-13 Ala-14, Ala-15), and also residues 1–4, interact favorably with water molecules. The hydrophilic C-terminal region (residues 17–23) still lies on the micellar surface exposed to solvent, but now FP residues Ser-17 to Gly-20 form a type-1 β-turn. The SDS detergent lipids are represented by green stick models; the FP backbone, as a red ribbon with the side chains for Ile-4, Leu-7, Phe-8, Leu-9, Phe-11, and Leu-12 as blue stick models; and the water molecules and sodium and chloride ions, by dots. (B) Top view of the same final configuration for the FP-SDS micellar system, indicating that the spherical micelle has been largely compacted by the presence of FP to form a more oblate conformation.
The conformation and topography of FP in the SDS micelle at the end of the 2.5-nsec dynamic simulation is shown in Figure 9 ▶, and the coordinates for the 19 lowest energy structures of FP in the SDS micelle system have been deposited in the PDB under the accession code 1P5A. The FP conformation in SDS micelles exhibits both similarities and differences with that of the initial FP structure (accession code: 1ERF). The N terminus of FP (residues 1–3) becomes significantly more frayed, although the central helical conformation for residues Ile-4 to Ala-15 remains largely intact if somewhat distorted (Fig. 9 ▶). Using definitions for helical structures that focus on the respective H-bonding patterns (Kabsch and Sander 1983), FP exhibits α-helix for residues Ile-4 to Leu-7, and the rarely seen α-helix for residues Phe-8 to Ala-15. FP also forms a type-1 β-turn for residues Ser-17 to Gly-20 (Fig. 9 ▶). Rather more dramatic changes occur in the overall topographical organization of FP in the SDS micelle. In the equilibrium conformation, the peptide has moved from the center to one end of the micelle (Fig. 9 ▶). The final structure demonstrates the amphipathic nature of the peptide, orienting its helix to promote strong interactions at the water–hydrophobic interface. The central hydrophobic core of FP remains buried in SDS, with its primary hydrophobic interactions with the micelle coming from the side chains of Ile-4, Leu-7, Phe-8, Leu-9, Phe-11, and Leu-12 (Fig. 9 ▶). On the other hand, Ala-6, Ala-14, and Ala-15 interact favorably with water molecules, as do residues Ala-1 to Gly-3. The hydrophilic residues Gly-16 to Ser-23 remain exposed to solvent, being in close association with the sulfate headgroup region of the SDS micelle. Of interest is the finding that the SDS micelle is substantially perturbed by the presence of FP, in which the initial, spherical micelle (Fig. 8 ▶) has been largely compacted at the end of the dynamics simulation to a more oblate conformation (Fig. 9 ▶). The fatty acyl chains of SDS molecules have reconfigured themselves to facilitate tight associations with the hydrophobic side of FP. There also seems to be a considerable splaying or disorganization of the individual SDS molecules that surround FP, with some SDS chains in vertical alignment with the long axis of the helix (Fig. 9 ▶).
Discussion
Our 13C-FTIR spectroscopic findings on FP in PBS permit the development of an aqueous structural model for FP based on the antiparallel β-sheet (Figs. 4 ▶,7 ▶). As noted above, conventional 12C-FTIR spectra of FP suspended in aqueous environments reflected elevated proportions of an-tiparallel β-sheet (Fig. 2 ▶; Table 1; Agirre et al. 2000; Gordon et al. 2002), yet could not identify those amino acids participating in the β-structures. Here, 13C-FTIR spectroscopy indicated the following conformational map for FP in aqueous PBS: antiparallel β-sheet for residues 1–12, with extended β-structures and random (or disordered) for residues 13–23 (Fig. 4 ▶). An idealized trimeric FP subunit based on this residue-specific information is shown in Figure 7B ▶, representing a model that maximizes H-bonding between opposing peptides (residues 1–12). Addition of FP peptides to either side of the trimeric FP subunit through H-bonding may create an “infinitely long” antiparallel β-sheet with a fibril axis perpendicular to the extended polypeptide chains. A second fibril axis (perpendicular to the plane of the antiparallel β-sheet) may be formed by successively layering these antiparallel β-sheets on top of one another, in a manner analogous to that proposed for a peptide fragment of the human islet amyloid polypeptide (IAPP; Ashburn et al. 1992). The first fibril axis stabilized by hydrogen bonding would be expected to be stronger than the second axis, which depends on weaker hydrophobic interactions between the aromatic and aliphatic side chains projecting from the extended FP chains (residues 1–12). Such a structural model, predicated on the trimeric FP motif in Figure 7B ▶, readily accounts for the large fibrillar structures seen in the present (Figs. S-3,S-4) and earlier (Slepushkin et al. 1992; Pereira et al. 1997) EM studies. It is also tempting to speculate that comparable antiparallel β-sheet structures (Figs. 4 ▶,7) may be associated with membranes, as prior CD and 12C-FTIR studies have frequently demonstrated β-sheet conformations for FP added to liposomes at high P/L ratios (Rafalski et al. 1990; Gordon et al. 1992; Mobley et al. 1999; Saez-Cirion and Nieva 2002). Experimental support for this hypothesis comes from a recent SS 13C-NMR analysis of FP bound to mixed liposomes indicating that residues Ala-1 to Ala-15 are extended β-strands (Yang et al. 2001). Furthermore, antiparallel β-sheet (residues 5–15) have been detected from the 13C-FTIR spectra of FPG5-A15 added to erythrocyte ghosts or lipids at high P/L ratios (Gordon et al. 2002).
The 13C-FTIR spectroscopic and TEM observations in this paper provide additional confirmation for proposals that the N-terminal domain of HIV-1 gp41 and prions belong to the same superfamily of proteins (Callebaut et al. 1994; Nieva et al. 2000; Tamm and Han 2000). In an early sequence homology and hydrophobic cluster analysis, Callebaut et al. (1994) reported that the N-terminal HIV-1 gp41 domain and the prion protein (PrP) each share similar hydrophobicity and unusually high levels of glycine and ala-nine, as well as serine and valine. Fernandez and Berry (2003) more recently suggested that enriched levels of amino acids with short side chains are unable to protect backbone amide–carbonyl H bonds (HB) from water, and may provide a molecular mechanism accounting for the formation of amyloid fibrils in aqueous media. When immersed in membrane environments where water has been excluded, amyloid peptides are likely to form α-helical or random coil structures. However, when exposed to water, amyloid peptides cannot properly “wrap” their HB intramolecularly, and instead aggregate in a supramolecular β-sheet structure dominated by intermolecular wrapping (Fernandez and Berry 2003). Similar to previous studies with such amyloid peptides derived from amyloid-forming protein (β/A4), IAPP, PrP, and SP-C (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999; Johansson 2001), we here find that FP in the membrane-mimic SDS micelles primarily folds as an α-helix, while FP in aqueous PBS buffer assumes a dominant β-sheet conformation (Figs. 4 ▶,7, ▶8, ▶9 ▶). Also consistent with FP belonging to the prion family are our FTIR spectral findings that 13C = O substitutions for residues participating in antiparallel β-sheet produce a characteristic split 1628 cm−1 peak (i.e., highfield 12C = O peak and an anomalously-intense low-field 13C = O peak; Figs. 5 ▶,6 ▶), in agreement with earlier 13C-FTIR spectra of amyloid peptides in water (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999).
Combining the present results with an earlier 13C-FTIR study of FP in environments of varying polarity (Gordon et al. 2002) immediately suggests a model for how FP converts from an α-helical conformation in membranes to an amyloid-like antiparallel β-sheet in PBS. Under conditions of high hydrophobicity, 13C-FTIR spectra of FP in POPG liposomes indicated α-helix for residue 1–16 (Gordon et al. 2002). With the slightly more polar HFIP solvent, both 13C-FTIR spectra and molecular modeling indicated that the α-helix was largely conserved, except for a limited fraying of the N-terminal Ala-1 and Val-2 residues. The molecular model of FP in HFIP (PDB accession code: 1ERF) also indicated that FP folded as an amphipathic α-helix, with a hydrophobic side (i.e., residues Ile-4, Leu-7, Phe-8, Leu-9, Phe-11, and Leu-12) and a more polar, hydrophilic side encompassing the “glycine stripe” (i.e., Gly-3, Gly-5, Gly-10, Gly-13, Ala-14, Ala-15, and Gly-16; Gordon et al. 2002). 13C-FTIR spectra demonstrated that a more polar TFE solvent further converted FP residues 1–7 into β- and random conformations, and also residues 13–16 into random conformations, while still leaving considerable α-helix for the hydrophobic core residues Leu-7 to Leu-12 (Gordon et al. 2002). In accord with Fernandez and Berry (2003), these results are explained by a progressive attack of water on the exposed “glycine stripe” and other underwrapped residues (e.g., Ala, Val) at the N- and C-terminal ends of the original α-helix with subsequent fraying of the α-helix, leaving only the wrapped hydrophobic core (residues 7–12) still helical. For FP in PBS buffer, even the hydrophobic core residues now shift from an α-helical conformation into antiparallel β-sheet (Figs. 4 ▶,7 ▶). This permits the N-terminal gp41 peptide in aqueous PBS to form characteristic “amyloid-like” fibrils that have been observed with EM (Figs. S-3,S-4; Slepushkin et al. 1992; Pereira et al. 1997), analogously to that reported for such other prion peptides as PrP (Pillot et al. 1997) and SP-C (Gustafsson et al. 1999). In this regard, it should be noted that the ability of FP to fuse and lyse model liposomes and cells (see Introduction) is shared by the amyloid peptides PrP, Alzheimer β-amyloid and IAPP (Lorenzo and Yankner 1994; Pillot et al. 1997; Tenidis et al. 2000). Given the observations here supporting the hypothesis that FP and prions belong to the same superfamily, it will be of interest to explore whether FP and other amyloid peptides also lyse and fuse cells and liposomes through common mechanisms.
It is also important to consider the relationship between the FP structure elucidated in SDS micelles using 13C-FTIR spectroscopy (Fig. 4 ▶) with that determined for this peptide in membrane environments. Although membrane-associated proteins and peptides are frequently investigated spectroscopically by first solubilizing them in detergent micelles such as SDS (Henry and Sykes 1994), there have been limited opportunities to directly compare the respective structures of proteins/peptides in detergent micelles and lipid bilayers. Such a comparison is possible here because of an earlier 13C-FTIR spectroscopic analysis of FP in POPG liposomes at low P/L (Gordon et al. 2002), which indicated α-helix for residues 1–16 and random and extended β-structures for residues 20–21. Consequently, FP in either SDS or POPG liposomes at low peptide loading share the same α-helical conformation for Gly-5 to Gly-16, with a glycine-based cap at Ala-15 known as the αL motif in which Gly-16 assumes a left-handed conformation and a single hydrogen bond occurs between the 〉N—H at Gly-16 and the 〉C = O at Leu-12 (Gordon et al. 2002). Based on these findings, it is tempting to propose that the α-helical motif (Gly-5 to Gly-16) observed in SDS is also responsible for the high α-helical levels reported for synthetic N-terminal gp41 peptides (no. 23 residues) in either model (Rafalski et al. 1990; Martin et al. 1996; Curtain et al. 1999; Mobley et al. 1999; Saez-Cirion and Nieva 2002) or erythrocyte ghost liposomes (Gordon et al. 1992) at low P/L ratios (< ~ 1/70). Support for this hypothesis comes from a recent 13C-FTIR spectral analysis of erythrocyte ghost liposomes with FPG5-A15 at a low P/L of 1/70, which confirmed that Gly-5 through Ala-15 is α-helical (Gordon et al. 2002). Similar 13C-FTIR spectroscopic observations for FPG5-A15 in erythrocyte ghosts at a low P/L of 1/70 (Gordon et al. 2002) raise the possibility that the hydrophobic core (residues Gly-5 to Ala-15) of the N-terminal gp41 peptide domain may also fold as α-helix when inserted into the surface membrane of target CD4+-cells during HIV infection.
Nevertheless, it should be pointed out the FP conformation in SDS micelles does not faithfully reproduce all of the structural features of FP in membrane lipid bilayers. Although 13C-FTIR spectroscopy showed that the α-helix for FP in POPG extended from residues 1–16 (Gordon et al. 2002), the corresponding 2D-NMR (Chang et al. 1997a) and 13C-FTIR (Fig. 4 ▶) spectral analyses for FP in SDS indicated fraying of the α-helix at the N-terminal residues Ala-1 to Ile-4. The participation of the N-terminal residues in the α-helix determined for FP in POPG (Gordon et al. 2002) may be relevant in HIV-cell fusion, because removal of as few as three residues from the N terminus of gp41 markedly diminished syncytia formation of CD4+ cells transfected with this deletion mutant (Schaal et al. 1995). In summary, the residue-specific structures determined for N-terminal gp41 peptides in SD micelles using either 2D-NMR (Chang et al. 1997a; Vidal et al. 1998) or 13C-FTIR (Fig. 4 ▶) spectroscopy appear to be first approximations of the FP structure in membrane environments at low P/L ratios. Accordingly, the conformational differences characterized here for FP in detergent micelles and bilayer lipids may serve as a more general caveat when extrapolating peptide/protein structures determined with detergents to those in membrane environments.
The conformational and topographical findings from our explicit dynamic simulations here for FP in SDS micelles provide a physical model that accounts for both the limitations and applicability of detergent micelles in emulating membrane environments for this peptide. Dynamic simulations indicate substantial changes from the initial FP conformation as the peptide migrates from the central core to one end of the micelle. Figure 9 ▶ shows that the N terminus of FP (residues 1–3) becomes more frayed, although the central helix for residues Ile-4 to Ala-15 remains largely intact albeit somewhat distorted, with a type-1 β-turn for Ser-17 to Gly-20 closely associated with the SDS micellar headgroup region. FP exhibits α-helix for residues Ile-4 to Leu-7, while residues Phe-8 to Ala-15 loosen to form α-helix, in which the backbone C = O of residue i hydrogen bonds to the backbone HN of residue i + 5 (PDB accession code: 1P5A). Thus, the αL motif that originally caps the α-helix is converted into a characteristic “π-bulge,” which forms the C-terminal end of the helix. Although infrequently observed (Weaver 2000), a recently modified α-helix definition algorithm (Fodje and Al-Karadaghi 2002) showed that the α-helix may be 10 times more prevalent than earlier reported. Results from physical experiments and other dynamic simulations provide independent confirmation for many, but not all, of the structural features of the dynamics simulation model for FP in SDS micelles (Fig. 9 ▶). The fraying of the N-terminal residues and the α-helix for residues Gly-5 to Leu-7 seen in Figure 9 ▶ is confirmed by both 2D-NMR (Chang et al. 1997a) and 13C-FTIR (Fig. 4 ▶) spectra. Furthermore, the type-1 β-turn for the hydrophilic residues Ser-17 to Gly-20 in Figure 9 ▶ was also observed from in vacuo dynamic simulations of FP (Chang et al. 1997a). However, there is no independent confirmation for the α-helix noted for residues Phe-8 to Ala-15 of FP (Fig. 9 ▶), as only an α-helix was determined in 2D-NMR (Chang et al. 1997a) and 13C-FTIR (Fig. 4 ▶) studies. The fraying of N-terminal residues and loosening of the helix at the C-terminal end in Figure 9 ▶ are each attributed to the aqueous exposure of the N-terminal residues, “glycine stripe” and GAAG helical cap, and subsequent attack by water on the underwrapped HB (Fernandez and Berry 2003). Interestingly, the related amyloid peptide SP-C, which is α-helical in a hydrophobic environment (Johansson et al. 1994), also exhibits a loosening into α-helix for C-terminal residues when exposed to water in dynamics simulations (Kovacs et al. 1995). It is tempting, then, to speculate that the α-helix may contribute a minor component to the overall conformational space allowed to FP in SDS micelles, to the extent that the FP helix becomes exposed to the aqueous media. In this regard, H/D exchange-NMR experiments for FP in SDS indicated that, after 30 min of exchange, resonances from amide-H due to Gly-5 through Leu-12 are still seen, while only the corresponding resonances from Phe-8, Leu-9 and Leu-12 are observed after 2 h of exchange (Chang et al. 1997a). These results are in agreement with the topographical model for FP in SDS (Fig. 9 ▶), which shows residues Gly-5 through Leu-12 relatively protected from H/D exchange by insertion of hydrophobic residues into the micel-lar core. Also, it is important to note that the dynamics-simulated FP-SDS model in Figure 9 ▶ proposes that Phe-8 will be buried in the hydrophobic interior of the micelle, and indeed, fluorescence spectra of an FP analog (Trp - Phe-8) with SDS demonstrate a blue-shift, in accord with this residue inserting into the micellar interior (Chang et al. 1997a).
Last, the structure assessed from dynamic simulations of FP in SDS micelles (Fig. 9 ▶) may be relevant to the corresponding structural models for FP in membranes. Earlier experiments showed an oblique intercalation of the α-helical N-terminal gp41 peptide into membrane lipids (Martin et al. 1993, 1996) with relatively deep penetration into the bilayer (Gordon et al. 1992), in agreement with recent Monte Carlo (Maddox and Longo 2002) and explicit (Kamath and Wong 2002) dynamic simulations. Additionally, Kamath and Wong (2002) reported that the amphipathic α-helix of the 16-residue FP orients its “glycine stripe” towards the polar headgroups of the bilayer and its hydrophobic side facing the bilayer core, reminiscent of the topography of the 23-residue FP structure in SDS micelles that buries the helical Phe-8 into the micellar core (Fig. 9 ▶). Experimental support for the membrane model of Kamath and Wong (2002) comes from the FP analog (Trp - Phe-8) exhibiting the characteristic blue-shift in fluorescence experiments, for peptide incorporated into lipid bilayers (Chang et al. 1997a; Saez-Cirion and Nieva 2002). A more detailed account of the interactions between FP and SDS is outside the scope of this article, and will be presented elsewhere (Y.N. Kaznessis, in prep.).
Materials and methods
Materials
Peptide synthesis reagents, included Fmoc amino acids and coupling solvents, were obtained from Applied Biosystems. SDS was from Avanti Polar Lipids. Deuterium oxide was supplied by Aldrich Chemical Co. Deuterated HFIP and formic acid were obtained from Cambridge Isotope Laboratories. Fmoc 13C-carbonyl alanine, glycine, leucine, and phenylalanine were purchased from Cambridge Isotope Laboratories. 13C-carbonyl phenylalanine was converted to the Fmoc derivative by AnaSpec. Uranyl acetate was purchased from Ted Pella. All organic solvents used for sample synthesis, purification and preparation were HPLC grade or better.
Solid-phase peptide synthesis, purification and characterization
The 23-amino acid N-terminal sequence of gp41 (FP; Fig. 1 ▶) of the HIV-1 strain LAV1a, was prepared with either an ABI 431A peptide synthesizer or a Protein Technologies Symphony7/Multiplex SPPS synthesizer, and purified by reverse phase HPLC; FP encompasses amino acid residues 519–541 of HIV-1 gp41 (Myers et al. 1991). The following 13C-carbonyl enhanced FP analogs were similarly prepared: FPA1/G3, FPA1/G3/G5/L7, FPG5/A6, FPG5-A16, FPL7/L9/L12, FPG13-G16, and FPG20/A21 (Fig. 1 ▶). After HPLC purification, the peptides were twice freeze-dried from 0.01 M HCl to remove any residual acetate counter ions that might interfere with FTIR measurements. The expected molecular masses of FP and isotope-enhanced FP analogs were obtained by fast-atom bombardment and electrospray ionization mass spectrometry (UCLA Center for Molecular and Medical Sciences Mass Spectrometry). Quantitative amino acid compositions for the peptides were determined at the Emory University Microchemical Facility.
Rationale for 13C-site–directed FP substitutions
To probe the secondary conformations within FP, FTIR spectroscopy was conducted with site-directed, isotope-enhanced peptides. Specifically, 13C-carbonyl groups were incorporated into multiple, neighboring amino acid residues of synthetic FP peptides (Fig. 1 ▶). Separate peptides were prepared with “cassettes” of multiply 13C-enhanced substitutions that were staggered to sequentially cover the peptide (Fig. 1 ▶). In prior investigations of various peptides including FP (Tadesse et al. 1991; Gordon et al. 2000; Gordon et al. 2002), cassettes of similarly 13C-enhanced peptides permitted local domain mapping of various secondary conformations (e.g., α-helix, β-sheet, β-turn, and random). The primary rationale behind these experiments is that the secondary structure within a peptide (or protein) usually extends over more than several adjacent residues.
FTIR spectroscopy
Infrared spectra were recorded at 25°C using either a Mattson Research Series FTIR spectrometer (Drew University) or Bruker Vector 22J FTIR spectrometers (California State Polytechnic University, Pomona, and Harbor-UCLA REI) equipped with DTGS detectors, averaged over 256 scans at a gain of 4 and a resolution of 2 cm−1 (Mobley et al. 1999; Gordon et al. 2000). For FTIR spectra of FP originally in solvents, peptide self-films were prepared by air drying peptide solutions in 100% HFIP onto 50 × 20 × 2 mm 45 degree ATR crystals fitted for either the Bruker (Pike Technologies) or Mattson (Spectral Solutions) spectrometers. The dried peptide self-films were then overlaid with solution containing deuterated solvents (i.e., deuterated HFIP:water:formic acid [70:30:0.1, v/v] or PBS [pH 7.4]) prior to spectral acquisition; control deuterated solvent samples were similarly prepared, but without peptide. Spectra of FP peptides in solvent were obtained by subtraction of the deuterated solvent spectrum from the peptide-deuterated solvent spectrum. For measurements with peptides in a lipid environment, peptides were suspended at 470 μM in 94 mM SDS and deuterated PBS at a peptide/lipid (P/L) ratio of 1 : 200. Spectra were recorded at 25°C on peptide that was first dried on the ATR from the SDS suspension, and then resolvated with deuterated PBS. The FP in SDS spectrum was obtained by subtracting the SDS with deuterated PBS spectrum from that of FP in SDS with deuterated PBS. P/L ratios were determined here using the earlier finding that the peptide concentration is proportional to the area (Samide) of the amide I band (1680–1600 cm−1; Martin et al. 1993), while the lipid–SDS concentration is proportional to the area (Sv[CH2]lipid) of the lipid–SDS CH2 stretching vibrations band (3100–2800 cm−1). Therefore, the peptide/detergent ratio is proportional to the following ratio: (Samide)/(Sv[CH2]lipid).
The amide I bands of conventional 12C-FTIR spectra of FP self-films and FP-SDS micellar samples were analyzed for the various secondary conformations (Gordon et al. 2000, 2002). The proportions of α-helix, β-turn, β-sheet, and disordered conformations were determined by Fourier self-deconvolutions for band narrowing and area calculations of component peaks determined with curve fitting software supplied by Mattson. The frequency limits for the different structures were as follows: α-helix (1662–1645 cm-1), β-sheet (1637–1613 and 1710–1682 cm−1), β-turns (1682–1662 cm−1), and disordered or random (1650–1637 cm−1; Byler and Susi 1986; Surewicz and Mantsch 1988; Goormaghtigh et al. 1999).
Enhancement of FP peptides with site-specific 13C-carbonyl groups permits the direct determination of those amino acid residues participating in secondary conformations. Because the stretching frequencies of the peptide backbone carbonyl groups are sensitive to local conformations, replacement of 12C with 13C should reduce the stretching frequency of an isolated carbonyl oscillator by ~37 cm−1 (Dwivedi and Krimm 1984; Tadesse et al. 1991). In the absence of significant transition dipole coupling (TDC) interactions (Moore and Krimm 1976a,b; Krimm and Bandekar 1986), the α-helix band should be lowered to (1625–1608 cm−1), β-turns to (1645–1625 cm−1) and disordered or random to (1613–1600 cm−1). These spectral shifts were detected by measuring FTIR spectra of the natural abundance and 13C-enhanced peptides in various environments, and noting the positions of any new peaks in the 13C-FTIR spectrum (Tadesse et al. 1991; Ludlam et al. 1996; Gordon et al. 2000). Subtle spectral shifts were also detected with difference FTIR spectra, obtained by subtracting the natural abundance spectrum from that of the isotopically enhanced peptide (Tadesse et al. 1991; Gordon et al. 2000). The difference FTIR spectra should show a negative peak at the original position of the conformational band, and a positive peak shifted from the original by ~37 cm−1.
Both 12C- and 13C-FTIR spectroscopy have also been used to identify antiparallel beta sheets. Without 13C isotopes, TDC interactions between intermolecular antiparallel β-sheets induce a splitting of the FTIR spectra into a weak high frequency band (~1695 cm−1) and a strong low-frequency band (~1626 cm−1). However, these TDC interactions may be disrupted by introducing 13C for 12C at the carbonyl groups. For 13C labels substituted into the β-sheet, the residual 12C = O low-frequency peak is shifted to higher frequencies and a new low-frequency peak appears due to the 13C = O absorbance (Halverson et al. 1991; Ashburn et al. 1992; Baldwin 1999). With greater interactions between 13C labels in adjacent strands, the frequency of the 13C = O peak will be lower (Ashburn et al. 1992).
All spectra were presented here using Harvard ChartXL 3.0 (Serif, Inc.; http://www.harvardgraphics.com).
Transmission Electron Microscopy
HIV fusion peptide was diluted in PBS (pH 7.4), to a final concentration of 0.5 mM, and incubated at room temperature directly on formvar-coated, carbon-stabilized 200-mesh copper grids (Ted Pella) for 1, 5, and 24 h. At the end of incubation, the grids were stained with 1% aqueous uranyl acetate for 5 min. Samples were then gently washed five times with ultrapure water and allowed to dry. Control grids were treated in exactly the same manner with the exception of peptide inclusion in PBS. To enhance staining of smaller structures, some samples were stained with 2% aqueous uranyl acetate for 10 min. Samples were examined in a Zeiss 10C transmission electron microscope at 80 kV. Images were collected on Kodak SO-163 film at 25,000× to 50,000×. The electron films were digitized at 1200 dpi, giving rise to a final pixel size of 0.42–0.84 nm.
Computational methods for modeling FP in PBS
The amino-terminal peptide (FP) of HIV-1 gp41 in PBS was modeled with an antiparallel β-sheet conformation using Insight/Discover 97.0 software (Molecular Simulations) running on a Silicon Graphics Indigo-2R10000 High Impact workstation (Beckman Research Institute City of Hope core facility).
Computational methods for modeling the FP–SDS micelle system
The interactions of FP in a sodium dodecyl sulfate (SDS) micelle were explicitly simulated with CHARMM version c28b2 (Brooks et al. 1983; Kaznessis et al. 2002), using supercomputing resources at the Minnesota Supercomputing Institute and the National Computational Science Alliance.
Accession code
The coordinates for the 19 lowest energy structures of FP in the SDS micelle system (i.e., peptide/lipid (P/L) ratio of 1 : 200, for 94 mM SDS suspended in PBS [pH 7.4]), together with a full list of restraints, have been deposited in the Protein Data Bank (PDB) under the accession code 1P5A.
Electronic supplemental material
Description of the supplementary figures included in Electronic Appendix: Figure S-1, difference FTIR spectra of FP peptides in SDS, obtained by subtracting the spectrum of the native 12C-carbonyl FP from those of 13C-enhanced FP peptides; Figure S-2, FTIR spectra of the 13C-labeled FPG5-A15 peptide in the HFIP or PBS environments at 25°C, each diluted with the unlabeled, native FP peptide; Figure S-3, transmission electron microscopy (TEM) of FP in phosphate-buffered saline (PBS); Figure S-4, transmission electron microscopy (TEM) of FP in PBS.
Acknowledgments
The City of Hope Molecular Modeling Core Facility was supported by Cancer Center Support Grant P30 CA33572. This study was supported by NIH MBRS Grants GM 08140 (to L.M.G. and A.J.W.) and GM 53933 (to P.W.M. and S.E.). The ABI 431A peptide synthesizer was obtained with NIH Small Equipment Grant GM50483 (to L.M.G. and A.J.W.); the Protein Technologies Symphony/Multiplex SPPS synthesizer was acquired with a NIH NCRR Shared Instrumentation Grant 1 S10 RR14867-01A1 (to M.R. Yeaman and A.J.W.); and the REI Bruker Vector 22 FTIR spectrometer was funded by a grant from the Harbor-UCLA REI Common Use and Replacement Equipment Program (to A.J.W.). Y.N.K. thanks the Biotechnology Institute and the Digital Technology Center at the University of Minnesota for financial support and the Minnesota Supercomputing Institute and the National Computational Science Alliance (Grant MCB20026N at the NCSA Origin 2000) for supercomputing resources.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
PBS, phosphate-buffered saline
HFIP, hexafluoroiso-propanol
TFE, trifluoroethanol
SDS, sodium dodecyl sulfate
ATR, attenuated-total-reflectance
FTIR, Fourier transform infrared spectroscopy
13C-FTIR, isotopically enhanced 13C-Fourier transform infrared spectroscopy
12C-FTIR, conventional FTIR spectroscopy on native peptides
CD, circular dichroism
TEM, transmission electron microscopy
EM, electron microscopy
2D-NMR, two-dimensional nuclear magnetic resonance
SS 13C-NMR, solid-state 13C-enhanced nuclear magnetic resonance
ESR, electron spin resonance
POPG, 1-palmitoyl-2-oleoyl phosphatidylglycerol
SP-B1–25, N-terminal peptide of human surfactant protein B, residues 1–25
P/L, peptide to lipid molar ratio
LUV, large unilamellar vesicle liposomes
HPLC, high-performance liquid chromatography
Fmoc, 9-Fluorenylmethyloxy-carbonyl
[Θ]MRE, mean residue ellipticity (deg cm2/dmole−1)
TDC, transition dipole coupling
gp41, HIV-1 glycoprotein 41,000
gp120, HIV-1 glycoprotein 120,000
HA2, influenza hemagglutinin protein
IAPP, islet amyloid polypeptide
PrP, prion protein
HB, backbone amide-carbonyl H bonds
Supplemental material: See www.proteinscience.org
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03407704.
References
- Agirre, A., Flach, C., Goni, F.M., Mendelsohn, R., Valpuesta, J.M., Wu, F., and Nieva, J.L. 2000. Interactions of the HIV-1 fusion peptide with large unilamellar vesicles and monolayers. A cryo-TEM and spectroscopic study. Biochim. Biophys. Acta 1467 153–164. [DOI] [PubMed] [Google Scholar]
- Aloia, R.C., Jensen, F.C., Curtain, C.C., Mobley, P.W., and Gordon, L.M. 1988. Lipid composition and fluidity of the human immunodeficiency virus type-1. Proc. Natl. Acad. Sci. 85 900–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, H.C. 1980. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 72 2384–2393. [Google Scholar]
- Ashburn, T.T., Auger, M., and Lansbury Jr., P.T. 1992. The structural basis of pancreatic amyloid formation: Isotope-edited spectroscopy in the solid state. J. Am. Chem. Soc. 114 790–791. [Google Scholar]
- Baldwin, M.A. 1999. Stable isotope-labeled peptides in study of protein aggregation. Methods Enzymol. 309 576–591. [DOI] [PubMed] [Google Scholar]
- Bergeron, L., Sullivan, N., and Sodroski, J. 1992. Target cell-specific determinants of membrane fusion within the human immunodeficiency virus type 1 gp120 third variable region and gp41 amino terminus. J. Virol. 66 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner, J.W., Dugan, C., and Mendelsohn, R. 2000. 13C isotope labeling of hydrophobic peptides. Origin of the anomalous intensity distribution in the infrared amide I spectral region of β-sheet structures. J. Am. Chem. Soc. 122 677–683. [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics simulations. J. Comp. Chem. 4 187–217. [Google Scholar]
- Bullough, P.A., Hughson, F.M., Skehel, J.J., and Wiley, D.C. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371 37–43. [DOI] [PubMed] [Google Scholar]
- Byler, D.M. and Susi, H. 1986. Examination of the secondary structure of protein by deconvolved FTIR spectra. Biopolymers 25 469–487. [DOI] [PubMed] [Google Scholar]
- Callebaut, I., Tasso, A., Brasseur, R., Burny, A., Portetelle, D., and Mornon, J.P. 1994. Common prevalence of alanine and glycine in mobile reactive centre loops of serpins and viral fusion peptides: Do prions possess a fusion peptide? J. Comput. Aided Mol. Des. 8 175–191. [DOI] [PubMed] [Google Scholar]
- Chang, D.-K., Cheng, S.-F., and Chien, W.-J. 1997a. The amino-terminal fusion domain peptide of Human Immunodeficiency Virus Type 1 gp41 inserts into the sodium dodecyl sulfate micelle primarily as a helix with a conserved glycine at the micelle–water interface. J. Virol. 71 6593–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, D.-K., Chien, W.-J., and Cheng, S.-F. 1997b. The FLG motif in the N-terminal region of glucoprotein 41 of human immunodeficiency virus type 1 adopts a type-1 β turn in aqueous solution and serves as the initiation site for helix formation. Eur. J. Biochem. 247 896–905. [DOI] [PubMed] [Google Scholar]
- Curtain, C.C., Separovic, F., Nielsen, K., Craik, D., Zhong, Y.C., and Kirk-patrick, A. 1999. The interactions of the N-terminal fusogenic peptide of HIV-1 gp41 with neutral phospholipids. Eur. Biophys. J. 28 427–436. [DOI] [PubMed] [Google Scholar]
- Delahunty, M.D., Rhee, I., Freed, E.O., and Bonifacino, J.S. 1996. Mutational analysis of the fusion peptide of the human immunodeficiency virus type 1: Identification of critical glycine residues. Virology 218 94–102. [DOI] [PubMed] [Google Scholar]
- Dimitrov, A.S., Xiao, X., Dimitrov, D.S., and Blumenthal, R. 2001. Early intermediates in HIV-1 envelope glycoprotein-mediated fusion triggered by CD4 and co-receptor complexes. J. Biol. Chem. 276 30335–30341. [DOI] [PubMed] [Google Scholar]
- Dwivedi, A.M. and Krimm, S. 1984. Vibrational analysis of peptides, polypeptides, and proteins. XXIV. Conformation of poly(α-aminoisobutyric acid). Biopolymers 23 2025–2065. [DOI] [PubMed] [Google Scholar]
- Essman, U., Perera, L., Berkowitz, M.L., Darden, T., Lee, H., and Pedersen, L.G. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103 8577–8593. [Google Scholar]
- Fernandez, A. and Berry, R.S. 2003. Proteins with H-bond packing defects are highly interactive with lipids bilayers: Implications for amyloidogenesis. Proc. Natl. Acad. Sci. 100 2391–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodje, M.N. and Al-Karadaghi, S. 2002. Occurrence, conformational features and amino acid propensities for the α-helix. Protein Eng. 15 353–358. [DOI] [PubMed] [Google Scholar]
- Freed, E.O., Myers, D.J., and Risser, R. 1990. Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc. Natl. Acad. Sci. 87 4650–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallaher, W.R. 1987. Detection of a fusion peptide sequence in the transmembrane protein of the human immunodeficiency virus. Cell 50 327–328. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano, F., Waxham, M.N., Ross, A.M., and Hoxie, J.A. 1987. Sequence similarities between human immunodeficiency virus gp41 and paramyxovirus fusion proteins. AIDS Res. Hum. Retroviruses 3 245–252. [DOI] [PubMed] [Google Scholar]
- Goormaghtigh, E., Raussens, V., and Ruysschaert, J.-M. 1999. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta 1422 105–185. [DOI] [PubMed] [Google Scholar]
- Gordon, L.M., Curtain, C.C., Zhong, Y.C., Kirkpatrick, A., Mobley, P.W., and Waring, A.J. 1992. The amino terminal peptide of HIV-1 glycoprotein 41 interacts with human erythrocyte membranes: Peptide conformation, orientation and aggregation. Biochim. Biophys. Acta 1139 257–274. [DOI] [PubMed] [Google Scholar]
- Gordon, L.M., Horvath, S., Longo, M.L., Zasadzinski, J.A., Taeusch, W., Faull, K., Leung, C., and Waring, A.J. 1996. Conformational and molecular topography of the N-terminal segment of surfactant protein B in structure-promoting environments. Protein Sci. 5 1662–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, L.M., Lee, K.Y.C., Zasadzinski, J.A., Walther, F.J., Sherman, M.A., and Waring, A.J. 2000. Conformational mapping of the N-terminal segment of surfactant protein B in lipid using 13C-enhanced Fourier transform infrared spectroscopy. J. Peptide Res. 55 330–347. [DOI] [PubMed] [Google Scholar]
- Gordon, L.M., Mobley, P.W., Pilpa, R., Sherman, M.A., and Waring, A.J. 2002. Conformational mapping of the N-terminal peptide of HIV-1 gp41 in membrane environments using 13C-enhanced Fourier transform infrared spectroscopy. Biochim. Biophys. Acta 1559 96–120. [DOI] [PubMed] [Google Scholar]
- Gustafsson, M., Thyberg, J., Naslund, J., Eliasson, E., and Johansson, J. 1999. Amyloid fibril formation by pulmonary surfactant protein C. FEBS Lett. 464 138–142. [DOI] [PubMed] [Google Scholar]
- Halverson, K.J., Sucholeiki, I., Ashburn, T.T., and Lansbury Jr., P.T. 1991. Location of β-sheet-forming sequences in amyloid proteins by FTIR. J. Am. Chem. Soc. 113 6701–6703. [Google Scholar]
- Haris, P.I. and Chapman, D. 1995. The conformational analysis of peptides using Fourier transform IR spectroscopy. Biopolymers 37 251–263. [DOI] [PubMed] [Google Scholar]
- Henry, G.D. and Sykes, B.D. 1994. Methods to study membrane protein structure in solution. Methods Enzymol. 239 515–535. [DOI] [PubMed] [Google Scholar]
- Hoover, W.H. 1985. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31 1695–1697. [DOI] [PubMed] [Google Scholar]
- Jiang, S., Zhao, Q., and Debnath, A.K. 2002. Peptide and non-peptide HIV fusion inhibitors. Curr. Pharm. Des. 8 563–580. [DOI] [PubMed] [Google Scholar]
- Johansson, J. 2001. Membrane properties and amyloid fibril formation of lung surfactant protein C. Biochem. Soc. Trans. 29 601–606. [DOI] [PubMed] [Google Scholar]
- Johansson, J., Szyperski, T., Curstedt, T., and Wuthrich, K. 1994. The NMR structure of the pulmonary surfactant-associated polypeptide SP-C in an apolar solvent contains a valyl-rich α-helix. Biochemistry 33 6015–6023. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L., Chandrasekhar, J., Medura, J.D., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential function for simulating liquid water. J. Chem. Phys. 79 926–935. [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kamath, S. and Wong, T.C. 2002. Membrane structure of the Human Immunodeficiency Virus gp41 fusion domain by molecular dynamics simulation. Biophys. J. 83 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaznessis, Y.N., Kim, S., and Larson, R.G. 2002. Specific mode of interaction between components of model pulmonary surfactants using computer simulations. J. Mol. Biol. 322 569–582. [DOI] [PubMed] [Google Scholar]
- Kliger, Y., Aharoni, A., Rapaport, D., Jones, P., Blumenthal, R., and Shai, Y. 1997. Fusion peptides derived from the HIV type 1 glycoprotein 41 associate within phospholipid membranes and inhibit cell-cell fusion. J. Biol. Chem. 272 13496–13505. [DOI] [PubMed] [Google Scholar]
- Kovacs, H., Mark, A.E., Johansson, J., and van Gusteren, W.F. 1995. The effect of environment on the stability of an integral membrane helix: Molecular dynamics simulations of surfactant protein C in chloroform, methanol and water. J. Mol. Biol. 247 808–822. [DOI] [PubMed] [Google Scholar]
- Krimm, S. and Bandekar, J. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38 181–364. [DOI] [PubMed] [Google Scholar]
- Lorenzo, A. and Yankner, B.A. 1994. α-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc. Natl. Acad. Sci. 91 12243–12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, M., Blacklow, S.C., and Kim, P.S. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 2 1075–1082. [DOI] [PubMed] [Google Scholar]
- Ludlam, C.F.C., Arkin, I.T., Liu, X.-M., Rothman, M.S., Rath, P., Aimoto, S., Smith, S.O., Engelman, D.M., and Rothschild, K.J. 1996. Fourier transform infrared spectroscopy and site-directed isotope labeling as a probe of local secondary structure in the transmembrane domain of phospholamban. Biophys. J. 70 1728–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, A. 1995. Molecular dynamics simulation analysis of a sodium do-decyl sulfate micelle in aqueous solution: Decreased fluidity of the micelle hydrophobic interior. J. Phys. Chem. 99 1846–1855. [Google Scholar]
- Maddox, M.W. and Longo, M.L. 2002. Conformational partitioning of the fusion peptide of HIV-1 gp41 and its structural analogs in bilayer membranes. Biophys. J. 83 3088–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, I., Defrise-Quertain, F., Decroly, E., Vandenbranden, M., Brasseur, R., and Ruysschaert, J.-M. 1993. Orientation and structure of the NH2-terminal HIV-1 gp41 peptide in fused and aggregated liposomes. Biochim. Biophys. Acta 1145 124–133. [DOI] [PubMed] [Google Scholar]
- Martin, I., Schaal, H., Scheid, A., and Ruysschaert, J.-M. 1996. Lipid membrane fusion induced by the Human Immunodeficiency Virus Type 1 gp41 N-terminal extremity is determined by its orientation in the lipid bilayer. J. Virol. 70 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune, J.M., Rabin, L.B., Feinberg, M.B., Lieberman, M., Kosek, J.C., Reyes, G.R., and Weisman, I.L. 1988. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 53 55–67. [DOI] [PubMed] [Google Scholar]
- Mobley, P.W., Curtain, C.C., Kirkpatrick, A., Rostamkhani, M., Waring, A.J., and Gordon, L.M. 1992. The amino-terminal peptide of HIV-1 glycoprotein 41 lyses human erythrocytes and CD4+ lymphocytes. Biochim. Biophys. Acta 1139 251–256. [DOI] [PubMed] [Google Scholar]
- Mobley, P.W., Lee, H.-F., Curtain, C.C., Kirkpatrick, A., Waring, A.J., and Gordon, L.M. 1995. The amino-terminal peptide of HIV-1 glycoprotein 41 fuses human erythrocytes. Biochim. Biophys. Acta 1271 304–315. [DOI] [PubMed] [Google Scholar]
- Mobley, P.W., Waring, A.J., Sherman, M.A., and Gordon, L.M. 1999. Membrane interactions of the synthetic N-terminal peptide of HIV-1 gp41 and its structural analogs. Biochim. Biophys. Acta 1418 1–18. [DOI] [PubMed] [Google Scholar]
- Mobley, P.W., Pilpa, R., Brown, C., Waring, A.J., and Gordon, L.M. 2001. Membrane-perturbing domains of HIV Type 1 glycoprotein 41. AIDS Res. Hum. Retroviruses 17 311–327. [DOI] [PubMed] [Google Scholar]
- Moore, W.H. and Krimm, S. 1976a. Vibrational analysis of peptides, polypep-tides, and proteins. I. Polyglycine I. Biopolymers 15 2439–2464. [DOI] [PubMed] [Google Scholar]
- ———. 1976b. Vibrational analysis of peptides, polypeptides, and proteins. II. β-poly(L-alanine) and β-poly(L-alanylglycine). Biopolymers 15 2465–2483. [DOI] [PubMed] [Google Scholar]
- Myers, G., Korber, B., Berzofsky, J.A., Smith, R.F., and Pavalakis, G.N. 1991. Human retroviruses and AIDS 1991: A compilation and analysis of nucleic acid and amino acid sequences, p. II-81. Los Alamos National Laboratory, Los Alamos, NM.
- Nieva, J.L., Nir, S., Muga, A., Goni, F.M., and Wilschut, J. 1994. Interaction of the HIV-1 fusion peptide with phospholipid vesicles: Different structural requirements for fusion and leakage. Biochemistry 33 3201–3209. [DOI] [PubMed] [Google Scholar]
- Nieva, J.L., Goni, F.M., Mason, A.L., Mock, A.R., Muga, A., Saez, A., and Gallaher, W.R. 2000. Similarities of prion protein to fusion/entry proteins of HIV and ebola: Membrane interactions of its putative fusion domain. Bio-phys. J. 78 412A. [Google Scholar]
- Pereira, F.B., Goni, F.M., and Nieva, J.L. 1995. Liposome destabilization induced by the HIV-1 fusion peptide: Effect of a single amino acid substitution. FEBS Lett. 362 243–246. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Permeabilization and fusion of uncharged lipid vesicles induced by the HIV-1 fusion peptide adopting an extended conformation: Dose and sequence effects. Biophys. J. 73 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillot, T., Lins, L., Goethals, M., Vanloo, B., Baert, J., Vandekerckhove, J., Rosseneu, M., and Brasseur, R. 1997. The 118–135 peptide of the human prion protein forms amyloid fibrils and induces liposome fusion. J. Mol. Biol. 274 381–393. [DOI] [PubMed] [Google Scholar]
- Rafalski, M., Lear, J.D., and DeGrado, W.F. 1990. Phospholipid interactions of synthetic peptides representing the N-terminus of HIV gp41. Biochemistry 29 7917–7922. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J.P., Ciccotti, G., and Berendesen, H.J.C. 1977. Numerical integration of the cartesian equations of motion for a system with constraints: Molecular dynamics of n-alkanes. J. Comp. Phys. 23 327–341. [Google Scholar]
- Saez-Cirion, A. and Nieva, J.L. 2002. Conformational transitions of membrane-bound HIV-1 fusion peptide. Biochim. Biophys. Acta 1564 57–65. [DOI] [PubMed] [Google Scholar]
- Schaal, H., Klein, M., Gehrmann, P., Adams, O., and Scheid, A. 1995. Requirement of N-terminal amino acid residues of gp41 for human immunodefi-ciency virus type 1-mediated cell fusion. J. Virol. 69 3308–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepushkin, V.A., Melikyan, G.B., Sidorova, M.V., Chumakov, V.M., Andreev, S.M., Manukyan, R.A., and Karamov, E.V. 1990. Interaction of human immunodeficiency virus (HIV-1) fusion peptide with artificial lipid membranes. Biochem. Biophys. Res. Commun. 172 952–957. [DOI] [PubMed] [Google Scholar]
- Slepushkin, V.A., Andreev, S.M., Sidorova, M.V., Melikyan, G.B., Grigoriev, V.B., Chumakov, V.M., Grinfeldt, A.E., Manukyan, R.A., and Karamov, E.V. 1992. Investigation of human immunodeficiency virus fusion peptides. Analysis of interrelations between their structure and function. AIDS Res. Hum. Retroviruses 8 9–18. [DOI] [PubMed] [Google Scholar]
- Surewicz, W.K. and Mantsch, H.H. 1988. New insight into protein secondary structure from resolution-enhanced infrared spectra. Biochim. Biophys. Acta 952 115–130. [DOI] [PubMed] [Google Scholar]
- Tadesse, L., Nazarbaghi, R., and Walters, L. 1991. Isotopically-enhanced infrared spectroscopy: A novel method for examining secondary structure at specific sites in conformationally heterogeneous peptides. J. Am. Chem. Soc. 113 7036–7037. [Google Scholar]
- Tamm, L.K. and Han, X. 2000. Viral fusion peptides: A tool set to disrupt and connect biological membranes. Biosci. Rep. 20 501–518. [DOI] [PubMed] [Google Scholar]
- Tenidis, K., Waldner, M., Bernhagen, J., Fischle, W., Bergmann, M., Weber, M., Merkle, M.-L., Voelter, W., Brunner, H., and Kapurniotu, A. 2000. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 295 1055–1071. [DOI] [PubMed] [Google Scholar]
- Vidal, P., Chaloin, L., Heitz, A., Van Mau, N., Mery, J., Divita, G., and Heitz, F. 1998. Interactions of primary amphipathic vector peptides with membranes. Conformational consequences and influence on cellular localization. J. Membr. Biol. 162 259–264. [DOI] [PubMed] [Google Scholar]
- Weaver, T.M. 2000. The α-helix translates structure into function. Protein Sci. 9 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn, W., Dessen, A., Harrison, S.C., Skehel, J.J., and Wiley, D.C. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387 426–430. [DOI] [PubMed] [Google Scholar]
- Yang, J., Gabrys, C.M., and Weliky, D.P. 2001. Solid-state nuclear magnetic resonance evidence for an extended β strand conformation of the membrane-bound HIV-1 fusion peptide. Biochemistry 40 8126–8137. [DOI] [PubMed] [Google Scholar]