

Abstract
The deletion mutant Δ(7-22) of α-sarcin, unlike its wild-type protein counterpart, lacks the specific ability to degrade rRNA in intact ribosomes and exhibits an increased unspecific ribonuclease activity and decreased interaction with lipid vesicles. In trying to shed light on these differences, we report here on the three-dimensional structure of the Δ(7-22) α-sarcin mutant using NMR methods. We also evaluated its dynamic properties on the basis of theoretical models and measured its correlation time (6.2 nsec) by time-resolved fluorescence anisotropy. The global fold characteristic of ribotoxins is preserved in the mutant. The most significant differences with respect to the α-sarcin structure are concentrated in (1) loop 2, (2) loop 3, which adopts a new orientation, and (3) loop 5, which shows multiple conformations and an altered dynamics. The interactions between loop 5 and the N-terminal hairpin are lost in the mutant, producing increased solvent accessibility of the active-site residues. The degree of solvent exposure of the catalytic His 137 is similar to that shown by His 92 in RNase T1. Additionally, the calculated order parameters of residues belonging to loop 5 in the mutant correspond to an internal dynamic behavior more similar to RNase T1 than α-sarcin. On the other hand, changes in the relative orientation of loop 3 move the lysine-rich region 111–114, crucial for substrate recognition, away from the active site. All of the structural and dynamic data presented here reveal that the mutant is a hybrid of ribotoxins and noncytotoxic ribonucleases, consistent with its biological properties.
Keywords: α-sarcin mutant, ribotoxins, NMR solution structure, ribonucleolytic activity, protein-RNA interaction
α-sarcin is the most well-characterized member of the fungal ribotoxin family. This extracellular 150-residue enzyme, secreted by the mold Aspergillus giganteus (Olson and Goerner 1965), has been extensively studied from a structural (Pérez-Cañadillas et al. 2000), dynamic (Pérez-Cañadillas et al. 2002), and electrostatic point of view (Pérez-Cañadillas et al. 1998) by NMR methods. α-sarcin associates with the cell membrane by electrostatic and hydrophobic interactions and is drawn into the cell via endocytosis without the help of known membrane receptors (Olmo et al. 2001). α-sarcin is a very specific ribonuclease that recognizes the ribosome and cleaves a single phosphodiester bond on the 3′ side of G4325 in the 28S rRNA. This bond is located in a highly conserved purine-rich 14-base sequence, known as the SRL (Endo and Wool 1982; Wool 1984; Glück and Wool 1996), targeted by ribosome-inactivating proteins. A bulged guanine, G4319, has been shown to be a crucial element for α-sarcin action (Glück et al. 1994; Glück and Wool 1996). The cleavage results in the release of a 400-bp fragment, known as the α-fragment (Endo and Wool 1982), and in the inactivation of the ribosome and inhibition of protein biosynthesis, leading to cell death via apoptosis. The ability of α-sarcin to enter cells, specifically recognize the ribosome, cleave the phosphodiester bond, and abort protein biosynthesis by the abolishment of the translational machinery is responsible for its potent cytotoxicity.
The family of ribotoxins is structurally similar to the family of smaller, noncytotoxic ribonucleases, represented by RNase T1 and RNase U2. These families share a common structural core composed of an α-helix packed nearly orthogonally on the back of a central β-sheet, with a conserved active site on the opposite concave face of the sheet. Structural differences between these two groups of proteins, ribotoxins and noncytotoxic RNases, include the length of the N-terminal β-hairpin and the length of the external loops that connect regular secondary structure elements. Despite their structural similarity, their biological properties differ considerably, in particular in terms of the specificity and cytotoxicity. The identification of the structural segments or motifs contributing to the different biological behaviors of these two families of ribonucleases is of great interest. as it would open the door to engineering proteins with desired properties. Inserting the targeting motifs of ribotoxins into other enzyme structures would allow the generation of specific targeted enzymes. In this sense, some groups have formed chimeric constructions, such as the insertion into the RNase T1 sequence of an α-sarcin segment known to be essential in the ribosome recognition process (Chen and Lin 2002); in addition, several deletion mutants of restrictocin (Nayak et al. 2000), α-sarcin (Hwu et al. 2000), and mitogillin (Kao and Davies 1999) were prepared to identify regions in fungal ribotoxins contributing to ribosome targeting and modulation of the catalytic activity. It was recently shown that the deletion of part of the long β-hairpin in α-sarcin produces an enzyme with interesting biological properties (García-Ortega et al. 2002): namely, the Δ(7-22) mutant of α-sarcin lacks the specific ability of the wild type to degrade rRNA in intact ribosomes and exhibits increased nonspecific ribonuclease activity and decreased interaction with lipid vesicles. This deletion transforms the cytotoxic α-sarcin into a noncytotoxic, but active, ribonuclease more closely related to the noncytotoxic microbial ribonuclease family. To shed light on the structural and molecular bases of the differences in biological activities between this deletion mutant and those of the wild type, and their implications in the distinctive properties of ribotoxins and noncytotoxic ribonucleases, we determined the 3D structure of the Δ(7-22) α-sarcin mutant using NMR methods. The NMR structural study was complemented by theoretical calculations, and the experimental determination of the global rotational correlation time of the mutant by time-resolved fluorescence spectroscopy.
Results
Assignment of the Δ(7-22) α-sarcin mutant spectra
NOESY spectra of the deletion mutant were assigned by the standard NOE-based sequential assignment methodology and by comparison with the previously assigned NOESY spectra of α-sarcin. The observed chemical shift differences for α and amide protons calculated as δmutant−δsarcin versus the amino acid sequence were recently reported (García-Ortega et al. 2002). This data analysis led us to conclude that the global fold and 3D structure of the wild-type protein are preserved in the mutant, and that only small local conformational changes occur, especially in the environment of residues spatially related to the deleted region.
Structure calculation of the α-sarcin Δ(7-22) mutant
The solution structure of Δ(7-22) was determined as described in Materials and Methods. Table 1 shows the number and classification of restraints used in the calculations. A total of 1990 distance restraints, an average of 14.6 restraints per residue, were extracted from the unambiguously assigned NOEs from the NOESY spectra recorded in D2O and H2O. Among the stereospecific assignments, 19 correspond to pairs of ββ′ protons (Cys 6, Tyr 25, Ser 34, His 35, His 36, Phe 52, Pro 68, Asp 75, Asp 77, Pro 80, His 82, Tyr 93, Phe 97, Pro 101, His 104, Phe 108, Tyr 126, Phe 131, Cys148); one to a pair of Qγ methyl groups (Val 122); two to a pair of δδ′ protons (Pro 101, Asn 54); and two to a pair of γγ′ protons (Glu 96, Ile 135) (α-sarcin numberings are used throughout the present paper).
Table 1.
NMR structural calculations summary
| Restraints used in the structure calculation | |
| Distance restraints | 1990 |
| φ angle restraints | 98 |
| Type of distance restraints | |
| Intraresidual (|i − j = 0|) | 452 |
| Sequential (|i − j = 1|) | 394 |
| Medium-range (2 ≤ |i − j| ≤ 5) | 273 |
| Long-range (|i − j| > 5) | 871 |
| Violation statistics | Mean | Minimum | Maximum |
| Target function | 2.32 | 1.82 | 2.82 |
| Maximum violation (Å) | 0.27 | 0.18 | 0.55 |
| Sum of violations (Å) | 13.4 | 11.8 | 14.9 |
| Energy | Mean (Kcal/mole) | Range (Kcal/mole) |
| Total | −725 | −1563 −285 |
| van der Waals | −558 | −818 −267 |
| Electrostatic | −10005 | −10784 −9698 |
| NOEs | 13 | 8 24 |
| PROCHEK analysis | |
| Favorable | 63% |
| Additional | 34% |
| Generous | 3% |
| Nonfavorable | 0% |
The final DYANA calculation produced 200 conformers from which the 25 conformers with the lowest target functions, ranging from 1.82 to 2.82, were selected as the most representative of the 3D structure in solution of Δ(7-22). These conformers were introduced in the AMBER 7 force field to perform energy minimization. The resulting structure is shown in Figure 1 ▶. The RMSD from the ideal bond lengths is 0.009 Å and from ideal bond angles is 2.5°.
Figure 1.

Representation of the solution structure of the Δ(7-22) α-sarcin mutant. (Left) Superposition of the 25 solution structures. (Right) Ribbon diagram of the Δ(7-22) α-sarcin mutant structure. Figs. 1–3 ▶ were produced with the program MOLMOL (Koradi et al. 1996).
The structure precision, as reflected by the RMSD values of atomic positions in the ensemble of 20 structures, is 0.88 ± 0.16 Å for the backbone atoms, and 1.42 ± 0.18 Å for all heavy atoms. The average pairwise RMSD values corresponding to the different structural segments are shown and compared to those of wild-type (WT) α-sarcin in Table 2 (comparative studies are carried out with the lowest 20 energy conformers). The precision of the overall structure is good, and the low values for the secondary regular structural elements indicate that they are very well defined. Moreover, most of the loops present in the molecule also show a high degree of definition. However, loop 5 and the shorter N-terminal β-hairpin of the mutant (residues 1–27, α-sarcin numbering) show a variety of different orientations relative to the structural core (Fig. 1 ▶). Their conformational variability in the mutant can be explained by the lack of the crucial long-range NOEs that were responsible for fixing their relative orientation in the wild-type protein.
Table 2.
Averaged pairwise RMSD values (Å) of wild-type α-sarcin and Δ(7-22) mutant
| α-sarcin Δ(7-22) | WT α-sarcina | |||
| Backbone | Heavy | Backbone | Heavy | |
| All residues | 0.88 ± 0.16 | 1.42 ± 0.18 | 0.86 ± 0.18 | 1.45 ± 0.19 |
| Residues 2° structure | 0.57 ± 0.20 | 1.12 ± 0.21 | 0.36 ± 0.06 | 0.85 ± 0.13 |
| Residues 1-27 | 0.73 ± 0.21 | 0.98 ± 0.18 | 0.60 ± 0.18 | 1.11 ± 0.19 |
| Loop 1, residues 38–49 | 0.29 ± 0.12 | 0.71 ± 0.18 | 0.19 ± 0.07 | 0.66 ± 0.17 |
| Loop 2, residues 53–93 | 0.69 ± 0.17 | 1.50 ± 0.29 | 1.06 ± 0.35 | 1.97 ± 0.41 |
| Loop 3, residues 98–119 | 0.50 ± 0.12 | 1.27 ± 0.27 | 0.28 ± 0.08 | 1.04 ± 0.16 |
| Loop 4, residues 125–132 | 0.12 ± 0.04 | 0.45 ± 0.12 | 0.15 ± 0.05 | 0.55 ± 0.14 |
| Loop 5, residues 139–143 | 0.33 ± 0.35 | 1.41 ± 0.61 | 0.43 ± 0.34 | 1.34 ± 0.30 |
a PDB: 1DE3.
Description of the structure
The 3D structure in solution of Δ(7-22) shows a preserved fold compared to the wild-type α-sarcin, as was previously deduced from a comparison of UV, CD, and fluorescence spectra (García-Ortega et al. 2002). The global fold consists of an α-helix (Glu 27-His 36) packed against a five-stranded β-sheet composed by strands β3 (His 50-Trp 51), β4 (Leu 95-Phe 97), β5 (Arg 121-Tyr 124), β6 (Gly 133-Ala 136), and β7 (Lys 146-Leu 147). The N-terminal end of the protein forms a short β-hairpin composed by strands β1 (Thr 3-Thr 5) and β2 (Leu 24-Asn 26) connected by a turn (Cys-Gly-Gly-Leu), similar to that found in ribonucleases T1 and U2. The rest of the residues are located in loops of different lengths connecting the regular secondary structural elements: loop 1 (Ala 37-Pro 49) connects the α-helix with the strand β3, loop 2 (Phe 52-Leu 94) links the strands β3 and β4, loop 3 (Pro 98-Ala 120) links the strands β4 and β5, loop 4 (Thr 125-Cys 132) links the strands β5 and β6, and loop 5 (His 137-Leu 145) connects strands β6 and β7. The spatial conformation of the loops is maintained by a network of interactions (hydrogen bonds, salt bridges, π-cation interactions, hydrophobic packing) as previously described for the native protein (Pérez-Cañadillas et al. 2000). The only remarkable exception is found in loop 5, whose orientation with respect to the whole molecule is poorly defined, and a larger conformational heterogeneity is observed as a consequence of the deletion that removes important interactions with residues in the native β-hairpin motif. Among the interactions lost are the hydrogen bond between the backbone oxygen of the catalytic His 137 and the side chain of Asn 8; the hydrogen bond established between the backbone atoms of Glu 140 and Asp 9; the salt bridges between the side chains of Glu 140-Lys 11 and Lys 139-Asp 9; and the π-cation interaction between the aromatic ring of Tyr 18 and the positive side chain of Lys 139. Two new backbone hydrogen bonds were detected in the structure of the mutant that help to maintain the internal conformation of loop 4, although the main basic patterns of interactions are essentially identical.
Structural comparison of wild type and α-sarcin Δ(7-22) mutant
A detailed comparison of the best superposition of conformers describing the structures of α-sarcin and the Δ(7-22) variant in solution was performed to identify the structural regions that undergo changes as a result of the deletion (Fig. 2 ▶). The RMSD values calculated for different regions of the mean structures of each enzyme are shown in Table 3, and the superposition of several regions corresponding to both structures are illustrated in Figure 2 ▶. The RMSD value for the best superposition of residues 1–6 and 23–150 in the two mean structures is 1.16 Å for backbone atoms and 1.67 Å for heavy atoms, and the larger values are observed in residues belonging to loops 2, 3, and 5. Most residues in these loops have local and global RMSD values above the corresponding mean value. When local and global RMSD values are depicted on a residue basis, two groups of residues belonging to loop 2 with RMSD values higher than the mean are identified: Gly 58-Phe 71 and Pro 80-Tyr 93. Residues with large RMSD values in loop 3 are in the segment Asp 109-Pro 117, which includes the lysine-rich region (Lys 111, Lys 112, Lys 114) known to be involved in the base recognition process of the bulged guanine 4319 in the ribosome, and residues from His 137 to Lys 146 in loop 5. These loop 3 and loop 5 regions show low local RMSD values but large global RMSD values. This indicates a large degree of similarity in the definition of their backbones but different overall orientations with respect to the rest of the protein (Fig. 2 ▶). A reduction of two residues in the length of the C-terminal end of strand β6 and the N-terminal end of strand β7 is observed compared to the native protein. This shortening could be associated with the increased flexibility of loop 5.
Figure 2.
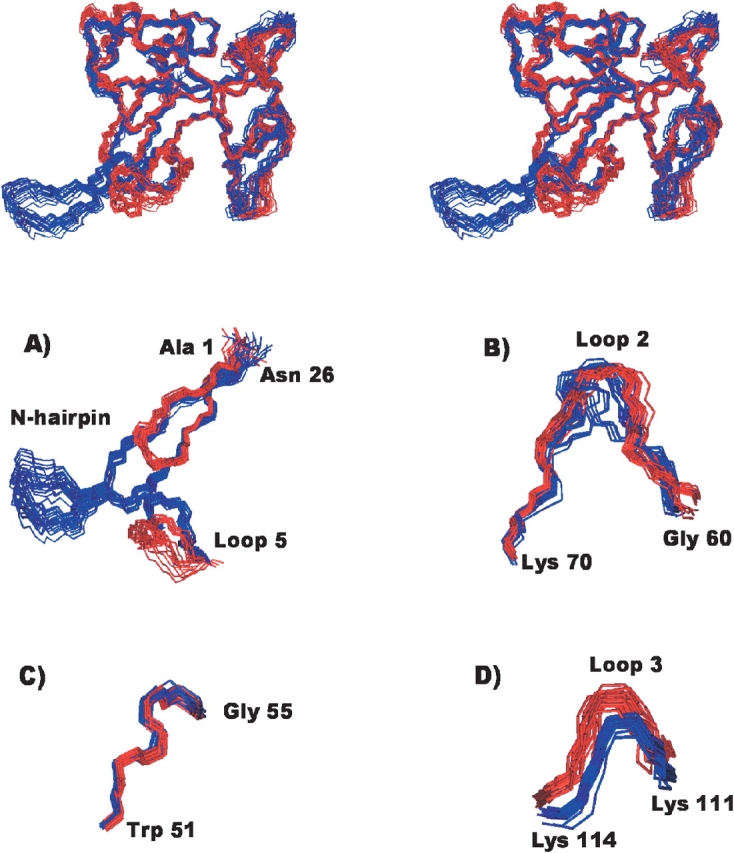
Structural comparison of the wild-type α-sarcin and the Δ(7-22) α-sarcin deletion mutant. (Top) Stereoscopic view of the superposition of the two ensembles of their 3D NMR structures; the 20 conformers of the wild type α-sarcin are in blue, and those of the Δ(7-22) mutant, in red. (Bottom) Detail of the superposition of the backbone atoms corresponding to some regions of the wild-type α-sarcin (blue) and the Δ(7-22) α-sarcin deletion mutant (red) structures. (A) Relative orientation of the N-terminal β-hairpin and loop 5, (B) segment of loop 2, (C) residues 51–55, and (D) lysine-rich fragment of loop 3.
Table 3.
Average pairwise RMSD (Å) of the best superimposition of the mean structures of the Δ(7-22) variant and the wild type α-sarcin
| Residues 1–6, 23–150 | Residues 1–6, 23–26 | Secondary structure | Loop 1 38–49 | Loop 2 53–93 | Loop 3 98–119 | Loop 4 125–132 | Loop 5 139–143 | |
| Backbone RMSD | 1.16 | 0.54 | 0.59 | 0.54 | 0.84 | 0.70 | 0.49 | 0.80 |
| Heavy RMSD | 1.67 | 1.20 | 1.07 | 0.97 | 1.53 | 1.12 | 1.14 | 1.87 |
PDB: 1DE3.
As loop 5 and the lysine-rich region of loop 3 are thought to participate in interactions with the rRNA, their altered orientations in the mutant would necessarily affect the enzyme biological activity. In particular, the lysine-rich fragment of loop 3 (that recognizes the bulged guanine in the ribosome) is farther away from the active-site residues (His 137 and Glu 96) in the mutant than in α-sarcin: the distances from the side-chain nitrogen atoms of Lys 111, Lys 112, and Lys 114 to the oxygen atoms of Glu 96 in Δ(7-22) are 20.5, 22.8, and 14.3 Å respectively, and 19.9, 21.9, and 12.7 Å in α-sarcin; distances from these Lys to the ɛ nitrogen of the imidazole ring of His 137 are 25.2, 25.3, and 16.1 Å in Δ(7-22), and 21.2, 22.8, and 13.3 Å in α-sarcin.
Active site of the α-sarcin Δ(7-22) mutant
Δ(7-22) lacks the wild-type α-sarcin ability to selectively cleave rRNA in intact ribosomes, but it does show increased nonspecific RNase activity (García-Ortega et al. 2002). A detailed comparative analysis at atomic level of the active sites of the wild-type α-sarcin and the deletion mutant Δ(7-22) is key to understanding their functional properties (Fig. 3 ▶). α-sarcin residues participating in the stabilization of the transition intermediate in the hydrolysis reaction of the phosphodiester bond, Arg 121, His 50 and probably Tyr 48, as well as the catalytic residue Glu 96, have very well-defined side chains in the mutant structure. The side chains of His 137 and Leu 145, found in loop 5, show a wide variety of orientations in Δ(7-22) that probably reflects an increased internal flexibility related to the loss of interactions with the deleted fragment of the N-terminal β-hairpin. A direct consequence of this higher mobility of loop 5 is an important increase in the degree of solvent exposure of these two residues. The solvent accessibility of Leu 145 increases from 13% to 20%, and that of the catalytic His 137 increases even more, rising from 7% to 22%. These observations indicate that the active site of the mutant is more accessible and more flexible than that of α-sarcin. The increased degree of solvent exposure and intrinsic mobility of the active site and the rest of the residues belonging to loop 5 would necessarily affect the mutant active site and most probably will be at the origin of its different biological activity. In this sense, calculations of the intrinsic mobility of the active-site residues might supply useful data yielding new important insights.
Figure 3.

Superposition of the active-site residues in 20 structures. (Left) Δ(7-22) α-sarcin deletion mutant. (Right) Wild-type α-sarcin (α-sarcin numbering). Backbone atoms are depicted in gray, and side chain atoms, in black.
Theoretical dynamic calculations
The backbone internal dynamics of α-sarcin (Pérez-Cañadillas et al. 2002) and RNase T1 (Fushman et al. 1994a) have been studied by heteronuclear 15N NMR spectroscopy. Here, we carried out theoretical calculations of the S2-order parameter, which describes the spatial restriction of the N-H vector motions.
In order to test the reliability of the contact model approximation for the calculation of S2-order parameters, S2-values were calculated in α-sarcin and then compared with those experimentally determined by NMR, as shown in Figure 4 ▶. The agreement between the calculated and experimental values is reasonably good. The model is able to qualitatively reproduce the global profile, and account correctly for the low S2-values of several highly disordered residues: Asn 8, Lys 14, Thr 15, Ala 37, Ser 40, Thr 44, Lys 61, Leu 62, Lys 64, Gly 65, Arg 66, Thr 67, Lys 73, Lys 114, Glu 144, and Leu 145, while it fails to identify the mobile Ala 30, Arg 78, and Ile 134, and the less flexible Thr 90 and Asp 102 residues. Despite these failings in individual residues, the model is reliable for identifying segments of the protein with high or low flexibility.
Figure 4.

Theoretical and experimental dynamic NH order parameters (S2) as a function of the α-sarcin sequence. Theoretical values were obtained with the application of the model described by Zhang and Brüschweiler (2002), and experimental values are taken from Pérez-Cañadillas et al. (2002).
A comparison of the theoretical S2 parameters for wild-type α-sarcin and Δ(7-22) is shown in Figure 5 ▶. As expected, most of the S2 difference values fall around zero, whereas residues with higher mobility in the native protein have positive values, and residues with higher mobility in the mutant have negative values. Interestingly, apart from sporadic differences in some individual residues (Val 2, Arg 66, Thr 67, Gly 86, Thr 90, and Glu 115), a higher flexibility is detected in the sequence of residues from Thr 138 to Glu 144, belonging to loop 5, and for some residues of the N-terminal β-hairpin flanking the deletion in the Δ(7-22) mutant when compared with the wild type.
Figure 5.

Comparison of the theoretical dynamic NH order parameters (S2) as a function of the sequence for the wild type and the Δ(7-22) α-sarcin mutant (α-sarcin numbering).
The global correlation time of the mutant was theoretically calculated using the program HYDRONMR. The input parameters were fixed to the experimental conditions used in its determination in the native protein and optimized to reproduce the experimental value (Pérez-Cañadillas et al. 2002) of 7.5 nsec obtained by NMR using the model-free formalism (Lipari and Szabo 1982). The calculation produced a value of 7.6 nsec for wild-type α-sarcin and 6.3 nsec for the mutant Δ(7-22), in agreement with the smaller size of Δ(7-22) and its more spherical shape caused by the absence of the deleted fragment in the N-terminal β-hairpin of the protein.
Rotational dynamics of the α-sarcin Δ(7-22) mutant, from tryptophan anisotropy fluorescence
The two most important experimental methods for studying the dynamics of biomolecules are those based on NMR relaxation and fluorescence anisotropy. Both methods have been applied to determine the global correlation time of a number of proteins and the results have been extensively compared (Damberg et al. 2002). Two of the major drawbacks of NMR methods are the need for labeled samples (13C and/or 15N) and the lengthy spectral assignment and data analysis processes. Thus, to obtain information about the global rotational dynamics of Δ(7-22), we used the fluorescence anisotropy methodology. This technique gives us a quicker answer, and it is more sensitive. However, the number of molecular sites available for investigation is limited to fluorophore groups. The recovered fluorescence lifetimes and anisotropy parameters of α-sarcin Δ(7-22) mutant are listed in Table 4. The total fluorescence, Im(t), was best fitted assuming a three-exponential-decay model, giving a mean lifetime of τm = 1.3 nsec (see Table 4). A long-lived component of ~3 nsec and two short-lived components of ~0.3 nsec and 1 nsec were observed. α-sarcin and its Δ(7-22) variant contain two tryptophan residues at positions 4 and 51. The three-exponential-decay represents a simplified description of the fluorescence decay of the two tryptophan residues of the α-sarcin Δ(7-22) mutant. The association of each lifetime with a specific Trp residue is not trivial in a multi-tryptophan protein. Although the shape of the emission spectrum of the α-sarcin Δ(7-22) mutant remains essentially unchanged upon (7-22) deletion, we observed changes in the lifetime distribution of both proteins, especially for the short-lived components (data not shown), which would indicate some local conformational changes in the environment of the tryptophan residues of the α-sarcin Δ(7-22) mutant with respect to the wild-type. Previous fluorescence steady-state studies of the two single tryptophan mutants of α-sarcin WT, the W4F and W51F mutants (de Antonio et al. 2000), showed that in WT the Trp-4 is the responsible for most of the tryptophan fluorescence of α-sarcin.
Table 4.
Time-resolved fluorescence and anisotropy parameters from tryptophan emissionaof the Δ(7-22) α-sarcin variantb
| Fluorescence decay parametersc | Hydrodynamic parameters | ||||||||
| α1 | τ1 (ns) | α2 | τ2 (ns) | α3 | τ3 (ns) | τmd (ns) | r(0)e | φGf (ns) | RHg (Å) |
| 0.82 (±0.01) | 0.3 (±0.1) | 0.13 (±0.01) | 1.07 (±0.05) | 0.05 (±0.01) | 3.06 (±0.05) | 1.3 | 0.22 (±0.005) | 6.2 (±0.7) | 20.6 (±0.8) |
a λexc = 297 nm; λem = 345 nm (8 nm band-pass) plus 325 nm cutoff filter.
b Buffer: 50 mM sodium phosphate, 0.2 M NaCl, pH = 7.0. T = 35°C. Protein concentration: 4.4 μM.
c The fluorescence lifetimes were obtained from analysis of the total fluorescence intensity decay, Im(t).
d The intensity average lifetime τm is defined as ∑αiτi2/∑αiτi.
e Zero-time limiting anisotropy.
f Global rotational correlation time.
g Hydrodynamic radius calculated from φG, assuming a rigid spherical model, by use of the Stokes-Einstein relationship: VH = kTφG/η. VH is the hydrodynamic volume, k is the Boltzmann’s constant, T is the absolute temperature, and η is the viscosity of the solution.
Numbers in parentheses represent the upper and lower limits of the recovered values (at the 67% confidence level) using rigorous error analysis as described in Beechem (1992).
The fluorescence anisotropy decay, r(t), showed a single exponential component. The global rotational correlation time, φG = 6.2 nsec, was estimated from r(t) assuming a nonassociative anisotropy decay model (Beechem et al. 1991) The tryptophan fluorescence from the α-sarcin Δ(7-22) mutant would only be depolarized by the overall global protein motion. We did not observe any internal tryptophan local motions in the psec–nsec range.
The effective hydrodynamic volume of α-sarcin Δ(7-22) mutant equals 3.7 × 104 Å3, which corresponds to a hydrated molecular radius of ~21 Å (see Table 4), calculated assuming a rigid spherical model.
Discussion
A detailed understanding of the function of macromolecules requires accurate knowledge of their three-dimensional structures and the characterization of their dynamics at atomic resolution. Comparison of the 3D structures of the noncytotoxic ribonuclease and ribotoxin families reveals strong structural similarities (Martínez-Ruiz et al. 1999). However, fungal ribotoxins have extra regions that are absent in the noncytotoxic ribonuclease structures. These extra sequences are located at the N terminus, and in the peripheral loops connecting the regular secondary structural elements. The N terminus of α-sarcin forms a β-hairpin composed of two short two-stranded β sheets connected by a hinge region exposed to the solvent, whereas only the inner β-sheet is conserved in RNases T1 and U2. These observations have led others to postulate that these additional sequences are inserted elements that contribute to the specific targeting and cytotoxicity of ribotoxins (Kao and Davies 1995). The right approach to understanding the roles of such elements in the ribotoxins’ functionality should consist of a careful study of the 3D structure of engineered proteins that lack one of these extra regions, combined with an appropriate biochemical and dynamic characterization.
The shape, size, and molecular flexibility of a biomolecule clearly determine its hydrodynamic properties and consequently its ability to diffuse in aqueous media and to interact with other molecules. α-sarcin and its Δ(7-22) variant have the ability to interact with RNA and phospholipid vesicles. However, differences in the kinetics and strength of these interactions have recently been described (García-Ortega et al. 2002). The rotational correlation time of 6.2 nsec for the α-sarcin Δ(7-22) mutant from TRFA is in good agreement with the value calculated by the program HYDRONMR. This means that the side chain of Trp 4 (the main contributor to fluorescence) that is close to the deleted region participates in the same dynamics as the backbone chain estimated by the theoretical model. The determined rotational time is shorter in Δ(7-22) than in the wild-type α-sarcin and approaches that determined for RNase T1 (Fushman et al. 1994b) by NMR methods (5.3 nsec). Apart from its different shape, the absence of the highly mobile N-terminal β-hairpin and the increased flexibility of loop 5 in the mutant could probably exert an influence on the observed differences of the global correlation time between WT and Δ(7-22) that are difficult to evaluate. It is also possible that these observed dynamical differences could affect in some way the electrostatic and hydrophobic interactions responsible for the α-sarcin -lipid or α-sarcin -RNA interactions (Gasset et al. 1989; Mancheño et al. 1995a). In the cell, these proteins translocate across membranes by endocytosis, with no specific membrane receptors as yet identified for such a process (Olmo et al. 2001), suggesting an active participation of some regions of the molecular structure. It is likely that, in the in vivo environment, the observed differences in the hydrodynamic properties between the WT and the deletion variant may have an enhanced effect that may affect the setting up of specific molecular interactions.
Apart from its dynamic properties, the protein structure is the result of a fine balance among many individual interactions. It has been reported that proteins respond to insertions or deletions with a much higher degree of local structural rearrangement than to single-substitution mutations (Shortle and Sondek 1995). Thus, structural consequences of a local modification, such as the β-hairpin deletion in α-sarcin, sometimes have related changes in regions that are distant in sequence from the one that has been deleted. On the other hand, a specific biological activity can be modulated by more than one structural unit, and frequently very slight modifications in structure can produce dramatic changes in activity (Koizumi et al. 2003). In general, all of these findings are difficult to predict on the basis of model compounds.
Structural implications of the N-terminal β-hairpin deletion
The present results clearly show that, in agreement with the known stability of this engineered protein (García-Ortega et al. 2002), the deletion of the outer part of the long β-hairpin does not break the ribotoxin structural integrity. Based on the Tm values of the thermal denaturation profiles, the mutant Δ(7-22) has been reported to have a decreased stability of 4 KJ/mole with respect to the wild-type α-sarcin. It should be noted that this is a small stability loss, taking into account all of the interactions that have been removed. Calculation of the global exposed surface to the solvent within the family of conformers shows that the desolvation energy cost per residue must be quite similar for both wild type and the mutant protein. Probably, the loss of some of these stabilizing interactions in terms of enthalpy are compensated for by an increase in entropy.
As the global fold is preserved, the different biological properties shown by the mutant arise from local structural variations or different hydrodynamic properties or both. The most significant differences between the wild type and the Δ(7-22) mutant are concentrated in loop 2, the dynamics of loop 5, and the new orientation of loop 3. A high RMSD value was observed for loop 2 when the mean structures of the α-sarcin and Δ(7-22) were compared (Fig. 2 ▶). Loop 2 is the longest loop present in the α-sarcin structure, is the most poorly defined (backbone RMSD more than 1 Å), and is also highly mobile. However, not all segments of this long loop demonstrate these properties equally. A short, rigid, and very well defined fragment of loop 2, Trp 51-Gly 55, has been proposed to specifically interact with rRNA near the scissile phosphodiester bond. No appreciable changes in orientation are observed for this fragment when the wild type and the mutant are compared (Fig. 2 ▶), indicating that the deletion of the N-terminal hairpin does not affect the orientation of residues proposed as the base recognition site of α-sarcin. The structural differences are concentrated in other parts of loop 2 for which there is no evidence of any involvement in substrate interaction. An extensive analysis of these changes does not reveal apparent clues for the modified biological properties of Δ(7-22).
Loop 5 increased flexibility: Creating a more flexible active site
Loop 5 of α-sarcin is strategically located at the entrance of the active site of the molecule, acting as a door to the active-site residues. α-sarcin’s loop 5 is the structural element that undergoes the most significant changes upon deletion of the N-terminal fragment (Leu 7-Arg 22). This is a direct consequence of the loss of structural interactions with some residues belonging to the deleted segment that were responsible for maintaining the backbone conformation of loop 5 and its spatial orientation relative to the structural core. In this respect, the loss of the hydrogen bond between the backbone of the catalytic His 137 (loop 5) and the side chain of Asn 8 is of special interest. Based on mutant studies, it was shown that the elimination of this specific interaction markedly increases the ribonuclease activity of mitogillin, a closely related ribotoxin (Kao and Davies 2000), suggesting that the loss of this H-bond increases the flexibility of loop 5 and reduces the strain at the active site, increasing its activity. The present results confirm this suggestion, reinforcing the importance of this key interaction in modulating the α-sarcin’s catalytic activity.
Changes in the orientation and flexibility of this part of the molecule produced by the loss of the native interactions with the N-terminal hairpin are likely to affect the solvent accessibility of the active-site residues, as well as the ability of substrates to reach the active site. It was recently shown that the low pKa of His 137 in the wild type, which is mainly due to desolvation (García-Mayoral et al. 2003), is a key determinant of its acting as a general acid in the catalytic mechanism. The variation in His 137 solvent accessibility in the mutant is expected to influence its electrostatic properties, leading to a relative stabilization of the imidazolium cation in the new, more solvated environment. In fact, the degree of solvent accessibility found in Δ(7-22) (22%) is similar to that determined for the catalytic His 92 from the NMR structure of RNase T1 (23%; Pfeiffer et al. 1997), and their pKas are also likely to be more similar. This could explain why the nonspecific ribonucleolytic activity of Δ(7-22) is more similar to that found in RNase T1 (see below).
Experimental 15N NMR relaxation studies have been also carried out to study the backbone dynamics of RNase T1 (Fushman et al. 1994a). That study showed that the largest amplitudes of local motion were located at residues belonging to loop 2 and loop 5. The order parameter values for these residues are considerably lower than those of the corresponding structural elements in α-sarcin. Additionally, a decreased flexibility in loop 5 of RNase T1 was detected upon binding to 2′ GMP, indicating that the mobility is restricted upon substrate binding. Although a quantitative comparison of internal motions cannot be done, it seems clear that mobility in loop 5 of Δ(7-22) is more similar to that present in RNase T1 than in wild-type α-sarcin.
Change in the loop 3 orientation
Following the change in the orientation of loop 5, the lysine-rich fragment of loop 3 is the segment most affected by the deletion. Changes in that segment are critical, because that region is involved in the recognition process of the ribosome by α-sarcin. Interestingly, studies performed on the RNase T1 variant with an inserted loop 3 corresponding to α-sarcin’s sequence concluded that although a similar structure was observed to be retained in the mutant, changes in the modified loop 3 were observed, and loop 5 also appeared slightly distorted. In particular, loop 3 was displaced towards the surface of the molecule (Chen and Lin 2002). According to our results, it seems also that loop 3 and loop 5 are closely related and that modifications in one of them are somehow propagated to the other. In the RNase T1 variant, the loop 5 distortion was explained in terms of steric hindrance effects due to the inserted segment in loop 3. In α-sarcin, this explanation is not reasonable, because residues from the lysine-rich region of loop 3 are far away from residues in loop 5. The change in the backbone orientation of loop 3 could be related to changes affecting its first residues, as Pro 101 interacts with hydrophobic residues at the open end of the N-terminal β-hairpin (Leu 23 and Tyr 25; Pérez-Cañadillas et al. 2000) in the native structure. These residues are close to the mutated region and are structurally affected by the deletion, which makes it impossible for them to maintain the same interactions with residues in loop 3. Additionally, a change in long-range electrostatic interactions may also contribute to the structural changes in loop 3.
Structural basis of the different ribonucleolytic activity of the α-sarcin Δ(7-22) mutant: Specific and unspecific substrates
Different docking studies performed on models of α-sarcin -RNA complexes (Szewczak et al. 1993; Szewczak and Moore 1995; Pérez-Cañadillas et al. 2000; Hwu et al. 2000), restrictocin-RNA complexes (Yang and Moffat 1996; Nayak et al. 2001; Yang et al. 2001), and kinetic studies on α-sarcin (Takeda et al. 1997) have suggested the participation of two distant regions in its specific interaction with the ribosome. First, the lysine-rich region of α-sarcin’s loop 3 participates in the substrate recognition by interacting with the negatively charged phosphodiester bond around the bulged G4319. Second, it has also been suggested that residues from Trp 51 to Gly 55 in loop 2 and residues from loop 5 interact with the ribosome at the conserved GAGA tetraloop (4323–4326) which is about 15 Å away from the bulged guanine (4319). Ribonucleolytic assays against a reticulocyte lysate showed that Δ(7-22) was unable to specifically cleave the phosphodiester bond in the ribosome; consequently, a third segment of α-sarcin, the N-terminal β-hairpin, was suggested to directly interact with some region of the ribosome (Hwu et al. 2000; García-Ortega et al. 2002). Ribonucleolytic assays against other substrates lacking the characteristic structural elements of native ribosomes, such as naked rRNA, SRL, poly(A), and small dinucleotides show that the mutant is able to more efficiently hydrolyze these ligands (García-Ortega et al. 2002).
The analysis of the present structure sheds new light on the lack of specific activity of the N-terminal deletion mutant and the increase of its nonspecific ribonucleolytic activity. It has been shown that the distance between bulged G4319 and the hydrolytic site G4325-A4326 in the substrate is critical, and that ribotoxin cleavage takes place at a fixed position in space separated from the recognition base G4319 (Glück and Wool 1996). To acquire their extreme specificity, ribotoxins appear to have evolved separate binding sites for recognizing each of these structural features of the SRL and the distances between them. In the mutant, the larger distances between lysines in loop 3 and the active site may lead to a mistaken positioning of the substrate with respect to the catalytic groups. Crystal structures of some restrictocin-inhibitor complexes have shown that ribotoxins can bind a substrate analog nonproductively (Yang et al. 2001). In these misdocked structures, the SRL analog does not sit as deeply in the active site as the bound structure does. This suggests that both substrate binding sites can be recognized and bound independently by the enzyme, although correct binding of both sites is needed for specific efficient catalysis. In the same way, it is possible that the G4319 base of the ribosome could be recognized by Δ(7-22) through lysines in loop 3 (as in the wild type), but the larger distances from these residues to the active-site groups and the lack of the N-terminal hairpin’s interactions with other parts of the ribosome hinder the formation of the productive complex and, consequently, abolish the hydrolysis of the SRL. Especially relevant is the structural context of the specific cleavage for the SRL in the ribosome, where the conformation and not the base sequence is the key determinant. In fact, the bulged guanine can be replaced by adenine and the structure of the SRL is retained, maintaining cell viability in vivo (Seggerson and Moore 1998). As the conformation is so important to the ribosome, it is reasonable that the same structural features should be critical for recognition by α-sarcin. Therefore, minor modifications in the α-sarcin structure dramatically affect its interaction with these features in the substrate and prevent cleavage.
In order to analyze the basis of the activity against substrates distinct from the ribosome, only the structural modifications of the enzyme should be taken into account. In these cases, as described above, the probable poorer fit of the ligand in the active site of the α-sarcin mutant results in an increase of the unspecific RNase activity.
On the other hand, the increase of nonspecific ribonucleolytic activity could also be related to the greater flexibility of the mutant’s active site, and the different electrostatic properties of the catalytic residues. It is well known that the distinct catalytic roles played by the active-site residues in each step of the transesterification reaction depend intimately upon their ionization states. Predictable changes in the His 137 pKa value in Δ(7-22) might affect the dependence of its activity on pH. The broader pH range of enzymatic activity of RNase T1 compared to α-sarcin was explained on the basis of the larger difference in the pKa values of the former’s catalytic residues (Pérez-Cañadillas et al. 1998; Kao and Davies 2000). The structural analysis of the α-sarcin deletion mutant leads us to foresee a shift in the pKa of His 137 to higher values, increasing the difference between the pKa values of the acid-base pair, which together with the larger accessibility and flexibility of the active site could explain the higher unspecific activity. In these terms, the mutant could be considered a hybrid between ribotoxins and noncytotoxic ribonucleases, with biological properties intermediate between these two families of ribonucleases.
Conclusions
This work focused on the NMR structural characterization of an engineered deletion variant of the protein α-sarcin removing one of the regions showing larger structural differences compared to the noncytotoxic ribonuclease structures: α-sarcin’s N-terminal β-hairpin. The deletion of α-sarcin’s N-terminal β-hairpin fragment, which is absent at the N-terminal ends of ribonucleases T1 and U2, generates a protein with altered enzymatic activity, making it unable to specifically cleave the ribosome while increasing its nonspecific ribonuclease activity. It also altered its global dynamical properties, being more similar to those described for RNase T1. We conclude that the larger flexibility of loop 5, the greater flexibility and the new electrostatic properties of the catalytic histidine resulting from the disruption of the interactions between loop 5 and the deleted β-hairpin, and the relative orientation of loop 3 with respect to the active site explain the modified biological function of the mutant.
Materials and methods
Design of the deletion mutant Δ(7-22) of α-sarcin
The mutant Δ(7-22) was designed to study the direct and/or indirect implications of the long N-terminal β-hairpin in the specificity and cytotoxicity of α-sarcin (García-Ortega et al. 2002). Thus, the sequence from Leu 7 to Arg 22 of the wild-type protein α-sarcin was deleted and substituted by a pair of glycine residues. This is the shortest sequence which can link the two remainder strands of the N-terminal β-sheet; moreover, two glycine residues occupy equivalent structural positions in RNase U2, the ribonuclease most closely related to α-sarcin (Mancheño et al. 1995b). The deletion mutant was cloned according to standard methods, produced in E. coli cells, and purified as described (García-Ortega et al. 2002).
NMR samples and spectra assignment
NMR samples were prepared by dissolving the mutant variant in aqueous solutions of either D2O or 90% H2O/10% D2O to a final concentration of 2 mM. DSS was used as the internal chemical shift reference. Two sets of NOESY and TOCSY spectra were recorded at pH 6.0 and 35°C with standard pulse sequences to carry out the assignment process. One set of experiments was acquired in 90% H2O/10% D2O on a Bruker AV-800 spectrometer. The other set, acquired in D2O on a Bruker AV-600 spectrometer, was helpful for the assignment of proton resonances near the solvent signal and allowed the discrimination of different degrees of protection of amide protons from solvent exchange. All of the spectra were processed using the XwinNMR/Bruker software package and analyzed with the ANSIG program (Kraulis 1989). The assignment of proton resonances was performed by the standard sequential assignment methodology from NOEs (Wüthrich 1986) facilitated by comparison with the corresponding spectra of the native α-sarcin acquired under identical experimental conditions (Campos-Olivas et al. 1996).
Structure calculations
The calculation of the 3D structure of the α-sarcin Δ(7-22) mutant in solution was performed with the program DYANA (Güntert et al. 1997) in two steps. Experimental NOEs from NOESY spectra recorded in H2O and D2O solutions were translated into upper distance restraints using the calibrating functions implemented in CALIBA within the DYANA package, with the default values for the weighting parameters. Distance restrictions from the two disulphide bridges in the molecule, Cys 6-Cys 148 and Cys 76-Cys 132 (α-sarcin numbering) were also introduced. Stereospecific assignments were generated from the analysis of the preliminary calculated structures with the program GLOMSA. Similarly, NOEs from the aromatic protons of the fast-flipping Phe and Tyr residues were assigned to one or other side of the ring. The structures determined with these constraints were compared to those of wild-type α-sarcin (WT) to identify regions that maintain the wild-type structure in the mutant. φ-Angle restraints derived from measured scalar couplings in the native protein (Pérez-Cañadillas et al. 2000) were introduced after checking that the backbone atoms in these structurally equivalent regions also shared equivalent torsion angle ranges. DYANA calculations were started from 200 random structures. First, a simulated annealing with a total of 4000 molecular dynamics (MD) steps, starting with 800 MD steps at temperature 8 followed by slow cooling down during 3200 MD steps to a final temperature of 0 was performed (temperature is measured in target function units per degree of freedom). Then, 1000 steps of conjugate gradient minimization were added. Finally, in order to further relax overlaps and constraint violations, several refinement steps were introduced following the main structure determination. They were performed with 111 conjugate minimization steps and 800 steps of simulated annealing, starting at a temperature of 0.01 and descending to a final temperature of 0.
The 25 conformers with the lowest target function values were energy minimized with the program AMBER 7 (Case et al. 2002) using a 500-cycle run of minimization based on 10 cycles of steepest descent minimization and 490 cycles of conjugate gradient minimization. The cut-off distance was 12.0 Å. No periodic boundary was applied. NMR upper distance restraints and stereo-specific assignments were introduced in the minimization run. The NMR restraint energy terms were considered with a fixed relative weight of 1. The coordinates of this ensemble were deposited in the RCSB Protein Data Bank under the accession code 1R4Y. The quality of the final 25 energy-minimized conformers was checked with the program PROCHECK_NMR (Laskowski et al. 1996). These conformers were visualized and globally characterized with the program MOLMOL (Koradi et al. 1996).
Dynamic and hydrodynamic calculations
A contact model (Zhang and Brüschweiler 2002) was used to predict N-H dynamic order parameters (S2) from the 3D NMR structures of the WT and Δ(7-22) mutant. The model allows the rapid estimation of the magnitude of fast time-scale backbone dynamics from the representative ensemble of high-resolution NMR structures. The data presented in this work are the result of the ensemble-averaging of the predicted values calculated for each of the 25 energy-minimized conformers of the WT and Δ(7-22).
Hydrodynamic calculations were performed with the program HYDRONMR (García de la Torre et al. 2000) with the PDB files of the 25 conformers describing the NMR structure of the mutant. We used the conditions that reproduce the experimental correlation time determined for the WT α-sarcin by NMR methods (Pérez-Cañadillas et al. 2002): temperature 308 K, solvent viscosity 0.72 cp, and atomic element radius 2.9 Å (including the thickness of the hydration layer).
Time-resolved tryptophan fluorescence anisotropy measurements (TRFA)
This technique was used to experimentally determine the global rotational correlation time of Δ(7-22), which is directly related to the hydrated volume of macromolecules. Time-resolved fluorescence intensity and depolarization measurements were performed by the time-correlated single photon counting technique. The experimental setup was similar to that described (Lillo et al. 2002). In brief, the tryptophan fluorophores of the enzyme (Trp 4 and Trp 51) were excited by means of vertically polarized light pulses from a Ti-sapphire picosecond laser (Tsunami, Spectra Physics), pumped with a 5W Nd:YVO4 laser (Millennia, Spectra Physics), and associated to a third harmonic generator tuned at 297 nm. Pulses of 1–2 μsec width were used with a repetition rate of 4 MHz, and ~20 μW of average power at the samples. The experimental samples (120 μL) were placed in thermostated 3×3 mm path length cuvettes. Data were stored in 4K channels, with a resolution of 6.6 psec/channel, up to ~5 × 106 total counts. The decay of the total fluorescence intensity, I(t)=Im(t), was recorded with the emission polarizer set at the magic angle (m = 54.7°), relative to the vertically polarized excitation beam. The two components of the fluorescence, polarized parallel Ivv(t) and perpendicular Ivh(t) to the plane of polarization of the excitation beam, were sequentially recorded by alternating the orientation of the emission polarizer every 2 min. Time-resolved anisotropy parameters were obtained from the simultaneous analysis of the parallel and perpendicular polarized decays, using the Globals Unlimited analysis program. Anisotropy decays were found to be adequately described by a monoexponential function, r(t) = r(0)exp(−t/φG), where r(0) is the zero-time limiting anisotropy, and φG is the overall global protein motion.
Samples were prepared at pH 7.0 by dissolving the protein in 50 mM sodium phosphate buffer, containing 0.1 M NaCl. Their final concentrations ranged from 3 to 5 μM as estimated by absorbance readings using the absorption coefficient (E2800.1%(Δ(7-22)) = 1.38). Measurements were performed at 35°C.
Acknowledgments
This work was supported by grants BIO2002-720 from the Ministerio de Ciencia y Tecnología, BMC2000-0551 from the Ministerio de Educación y Cultura, and BQU2000-1500 from the Dirección General de Enseñanza Superior e Investigación (Spain). F.G.-M. and L.G.-O. are recipients of fellowships from the Comunidad Autónoma de Madrid, and Ministerio de Educación y Cultura (Spain), respectively. M.P. L. thanks Dr. A.U. Acuña for his help with the laser time-resolved instrument. We thank Dr. M. Gairi (Unitat RMN, Parc Cientific de Barcelona) for technical assistance during data acquisition at 800 MHz.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
Δ(7-22), the α-sarcin deletion mutant in which residues 7–22 have been removed and replaced by a pair of glycine residues
TRFA, time-resolved fluorescence anisotropy
SRL, sarcin ricin loop
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03532204.
References
- Beechem, J.M. 1992. Global analysis of biochemical and biophysical data. Methods Enzymol. 210 37–54. [DOI] [PubMed] [Google Scholar]
- Beechem, J.M., Gratton, E., Ameloot, M., Knutson, J.R., and Brand, L. 1991. Topics in fluorescence spectroscopy: Principles II (ed. J.R. Lakowicz), pp. 241–305. Plenum, New York.
- Campos-Olivas, R., Bruix, M., Santoro, J., Martínez del Pozo, A., Lacadena, J., Gavilanes, J.G., and Rico, M. 1996. 1H and 15N nuclear magnetic resonance assignment and secondary structure of the cytotoxic ribonuclease α-sarcin. Protein Sci. 5 969–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D.A., Pearlman, D.A., Caldwell, J.W., Cheatham III, T.E., Wang, J., Ross, W.S., Simmerling, C.L., Darden, T.A., Merz, K.M., Stanton, R.V., Cheng, A.L., et al. 2002. AMBER 7. University of San Francisco, CA.
- Chen, D.T., and Lin, A. 2002. Domain swapping in ribonuclease T1 allows the acquisition of double-stranded activity. Protein Eng. 15 997–1003. [DOI] [PubMed] [Google Scholar]
- Damberg, P., Jarvet, J., Allard, P., Mets, U., Rigler, R., and Graslund, A. 2002. (13)C-(1)H NMR relaxation and fluorescence anisotropy decay study of tyrosine dynamics in motilin. Biophys. J. 83 2812–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Antonio, C., Martínez del Pozo, A., Mancheño, J.M., Oñaderra, M., Lacadena, J., Martínez-Ruiz, A., Pérez-Cañadillas, J.M., Bruix, M., and Gavilanes, J.G. 2000. Assignment of the contribution of the tryptophan residues to the spectroscopic and functional properties of the ribotoxin α-sarcin. Proteins 41 350–361. [DOI] [PubMed] [Google Scholar]
- Endo, Y. and Wool, I.G. 1982. The site of action of α-sarcin on eukaryotic ribosomes. The sequence at the α-sarcin cleavage site in 28 S ribosomal ribonucleic acid. J. Biol. Chem. 257 9054–9060. [PubMed] [Google Scholar]
- Fushman, D., Ohlenschlager, O., and Ruterjans, H. 1994a. Determination of the backbone mobility of ribonuclease T1 and its 2′GMP complex using molecular dynamics simulations and NMR relaxation data. J. Biomol. Struct. Dyn. 11 1377–1402. [DOI] [PubMed] [Google Scholar]
- Fushman, D., Weisemann, R., Thüring, H., and Rüterjans, H. 1994b. Backbone dynamics of ribonuclease T1 and its complex with 2′GMP studied by two-dimensional heteronuclear NMR spectroscopy. J. Biomol. NMR 4 61–78. [DOI] [PubMed] [Google Scholar]
- García de la Torre, J., Huertas, M.L., and Carrasco, B. 2000. HYDRONMR: Prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson. 147 138–146. [DOI] [PubMed] [Google Scholar]
- García-Mayoral, M.F., Pérez-Cañadillas, J.M., Santoro, J., Ibarra, B., Sánchez-Ruiz, J.M., Lacadena, J., Martínez del Pozo, A., Gavilanes, J.G., Rico, M., and Bruix, M. 2003. Dissecting structural and electrostatic interactions of charged groups in α-sarcin. An NMR study of some mutants involving the catalytic residues. Biochemistry 42 13122–13133. [DOI] [PubMed] [Google Scholar]
- García-Ortega, L., Masip, M., Mancheño, J.M., Oñaderra, M., Lizarbe, M.A., García-Mayoral, M.F., Bruix, M., Martínez del Pozo, A., and Gavilanes, J.G. 2002. Deletion of the NH2-terminal β-hairpin of the ribotoxin α-sarcin produces a nontoxic but active ribonuclease. J. Biol. Chem. 277 18632–18639. [DOI] [PubMed] [Google Scholar]
- Gasset, M., Martínez del Pozo, A., Oñaderra, M., and Gavilanes, J.G. 1989. Study of the interaction between the antitumour protein α-sarcin and phospholipid vesicles. Biochem. J. 258 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück, A. and Wool, I.G. 1996. Determination of the 28 S ribosomal RNA identity element (G4319) for α-sarcin and the relationship of recognition to the selection of the catalytic site. J. Mol. Biol. 256 838–848. [DOI] [PubMed] [Google Scholar]
- Glück, A., Endo, Y., and Wool, I.G. 1994. The ribosomal RNA identity elements for ricin and for α-sarcin: Mutations in the putative CG pair that closes a GAGA tetraloop. Nucleic Acids Res. 22 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güntert, P., Mumenthaler, C., and Wüthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273 283–298. [DOI] [PubMed] [Google Scholar]
- Hwu, L., Huang, K., Chen, D., and Lin, A. 2000. The action mode of the ribosome-inactivating protein α-sarcin. J. Biomed. Sci. 7 420–428. [DOI] [PubMed] [Google Scholar]
- Kao, R. and Davies, J. 1995. Fungal ribotoxins: A family of naturally engineered targeted toxins? Biochem. Cell Biol. 73 1151–1159. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Molecular dissection of mitogillin reveals that the fungal ribotoxins are a family of natural genetically engineered ribonucleases. J. Biol. Chem. 274 12576–12582. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Single amino acid substitutions affecting the specificity of the fungal ribotoxin mitogillin. FEBS Lett. 466 87–90. [DOI] [PubMed] [Google Scholar]
- Koizumi, K., Lintas, C., Nirenberg, M., Maeng, J.S., Ju, J.H., Mack, J.W., Gruschus, J.M., Odenwald, W.F., and Ferretti, J.A. 2003. Mutations that affect the ability of the vnd/NK-2 homeoprotein to regulate gene expression: Transgenic alterations and tertiary structure. Proc. Natl. Acad. Sci. 100 3119–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 29–32, 51–55. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1989. ANSIG: A program for the assignment of protein 1H 2D NMR spectra by interactive computer graphics. J. Magn. Reson. 24 627–633. [Google Scholar]
- Laskowski, R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Lillo, M.P., Cañadas, O., Dale, R.E., and Acuña, A.U. 2002. Location and properties of the taxol binding center in microtubules: A picosecond laser study with fluorescent taxoids. Biochemistry 41 12436–12449. [DOI] [PubMed] [Google Scholar]
- Lipari, G. and Szabo, A. 1982. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104 4546–4559. [Google Scholar]
- Mancheño, J.M., Gasset, M., Albar, J.P., Lacadena, J., Martínez del Pozo, A., Oñaderra, M., and Gavilanes, J.G. 1995a. Membrane interaction of a β-structure-forming synthetic peptide comprising the 116–139th sequence region of the cytotoxic protein α-sarcin. Biophys. J. 68 2387–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancheño, J.M., Gasset, M., Lacadena, J., Martínez del Pozo, A., Oñaderra, M., and Gavilanes, J.G. 1995b. Predictive study of the conformation of the cytotoxic protein α-sarcin: A structural model to explain α-sarcin -membrane interaction. J. Theor. Biol. 172 259–267. [DOI] [PubMed] [Google Scholar]
- Martínez-Ruiz, A., Kao, R., Davies, J., and Martínez del Pozo, A. 1999. Ribotoxins are a more widespread group of proteins within the filamentous fungi than previously believed. Toxicon 37 1549–1563. [DOI] [PubMed] [Google Scholar]
- Nayak, S.K., Shveta, and Batra, J.K. 2000. Localization of the catalytic activity in restrictocin molecule by deletion mutagenesis. Eur. J. Biochem. 267 1777–1783. [DOI] [PubMed] [Google Scholar]
- Nayak, S.K., Bagga, S., Gaur, D., Nair, D.T., Salunke, D.M., and Batra, J.K. 2001. Mechanism of specific target recognition and RNA hydrolysis by ribonucleolytic toxin restrictocin. Biochemistry 40 9115–9124. [DOI] [PubMed] [Google Scholar]
- Olmo, N., Turnay, J., González de Buitrago, G., López de Silanes, I., Gavilanes, J.G., and Lizarbe, M.A. 2001. Cytotoxic mechanism of the ribotoxin α-sarcin. Induction of cell death via apoptosis. Eur. J. Biochem. 268 2113–2123. [DOI] [PubMed] [Google Scholar]
- Olson, B.H. and Goerner, G.L. 1965. α-sarcin, a new antitumor agent. I. Isolation, purification, chemical composition, and the identity of a new amino acid. Appl. Microbiol. 13 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Cañadillas, J.M., Campos-Olivas, R., Lacadena, J., Martínez del Pozo, A., Gavilanes, J.G., Santoro, J., Rico, M., and Bruix, M. 1998. Characterization of pKa values and titration shifts in the cytotoxic ribonuclease α-sarcin by NMR. Relationship between electrostatic interactions, structure, and catalytic function. Biochemistry 37 15865–15876. [DOI] [PubMed] [Google Scholar]
- Pérez-Cañadillas, J.M., Santoro, J., Campos-Olivas, R., Lacadena, J., Martínez del Pozo, A., Gavilanes, J.G., Rico, M., and Bruix, M. 2000. The highly refined solution structure of the cytotoxic ribonuclease α-sarcin reveals the structural requirements for substrate recognition and ribonucleolytic activity. J. Mol. Biol. 299 1061–1073. [DOI] [PubMed] [Google Scholar]
- Pérez-Cañadillas, J.M., Guenneugues, M., Campos-Olivas, R., Santoro, J., Martínez del Pozo, A., Gavilanes, J.G., Rico, M., and Bruix, M. 2002. Backbone dynamics of the cytotoxic ribonuclease α-sarcin by 15N NMR relaxation methods. J. Biomol. NMR 24 301–316. [DOI] [PubMed] [Google Scholar]
- Pfeiffer, S., Karimi-Nejad, Y., and Ruterjans, H. 1997. Limits of NMR structure determination using variable target function calculations: Ribonuclease T1, a case study. J. Mol. Biol. 266 400–423. [DOI] [PubMed] [Google Scholar]
- Seggerson, K. and Moore, P.B. 1998. Structure and stability of variants of the sarcin-ricin loop of 28S rRNA: NMR studies of the prokaryotic SRL and a functional mutant. RNA 4 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortle, D. and Sondek, J. 1995. The emerging role of insertions and deletions in protein engineering. Curr. Opin. Biotechnol. 6 387–393. [DOI] [PubMed] [Google Scholar]
- Szewczak, A.A. and Moore, P.B. 1995. The sarcin/ricin loop, a modular RNA. J. Mol. Biol. 247 81–98. [DOI] [PubMed] [Google Scholar]
- Szewczak, A.A., Moore, P.B., Chang, Y.L., and Wool, I.G. 1993. The conformation of the sarcin/ricin loop from 28S ribosomal RNA. Proc. Natl. Acad. Sci. 90 9581–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda, E., Bi, X., Yoshinari, S., and Endo, Y. 1997. Mechanism of substrate recognition by the ribotoxin, α-sarcin. Nucleic Acids Symp. Ser. 37 131–132. [PubMed] [Google Scholar]
- Wool, I.G. 1984. The mechanism of action of the cytotoxic nuclease α-sarcin and its use to analyse ribosome structure. Trends Biochem. Sci. 9 14–17. [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids, pp. 117–199. Wiley, New York.
- Yang, X. and Moffat, K. 1996. Insights into specificity of cleavage and mechanism of cell entry from the crystal structure of the highly specific Aspergillus ribotoxin, restrictocin. Structure 4 837–852. [DOI] [PubMed] [Google Scholar]
- Yang, X., Gerczei, T., Glover, L.T., and Correll, C.C. 2001. Crystal structures of restrictocin-inhibitor complexes with implications for RNA recognition and base flipping. Nat. Struct. Biol. 8 968–973. [DOI] [PubMed] [Google Scholar]
- Zhang, F. and Brüschweiler, R. 2002. Contact model for the prediction of NMR N-H order parameters in globular proteins. J. Am. Chem. Soc. 124 12654–12655. [DOI] [PubMed] [Google Scholar]